

Summary
The cardiac outflow tract (OFT) is a developmentally complex structure derived from multiple lineages and is often defective in human congenital anomalies. While emerging evidence shows that the fibroblast growth factor (FGF) is essential for OFT development, the downstream pathways mediating FGF-signaling in cardiac progenitors remain poorly understood. Here, we report that FRS2α, an adaptor protein that links FGF receptor kinases to multiple signaling pathways, mediates critical aspects of FGF-dependent OFT development. Ablation of Frs2α in mesodermal OFT progenitor cells that originate in the second heart field (SHF) affects their expansion into the OFT myocardium, resulting in OFT misalignment and hypoplasia. Moreover, Frs2α mutants had defective endothelial-mesenchymal-transition and neural crest cell recruitment into the OFT cushions, resulting in OFT septation defects. The results provide new insight into the signaling molecules downstream of FGF receptor tyrosine kinases in cardiac progenitors.
Keywords: receptor tyrosine kinase, cell signaling, heart development, second heart field, mouse model
Introduction
Congenital heart disease (CHD) represents the most common birth defect, affecting nearly 1% of newborns. Since more than 30% of CHD patients have OFT defects (Rosamond et al., 2007), understanding how OFT development is controlled at the molecular level is of great interest. The OFT initially develops as an unseptated myocardial tube lined with endothelial cells, the endocardium. The OFT myocardium is comprised of cells deployed from a population of mesodermal cells residing in the pharyngeal and splanchnic mesoderm called the SHF after formation of the primary heart tube (Buckingham et al., 2005; Srivastava, 2006). The OFT then undergoes dramatic remodeling and is divided into the right ventricular/pulmonary and left ventricular/aortic outflows at the arterial pole by the fusion and remodeling of the OFT cushions (Lamers and Moorman, 2002). This is a critical process for separating the pulmonary and systemic circulations postnatally. First, OFT myocardial cells secrete extracellular matrix molecules to form the OFT cushions. Subsequently, the matrix is invaded by cells from two sources: OFT endocardial cells that undergo an endothelial-mesenchymal-transition (EMT), and neural crest cells (NCCs) that migrate from pharyngeal arches 3, 4 and 6. Disruption of either endocardial EMT or the contribution of NCCs to the OFT cushions can cause OFT defects (Armstrong and Bischoff, 2004; Hutson and Kirby, 2003; Moon et al., 2006).
The FGF family of regulatory polypeptides controls a broad spectrum of normal cellular processes during development and through adulthood (Eswarakumar et al., 2005; McKeehan et al., 1998). Emerging evidence demonstrates that several FGF ligands are involved in OFT development. Fgf8 and Fgf10 have been shown to be expressed in a temporally- and spatially-specific manner in the SHF progenitor cells residing in the splanchnic mesoderm (SM) and pharyngeal mesoderm core. Fgf8 is also expressed in the pharyngeal ectoderm and endoderm. Fgf15 is expressed in the pharyngeal endoderm (PE) and pharyngeal mesenchyme. Tissue-specific ablation of Fgf8 disrupts OFT remodeling, resulting in both alignment and septation defects (Ilagan et al., 2006; Park et al., 2006). Fgf15 loss-of-function also causes OFT alignment defects (Vincentz et al., 2005). Furthermore, the FGF signaling axis genetically interacts with Tbx1 pathways that regulate development of OFT and pharyngeal arch derivatives in a tissue-specific manner (Aggarwal et al., 2006; Vitelli et al., 2006) Disruption of Tbx1 function is associated with DiGeorge and other syndromes associated with the microdeletion of chromosome 22q11.2 (Lindsay et al., 2001).
The FGFs exert their regulatory activity by activating FGF receptor (FGFR) tyrosine kinases encoded by four highly homologous genes, in partnership with peri-cellular matrix heparan sulfate proteoglycans (McKeehan et al., 1998; Ornitz, 2000). Thus far, only a few intracellular signaling molecules have been shown to bind to FGFRs directly. These include phospholipase Cγ (Mohammadi et al., 1992; Peters et al., 1992), Crk (Larsson et al., 1999), Crkl (Moon et al., 2006), FRS2α, and FRS2β (Kouhara et al., 1997; Ong et al., 1996). FRS2α, also designated as SNT1, is a proximal-interactive adaptor protein that has 6 tyrosine phosphorylation sites that are phosphorylated by FGFRs upon activation by FGF ligands. Among them, phosphorylated Y196, Y306, Y349, and Y392 are Grb2 binding sites that link FGFR kinases to the PI-3 kinase pathway; phosphorylated Y436 and Y471 are Shp2 binding sites that link FGFR kinases to the MAP kinase pathway (Kouhara et al., 1997; Ong et al., 2000; Zhang et al., 2007). Deleting the FRS2α-binding VT (valine and threonine) dipeptide motif in the intracellular juxtamembrane domain of FGFR1 in mice leads to defects in multiple organs (Hoch and Soriano, 2006). In addition to FGFRs, several other receptor tyrosine kinases have been reported to phosphorylate FRS2α, although the roles of FRS2α in these signaling pathways remain not well defined (Avery et al., 2007; Ong et al., 2000).
Frs2α is expressed in mouse embryos during early embryogenesis and almost ubiquitously in all fetal and adult tissues (McDougall et al., 2001). Complete disruption of Frs2α function abrogates FGF-induced activation of MAP and PI-3 kinases, chemotactic responses, and cell proliferation, and also causes embryonic lethality at embryonic day (E) 7-E7.5 (Hadari et al., 2001). Mice carrying mutations in the two Shp2 binding sites exhibit a variety of developmental defects in many organs, while mice carrying mutations in the four Grb2 binding sites generally have less severe phenotypes (Yamamoto et al., 2005).
In order to circumvent the early embryonic lethality resulting from Frs2α ablation and examine the roles of FRS2α-mediated signals in heart morphogenesis, we employed the LoxP/Cre recombination system to tissue-specifically inactivate Frs2α alleles in heart progenitor cells. Ablation of Frs2α in the SHF caused OFT misalignment, including overriding aorta (OA) and double outlet right ventricle (DORV), by compromising SHF progenitor cell proliferation, therefore reducing contribution of the SHF-derived mesodermal cells to the OFT myocardium. In addition, ablation of Frs2α in the OFT myocardium increased VEGF expression and disrupted endocardial EMT. Deletion of Frs2α in the SHF and PE reduced Bmp4 expression and disrupted NCC invasion of the OFT cushions, resulting in persistent truncus arteriosus (PTA), a severe OFT septation defect. Furthermore, double ablation of Fgfr1 and Fgfr2 phenocopied ablation of Frs2α. The results suggest that FRS2α-mediated signals in OFT myocardial cells promote endocardial EMT by downregulating VEGF expression, whereas in SM and PE, FRS2α-mediated signals upregulate BMP4 expression and promote NCCs contribution to the OFT. Together, the results delineate a molecular mechanism underlying how FGF elicits tissue-specific signals to regulate OFT development.
Experimental Procedures
Animals
All animals were housed in the Program for Animal Resources of the Institute of Biosciences & Technology, and all experimental procedures were approved by the Institutional Animal Care and Use Committee. The mice carrying Frs2αflox, Fgfr1flox, Fgfr2flox, Nkx2.5Cre knock-in alleles, Mef2ccre, Wnt1Cre, and Tie2cre transgenic alleles were bred and genotyped as described (Danielian et al., 1998; Kisanuki et al., 2001; Lin et al., 2007b; Moses et al., 2001; Trokovic et al., 2003; Verzi et al., 2005; Yu et al., 2003). All mice are on a mixed background (including C57/BL6, SV129, and ICR) and all embryos arising from each cross are outbred. The hearts were excised at the stages indicated in the text, fixed with 4% paraformaldehyde-PBS for 4 hours, and paraffin embedded. The sections were re-hydrated and H&E stained for histological analyses.
Immunohistostaining
Immunostaining was performed on 5 μm sections mounted on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA). The antigens were retrieved by incubation in the citrate buffer (10 mM) for 20 minutes at 100 °C or as suggested by manufacturers of the antibodies. The source and concentration of primary antibodies are: mouse anti-AP2α (1:10 dilution) and anti-Isl1 (1:500 dilution) from Development Studies Hybridoma Bank; anti-pERK (1:100 dilution), anti-pAKT (1:100 dilution) and anti-phosphorylated Smad1/5/8 (1:500 dilution) from cell Signaling (Danvers, MA); anti-FRS2α (1:100 dilution), anti-NFATc1 (1:100 dilution), anti-phosphorylated histone 3 (1:100 dilution), anti-VEGF-A (1:100 dilution), and anti-PECAM (1:100 dilution) from Santa Cruz (Santa Cruz, CA). The specifically bound antibodies were detected with HRP-conjugated secondary antibody (Bio-Rad Co., Hercules, CA) and visualized using TSA™ Plus Fluorescence Systems from PerkinElmer (Boston, MA) on a Zeiss LSM 510 Confocal Microscope. For TUNEL assays, tissues were fixed and sectioned, and the apoptotic cells were detected with the ApopTag Peroxidase In Situ Kit from Chemicon, Inc. (Temecula, CA).
In situ hybridization
Whole-mount in situ hybridization was performed as previously described (Edmondson et al., 1994). Briefly, after fixation in 4% paraformaldehyde-PBS overnight, embryos were treated with 10 μg/ml protease K for 5-15 minutes at room temperature, and post fixed with 4% paraformaldehyde-PBS for 20 minutes. After prehybridization at 70 °C for 2 hours, the hybridization was carried out by overnight incubation at 70 °C. Following the hybridization, embryos were washed with the TBST solution (0.25 M Tris-HCl buffered saline, PH 7.5, 0.01% Triton-X100, 2 mM Levamisole), blocked with 10% sheep serum in TBST, and then rocked overnight at 4 °C in a 1/4000 dilution of alkaline phosphatase conjugated anti-digoxigenin antibody (Roche, Indianapolis, IN) in blocking buffer. After being washed 8 times with TBST, specifically bound antibodies were visualized by alkaline phosphatase staining. The embryos were post hybridization fixed with 4% paraformaldehyde-PBS. At least three mutants and three control embryos were analyzed for each probe.
Short-term mouse embryo culture
Short-term mouse embryo culture was performed as previously described (Grego-Bessa et al., 2007). Briefly, E8.5 mouse embryos were dissected in PBS and cultured in 12-well plates containing 1% agarose to avoid embryo attachment; 1 ml DMEM medium containing 50% FBS and antibiotics (penicillin and streptomycin, 100 U/ml each) was added on top of the agarose. Inhibitors for pERK and PI3K (LY294002, Calbiochem, Darmstadt, Germany) were added to the media to a final concentration of 10 μM. Embryos were cultured in a 5% CO2 incubator at 37 °C for 16 hours. The embryos were then dissected in PBS, and fixed in 4% paraformaldehyde-PBS for further analyses.
Results
Ablation of Frs2α in the second heart field and pharyngeal endoderm
Expression of Frs2α in the cardiac mesoderm could be detected as early as at 0 somite stage (ss) by immunostaining (Fig. 1A). To investigate FRS2α function in heart development, we ablated Frs2α in cardiac progenitors by crossing mice bearing the Frs2α conditional null (Frs2αflox) allele (Lin et al., 2007b) and mice carrying the Nkx2.5cre knock-in allele (Moses et al., 2001) or the Mef2ccre transgene (Verzi et al., 2005). Nkx2.5Cre is expressed at 0 somite stage (ss) and directs Cre activity in the first heart field (FHF), SHF, and the PE. Mef2ccre is active at 0-1 ss in the specified SHF, but not the FHF and PE.
Fig.1. Diminished Frs2α expression in Frs2αcn outflow tract and SHF.

Expression of Frs2α in 0 somite stage (ss) (A) or E9.5 (B) embryos was assessed by immunostaining with anti-FRS2α antibody (green) on paraffin sections. Nuclei were stained with To-Pro3 (red). a1-3, b1-3, and c1-3 were high magnification views of the boxed areas in panels a, b, and c, respectively. Note that in Frs2αcn/Nkx mutants, Frs2α expression was diminished in ventricular myocardium (V-M), OFT myocardium (OFT-M) and endocardium (OFT-E), pharyngeal endoderm (PE), and splanchnic mesoderm (SM). In Frs2αcn/Mef mutants, Frs2α expression was disrupted in V-M, OFT-M, and SM, but was intact in the OFT-E and PE.
At E9.5, Frs2α was highly expressed in the PE and SM (Fig. 1Ba). In Nkx2.5cre;Frs2αf/f embryos (designated Frs2αcn/Nkx), Frs2α expression was efficiently disrupted in the OFT myocardium and endocardium, SM, and PE, as well as the atrial and ventricular myocardium of E9.5 embryos (Fig. 1Bb). In Mef2ccre;Frs2αf/f (Frs2αcn/Mef) embryos, disruption of Frs2α expression was complete in the SM, OFT myocardium, and right ventricle myocardium (Fig. 1Bc), mosaically in the OFT endocardium. Detailed timecourse analyses showed that the FRS2α protein level was significantly reduced by the 8-9 ss in Frs2αcn/Nkx embryos, and by 11-12 ss in Frs2αcn/Mef embryos (Supplemental Fig. 1), which is consistent with the relatively delayed activity of the Mef2ccre driver (Verzi et al., 2005). Together, these data indicate that the Frs2αcn/Nkx embryos had an efficient and widespread Frs2α deletion, while the Frs2αcn/Mef embryos had a more restricted and delayed Frs2α deletion. These two different conditional mutants provide an opportunity to dissect the tissue-specific roles of FRS2α in the OFT.
The two classes of Frs2α conditional mutants have disrupted OFT morphogenesis
The majority of Frs2αcn/Nkx or Frs2αcn/Mef embryos died neonatally with severe OFT malformations (Table 1-3). The few surviving neonates died within 3 weeks after birth. We examined hearts from E14.5 fetuses and found that both Frs2αcn/Nkx and Frs2αcn/Mef hearts exhibited OFT alignment defects, including OA (Fig. 2a-c) and DORV (Fig. 2d-f). Frequently, the Frs2αcn/Nkx OFT was completely unseptated or partially septated into aorta and pulmonary artery (Fig. 2h), which is classified PTA. In contrast, none of the Frs2αcn/Mef fetuses exhibited septation defects, suggesting that FRS2α-mediated signaling in the OFT myocardium and SM within the Mef2ccre lineage is not obligatory for OFT septation.
Table 1.
Survival analyses of Frs2αcn/Nkx embryos and postnatal pups
| Age | Total | Frs2αcn/Nkx | Expected |
|---|---|---|---|
| E8.5-E17.5 | 986 | 247 | 247 |
| Postnatal | 186 | 33* | 47 |
20 died perinatally, 6 died neonatally, 7 survived for more than 2 weeks.
Table 3.
Comparison of cardiovascular defects (E14.5-P1)
| Type of Defects | Frs2αcn/Nkx (n=29) | Frs2αcn/Mef (n=12) | Fgfr1cn/Nkx (n=14#) | Fgfr2cn/Nkx (n=8) | Fgfr1/r2cn/Nkx (n=7) |
|---|---|---|---|---|---|
| OA or DORV | 16 | 5 | 0 | 3 | 5 |
| TA | 11 | 0 | 0 | 0 | 4 |
| VSD | 19 | 3 | 1 | 5 | 6 |
7 are R1f/fR2f/wNkx2.5cre,
Fig. 2. OFT alignment and septation defects in Frs2αcn mutants.
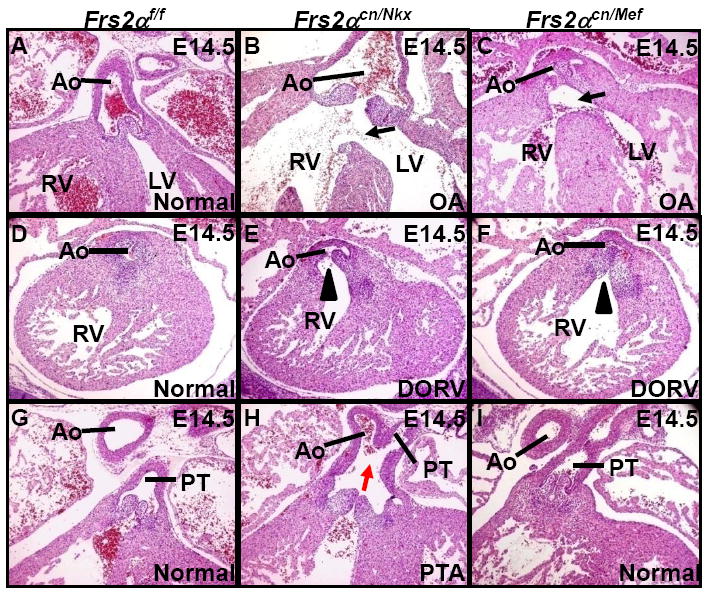
H&E staining of E14.5 embryonic heart sections demonstrates overriding aorta (a-c) and double outlet right ventricle (d-f) defects in Frs2αcn/Nkx and Frs2αcn/Mef mutants, and persistent truncus arteriosus defects in Frs2αcn/Nkx mutant (g-i). Ao, aorta; DORV, double outlet right ventricle; LV, left ventricle; OA, overriding aorta; RV, right ventricle; PT, pulmonary trunk; PTA, persistent truncus arteriosus. Black arrows denote OA-associated ventricular septal defects; red arrow denotes the persistent truncus arteriosus; arrowheads denote double outlet right ventricle.
In addition to the OFT myocardium and SHF, Nkx2.5cre also mediated Frs2α ablation in the OFT endocardium (Fig. 1Bb2) and the PE (Fig. 1Bb3), suggesting that FRS2α-mediated signals in the PE and OFT endocardium are required for OFT septation. However, ablation of Frs2α in the OFT endocardium with Tie2Cre (Kisanuki et al., 2001) did not disrupt OFT morphogenesis (Supplemental Fig. 2). Therefore, the results suggest that FRS2α mediated events in the PE, but not the endocardium, are required for OFT septation. Since Nkx2.5cre is expressed slightly earlier than Mef2ccre, the different OFT phenotypes in Frs2αcn/Nkx and Frs2αcn/Mef mutants also suggest the need for FRS2α-mediated signals in OFT septation occurs early in the SHF. Although Nkx2.5cre knockin mice have no OFT defect (Moses et al., 2001), the data do not rule out the possibility that the phenotype in Frs2αcn/Nkx OFT is a synergistic effect of FRS2α deficiency and Nkx2.5 haploid insufficiency.
Impaired SHF patterning and compromised activation of the MAP kinase pathway in the Frs2α mutant SHF and OFT
During early OFT development, cells are deployed from the SHF to the OFT starting at approximately E8.25; ablation of the SHF in chicken embryos has been shown to cause OFT misalignment (Ward et al., 2005). The SHF is the common domain of Nkx2.5cre and Mef2ccre-mediated ablation, and the common OFT misalignment phenotypes in these two mutant classes suggest that FRS2α-mediated signals in the SHF promote the progenitor cells residing in the SHF to contribute to the OFT myocardium. To test this possibility, immunostaining was employed to assess the number of cells expressing Isl1, a transcription factor that is expressed in the SHF and PE and is critical for the ability of these progenitor cells to proliferate and ultimately contribute to the OFT (Cai et al., 2003; Park et al., 2006).
Both Frs2αcn/Nkx and Frs2αcn/Mef embryos had a reduction in Isl1+ cells in the distal OFT myocardium at E9.5; Frs2αcn/Nkx embryos also had fewer Isl1+ cells in the OFT endocardium (Supplemental Fig. 3). To test whether the decrease in Isl1+ cells was due to a proliferation defect, double staining for phospho-Histone H3 (pHH3) and Isl1 was carried out at the 11-12 ss (E8.5). The results revealed that the numbers of both total Isl1+ cells and proliferating Isl1+ cells were significantly reduced in the OFT and SM/SHF of Frs2αcn/Nkx and Frs2αcn/Mef mutants (Fig. 3A). Consistently, both Frs2αcn/Nkx and Frs2αcn/Mef OFTs were shorter than the controls (Fig. 3Ba-d). Lineage-tracing with the R26R-lacZ reporter revealed that the β-galactosidase stained tissue in Frs2αcn/Mef hearts was smaller than that in the heterozygous littermates (Fig. 3Be,f), suggesting that contribution of the Frs2α-deficient SHF to the OFT and right ventricle was compromised. No clear difference in apoptosis was observed in the SM/SHF of Frs2αcn/Nkx or Frs2αcn/Mef embryos, although we detected increased apoptosis in the PE of Frs2αcn/Nkx embryos (Supplemental Fig.4). Together, the results suggest that the shared alignment defects in these two classes of mutants result from proliferation defects in the SHF.
Fig. 3. Ablation of Frs2α compromises expansion of SHF progenitor cells to the OFT myocardium.

A. Embryo sections were immunostained with anti-Isl1 (green) and anti-phosphorylated histone H3 (red) antibodies. Nuclei were counterstained with To-Pro3 (blue). Total and proliferating Isl1+ cells in the OFT and SM from four individuals are shown in panels d&e as mean ± sd.
B. Reduced OFT length and SHF lineage in Frs2α mutants. The OFT lengths at E9.5 measured from 6 individuals as indicated in panels a-c are presented in panel d as mean ± sd. Panels e&f, X-Gal staining. The blue staining represents cells from SHF progenitors in which the R26R reporter was activated. The outline of X-Gal stained area in the control was superimposed on the mutant to better illustrate the difference. Inserts are sections from the same tissues demonstrating OFT myocardium derived from the Mef2ccre lineage (indicated by black arrows).
C. Compromised ERK1/2, but not AKT, phosphorylation in the Frs2αcn/Nkx OFT and SM. Embryo sections were immunostained with the anti-phosphorylated ERK1/2 (a-c) or phosphorylated AKT (d-f) antibodies (green). Nuclei were counterstained with To-Pro3 (red).
D. Inhibition of the MAP kinase, but not PI3K/AKT, pathway reduces contribution of Isl1+ cells to the OFT. Panels a-c, short-term (24 hours) cultures of E8.5 embryos were sectioned and stained by anti-Isl1 antibody (green) and To-Pro3 (red). Inserts are high magnification views of the same sections. Panels d, cultured E8.5 embryos were treated with ERK1/2 or PI3K inhibitors as indicated. Adjacent sections from the same embryos were immunostained with anti-phosphorylated ERK1/2 or AKT (green) and To-Pro3 (red), demonstrating the specificity and efficacy of the ERK1/2 and PI3K inhibitors.
Since FRS2α has multiple tyrosine phosphorylation sites that link the MAP kinase and PI3K/AKT pathways to FGFR kinases (Kouhara et al., 1997; Ong et al., 2000), we used immunostaining to investigate which FRS2α-mediated downstream signaling pathway was interrupted in the mutant mice. Less phosphorylated ERK was detected in the OFT, SM, and PE of Frs2αcn/Nkx embryos at 11-12 ss, and in the OFT and SM of Frs2αcn/Mef embryos at the same stage (Fig. 3Ca-c). No significant difference was observed in anti-phosphorylated AKT staining at the same stage (Fig. 3Cd-f). Consistent with these findings, treating cultured E8.5 embryos with an ERK1/2, but not PI3K/AKT, inhibitor significantly reduced the number of Isl1+ cells in the SM, PE, and OFT (Fig. 3Da-c), although both inhibitors effectively and specifically inhibited ERK1/2 and AKT phosphorylation, respectively (Fig. 3Dd). These data indicate that the MAP kinase, but not the PI3K/AKT, pathway is essential for behavior of SHF cells. The data are consistent with the previous report that the number of Isl1+ cells in the SHF is rapidly reduced after Fgf8 ablation in the SHF (Park et al., 2006). Pitx2 is a homeobox transcription factor essential for OFT rotation and alignment (Ai et al., 2006). Whole-mount in situ hybridization showed that expression of Pitx2 was unchanged in Frs2αcn/Nkx and Frs2αcn/Mef hearts (Supplemental Fig. 5), suggesting that the alignment defects in these mutants are not the result of altered Pitx2 expression.
Impaired cushion development in Frs2αcn/Nkx OFT is a compound result of defects in endocardial EMT and NCC invasion
OFT cushion formation and remodeling is a major process in OFT septation. H&E staining revealed that cellularity was reduced in both the proximal and distal segments of Frs2αcn/Nkx, but not Frs2αcn/Mef, OFT cushions at E10.5 (Fig. 4A,B). At E11.5, the OFT cushions were hypoplastic and failed to fuse at the distal part (Fig. 4C). The results suggest that both EMT and NCC invasion were affected in Frs2αcn/Nkx embryos, which collectively contribute to the OFT septation defect.
Fig. 4. Ablation of Frs2α disrupts OFT cushion formation by inhibiting endocardial EMT and NCC contribution.
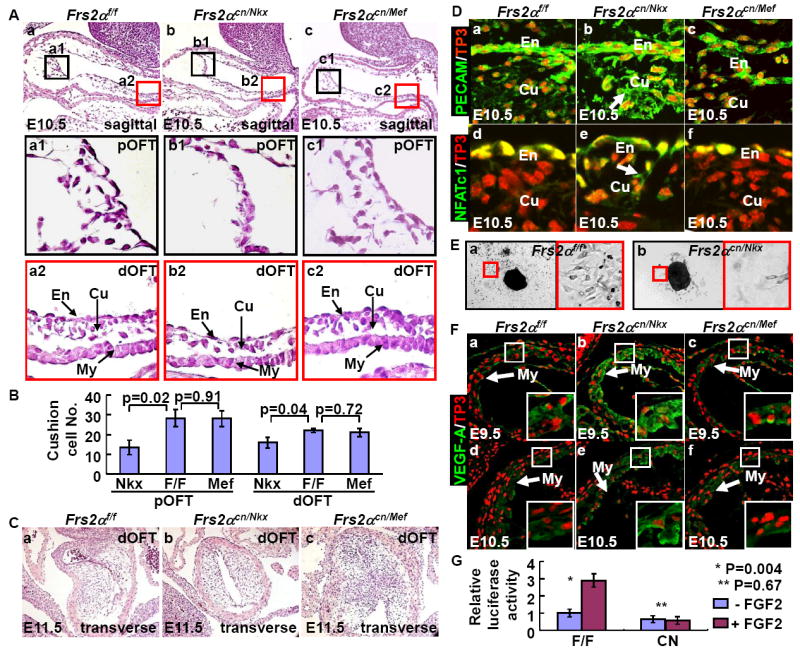
A&B. Reduced cellularity in Frs2α mutant OFT cushions. Sagittal sections of E10.5 embryos were H&E stained. High-magnification views of boxed areas representing proximal and distal OFT are shown as indicated. Cell numbers in the proximal and distal cushions (Cu) were assessed and statistical data from 4 individuals are presented as means±sd (B). Note that Frs2αcn/Nkx, but not Frs2αcn/Mef, OFT cushions had decreased cellularity. En, endocardium.
C. H&E staining of transverse sections of E11.5 distal OFT showing the fusion defect in Frs2αcn/Nkx OFT cushions.
D. Immunostaining for PECAM (a-c) and NFATc1 (d-f) shows compromised EMT in the Frs2αcn/Nkx proximal OFT cushions. Note that both PECAM and NFATc1 are still expressed in Frs2αcn/Nkx cushion cells as indicated by arrows.
E. Ex vivo culture of E9.5 OFTs shows compromised EMT in Frs2αcn OFT myocardium.
F. Increased immunostaining for VEGF-A in the Frs2αcn/Nkx OFT myocardium (My). Expression of VEGF-A in E9.5 and E10.5 embryos. Nuclei were stained with To-Pro3. Boxed inserts are high magnification views of the myocardium. Note that VEGF expression in Frs2αcn/Nkx myocardial cells is significantly increased.
G. FRS2α is essential for FGF2 to activate NFAT transcriptional activity. Mouse embryonic fibroblasts carrying homozygous Frs2αflox alleles were transfected with an NFAT-dependent luciferase reporter with or without Cre coexpression. The cells were cultured in the presence or absence of 2 ng/ml FGF2 as indicated. Luciferase activity was then assessed. Data are mean ± sd of triplicate samples.
PECAM and NFATc1 are expressed in the endocardium. Downregulation of PECAM in the endocardium is a prerequisite for the EMT (Enciso et al., 2003). Immunostaining with anti-PECAM and anti-NFATc1 revealed that both PECAM and NFATc1 were present in the endocardium of control and Frs2αcn/Mef OFT, and were diminished after the cells underwent EMT and invaded the cushion. In contrast, expression of PECAM and NFATc1 was sustained in Frs2αcn/Nkx endocardial cells even after the cells had invaded the OFT cushion (Fig. 4D). This indicates that although some cells were successfully activated and could invade the matrix, they failed to complete EMT. Ex vivo culture experiments with E9.5 OFTs further demonstrated the EMT defects in the Frs2αcn/Nkx myocardium (Fig. 4E). Interestingly, ablation of Frs2α alleles in the OFT endocardium with Tie2cre did not cause EMT and OFT defects (Supplemental Fig. 2), suggesting that FRS2α-mediated signals in the OFT endocardium are not essential for the EMT and that FRS2α-mediated signals regulate OFT endocardial EMT indirectly.
Given that OFT endocardial EMT is critically dependent on signaling molecules secreted by the OFT myocardium (Armstrong and Bischoff, 2004), and that the quantity of FRS2α protein is diminished in the SHF and just the forming OFT myocardium at the 8-9 ss in Frs2αcn/Nkx, but not Frs2αcn/Mef mutants (Supplemental Fig. 1), it is likely that FRS2α-mediated signals in the myocardial precursors are required upstream of the myocardial signaling cascade that initiates and supports endocardial EMT before the 8-9 ss.
We found increased immunostaining for VEGF-A in Frs2αcn/Nkx, but not the Frs2αcn/Mef, OFT myocardium (Fig. 4F), which is consistent with the report that overexpression of VEGF-A prevents nascent cushion endothelial cells from undergoing the EMT (Armstrong and Bischoff, 2004). It has been shown that myocardial NFATc2/3/4 promotes EMT by suppressing VEGF-A expression (Chang et al., 2004). In vitro assay of mouse embryonic fibroblasts revealed that transcriptional activity of NFAT was regulated by FGF in an FRS2α-dependent manner (Fig. 4G).These results suggest that FRS2α-mediated signals in the OFT myocardium repress VEGF-A expression and promote the OFT endocardial EMT, likely through regulating the transcription activity of NFAT. No defects were found in the atrioventricular (AV) cushions of either Frs2αcn/Nkx or Frs2αcn/Mef hearts (Supplemental Fig. 6). It is possible that either FRS2α-mediated signals do not regulate the AV cushion formation or other pathways redundantly regulate the process, since the AV canal also consist of cells from other lineages (Galli et al., 2008; Hutson et al., 2006; Liao et al., 2004).
Because NCCs make critical contributions to the distal OFT cushions, we examined migrating NCCs at E9.5 by immunostaining with anti-AP2α antibody, which labels migrating NCCs and the ectoderm). The results showed that the number of migrating NCCs (Ap2α+) in pharyngeal arches 3 and 4/6 and around the aortic sac in Frs2αcn/Nkx embryos were reduced (Fig. 5A, B). In contrast, no difference in the number of migrating NCCs was detected in Frs2αcn/Mef embryos. Coimmunostaining with anti-Ap2α and anti-pHH3 antibodies revealed significantly decreased proliferation in AP2α+ cells of Frs2αcn/Nkx embryos (Fig. 5C, D). We did not detect an increase in apoptosis in NCCs of Frs2αcn/Nkx embryos at E10.5 (Supplemental Fig. 4). These data suggest that the reduced number of NCCs in the OFT is a result of defective NCC proliferation, although NCC migration may also be affected. These defects are not attributable to Frs2α loss of function in NCCs because the Nkx2.5cre expression domain does not include the neural crest (Moses et al., 2001). Furthermore, ablation of Frs2α in NCCs with Wnt1-Cre (Danielian et al., 1998) did not cause OFT defects (data not shown). These findings indicate that FRS2α-mediated signals in NCCs are not required for NCC deployment and that the loss of FRS2α-mediated signals from the SM and/or PE causes secondary defects in the ability of NCC to contribute to the OFT.
Fig. 5. Compromised NCC contribution to the Frs2αcn/Nkx OFT cushions.

A&B. Immunostaining with anti-AP2α antibody reveals reduced numbers of migrating NCCs in aortic sac and pharyngeal arch 3 and 4/6 in Frs2αcn/Nkx, but not in Frs2αcn/Mef, embryos at E9.5 (A). Numbers of AP2α+ cells in the aortic sac (AS) and pharyngeal arches (PA) 3 and 4/6 were scored from 5 individuals and presented as mean ± sd (B).
C&D. Coimmunostaining reveals that proliferation of cardiac NCCs is compromised in Frs2αcn/Nkx embryos (C). Proliferating cells are labeled with anti-phosphorylated histone H3 antibody (red), NCC cells with anti-AP2α□ antibody (green), and nuclei with To-Pro3 (blue). Triple positive cells are indicated by arrows. Statistical analyses of proliferating cardiac NCCs from 5 individuals are presented as mean ± sd in D.
BMPs from the OFT myocardium, SM, and PE control NCC patterning (Liu et al., 2004; Ma et al., 2005; Stottmann et al., 2004), and their expression is positively regulated by FGFs (Choi et al., 2005; Gotoh et al., 2005). Whole mount in situ hybridization demonstrated that Bmp4 expression was reduced in the OFT, SM, and PE of Frs2αcn/Nkx embryos (Fig. 6h,k). In contrast, after ablation of Frs2α with Mef2ccre, Bmp4 expression was reduced in the OFT and SM, but not the PE (Fig. 6A). Consistently, phosphorylation of Smad1/5/8, downstream targets of the BMP receptor, was reduced in the same domains (Fig. 6B). These data suggest that in Frs2αcn/Mef embryos, the net level of BMP signaling in the pharynx and OFT, although decreased, is sufficient to support NCC proliferation and migration into the OFT cushions, whereas in Frs2αcn/Nkx mutants, BMP signaling falls below a critical threshold.
Fig. 6. Compromised BMP4 signaling in Frs2α mutant embryos.

A. Whole-mount in situ hybridization with antisense Bmp4 probe on E9.5 (a-c) or E10.5 embryos (d-f). Sections confirm the decreased Bmp4 expression in the Frs2αcn OFT and pharyngeal arches (g-l).
B. Phosphorylated Smad1/5/8 assessed in E9.5 embryos. High magnification views of boxed areas are shown as indicated.
Ablation of both Fgfr1 and Fgfr2 phenocopies the Frs2α-deficiency in OFT morphogenesis
Among the four FGF receptors, Fgfr1 and Fgfr2 are broadly expressed in the pharyngeal region at E8.5 and Fgfr3 is only expressed in pharyngeal arch 1 (Trokovic et al., 2005; Wright et al., 2003). At E9.5, Fgfr2 expression persists in the PE and SM. At the same stage, Fgfr1 was expressed in the PE, but not in the SM (Fig. 7A). To test whether FGFR1 and FGFR2 synergistically regulate OFT development, we conditionally inactivated Fgfr1 and Fgfr2 individually or simultaneously by Nkx2.5cre, denoted as Fgfr1cn/Nkx, Fgfr2cn/Nkx, and Fgfr1/r2cn/Nkx, respectively. Histological analyses showed that Fgfr1cn/Nkx embryos had no obvious OFT defects, whereas both Fgfr2cn/Nkx and Fgfr1/r2cn/Nkx mutants often developed OA and DORV (Table 3 and Fig. 7B). In addition, Fgfr1/r2cn/Nkx double mutants also exhibited the PTA phenotype (Fig. 7 and Fig. S7 in the supplementary material). Similar to our findings in Frs2αcn/Nkx mutant OFTs, both the proximal and distal segments of Fgfr1/r2cn/Nkx OFT cushions were hypocellular (Fig. 8A). Immunostaining revealed sustained PECAM expression in cushion cells, increased VEGF-A expression in the OFT myocardium, and reduced Ap2α+ cell proliferation in the aortic sac and pharyngeal arches in Fgfr1/r2cn/Nkx double mutants (Fig. 8B,C). The finding that Fgfr1/r2cn/Nkx and Frs2αcn/Nkx mutants had similar defects in endocardial EMT and NCC contribution, suggesting that FGFR1 and FGFR2 redundantly regulate OFT remodeling via FRS2α-dependent pathways. In addition, Fgfr1/r2cn/Nkx embryos also exhibited reduced total and proliferating Isl1+ cells and compromised activation of MAP kinase, but not AKT, pathways (Fig. 8D), indicating that the FGFR1/2-FRS2α-MAP kinase signaling axis in the SM is required for regulating the accrual of SHF cells to the OFT myocardium.
Fig. 7. OFT defects in Fgfr1 and Fgfr2 double conditional null embryos.

A. Whole-mount in situ hybridization demonstrating that Fgfr1 (a) and Fgfr2 (d) expression in E9.5 embryos. Cryosections reveal detailed expression patterns of Fgfr1 (b-c) and Fgfr2 (d-e). b1, and e1 are higher magnification views of b and e, respectively.
B. H&E staining of E14.5 embryo sections demonstrates that both Fgfr2 conditional mutant and Fgfr1/r2 double conditional mutants have VSD (a-d) and DROV (e-f), Fgfr1/r2 double conditional mutants have PTA, whereas Fgfr1 mutants have normal OFTs. Black arrowheads denote OA associated ventricular septal defects, white arrowheads denote DROV, and red arrow denotes PTA.
Fig. 8. Double ablation of Fgfr1/Fgfr2 disrupts OFT cushion formation.
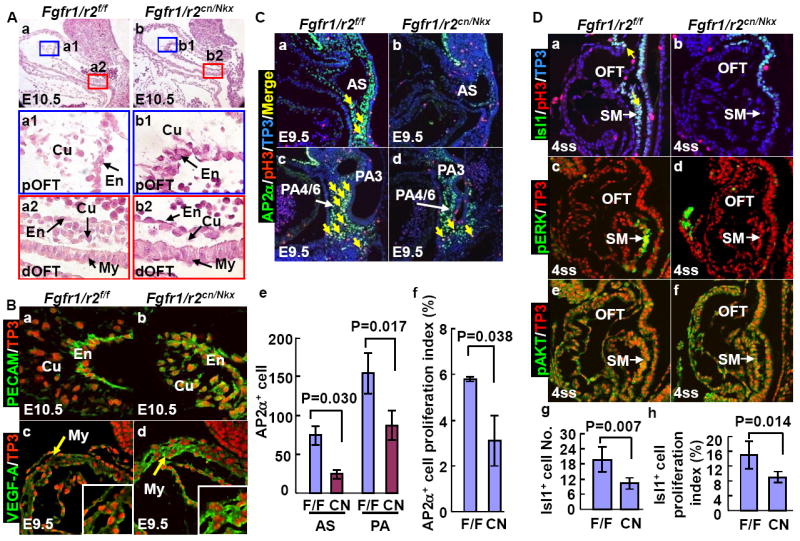
A. H&E staining of sagittal sections of E10.5 OFTs. High-magnification views of the boxed areas representing proximal and distal OFT are shown. Note reduced cellularity in both distal and proximal parts of mutant OFT cushions.
B. Compromised EMT in the Fgfr1/Fgfr2cn/Nkx OFT endocardium. Immunostaining revealed sustained PECAM expression in mutant OFT cushion cells (a-b) and increased VEGF-A expression in the mutant OFT myocardium (c-b). Inserts are high magnification views of the same section.
C. Reduced proliferation of the cardiac NCCs. Proliferating cells were labeled with anti-phosphorylated histone 3 (pH3, red); NCCs were labeled with anti-AP2α+ antibody (green), and nuclei were labeled with To-Pro3 (blue). Triple positive cells are indicated by arrows. Mean ± sd number of AP2α positive and triple positive cells from 4 individuals are shown in panels e and f.
D. Embryos at the 4 ss stained with the indicated antibodies and To-Pro3, demonstrate that total and proliferating Isl1+ cells in the SHF are reduced in mutants. Note that only MAP kinase, but not AKT, phosphorylation is compromised in the mutants. Yellow arrows indicate proliferating Isl1+ cells. Mean ± sd of Isl1+ cells and proliferating Isl1+ cells in the OFT and SM are shown in panels g and h. F/F, Fgfr1/r2 double floxed embryos; CN, Fgfr1/r2 double conditional null mutants.
Furthermore, the absence of OFT phenotypes in Fgfr1cn/Nkx and all mutants carrying one conditional null allele indicates that the aforementioned defects of Frs2αcn/Nkx, Fgfr2cn/Nkx, or Fgfr1/r2cn/Nkx mutants are not caused by Nkx2.5 heterozygosity since all these mutants had one Nkx2.5 null allele due to the Cre knockin.
Discussion
Here we report that FRS2α-mediated signaling pathways in OFT myocardial precursor cells that reside in the SHF are required for normal expansion and deployment of these cells to the OFT myocardium. These FRS2α-mediated pathways are also required to indirectly regulate endocardial EMT and recruitment of neural crest cells into the OFT cushions. Ablation of Frs2α in the SHF compromised MAP kinase activation and caused OFT alignment and septation defects. Our results are consistent with those presented in a companion study (Park et. al.), in which disrupting FGF signaling either by ablation of Fgfr1 and Fgfr2 or by overexpression of the FGF antagonist, Sprouty2, in the SHF causes arterial pole defects. Frs2αcn/Nkx hearts also exhibited defects in the atria and ventricles. Due to length limitations, this report focuses on the OFT defects.
FRS2α in mediating FGF signals for the OFT morphogenesis
Among the 22 FGF homologues, Fgf8 and Fgf10 are expressed in the SHF (Ilagan et al., 2006; Kelly et al., 2001; Park et al., 2006). Deficient or excessive FGF8 results in abnormal SHF development and OFT defects in a dosage- and spatiotemporal-specific manner (Hutson et al., 2006; Ilagan et al., 2006; Park et al., 2006). Ablation of Fgf10 results in malposition of the heart in the thoracic cavity (Marguerie et al., 2006). Although Fgf15 is expressed in the PE, it is unclear whether this mesodermal domain in the pharyngeal arches includes the SHF (Vincentz et al., 2005); loss of Fgf15 also causes OFT alignment defects. The OFT hypoplasia and misalignment we observed after ablation of Frs2α in the SHF are consistent with the requirements for these ligands during OFT morphogenesis and with the report that ablation of the Fgfr2IIIb isoform causes OFT alignment and ventricular septal defects (Marguerie et al., 2006). Ablation of Fgf8 function in heart precursors while they still reside in the primitive streak prevents formation of the OFT and right ventricle (Park et al., 2006). However, we did not observe this after ablation of Frs2α with Nkx2.5cre, suggesting either that onset of Nkx2.5cre is too late to disrupt transduction of the FGF8 signals that regulate the earliest phases of SHF development, or that these early signals are not FRS2α-dependent. In addition, ablation of Fgf8 with Nkx2.5cre also results in a significant loss of the Nkx2.5cre lineage and severe OFT and right ventricle truncation by E9.5. Fgf8;Nkx2.5cre mutants have significant decreases in cell proliferation and increases in cell death in both the PE and SM (Ilagan et al., 2006). Although Frs2αcn/Nkx mutants also had a significant decrease in cell proliferation in the PE and SM, no increase in cell death was found associated with Frs2αcn/Nkx. Thus, the FGF8 signals that promote SHF cell proliferation are likely mediated via FRS2α, and those that prevent cell death in these two domains are likely elicited via FRS2α-independent pathways.
Here we show that ablation of Frs2α reduced SHF cell proliferation by 50%, which is significant and is reflected in the shortened OFT. Similar correlation of compromised proliferation, shorted OFTs, and disrupted OFT alignment and rotation has been reported elsewhere (Ilagan et al., 2006; Park et al., 2006). In addition, since Frs2α ablation did not prevent all SHF cell proliferation, it is likely that other signaling pathways also regulate SHF cell proliferation. It has been shown that Wnt signaling can regulate SHF cell proliferation via both FGF-dependent and FGF-independent pathways (Cohen et al., 2007; Lin et al., 2007a). Therefore, further efforts are needed to clarify this issue. No heart misposition phenotype was observed in Frs2αcn embryos, implying that FGF10 regulates heart position independent of FRS2α-mediated signaling, although the data do not rule out the possibility that Nkx2.5Cre and Mef2ccre are not expressed in the cells that regulate heart position.
In mice, Fgf8 deficiency phenocopies syndromes associated with 22q11 deletions in humans, characterized by cardiovascular, thymic, parathyroid, and craniofacial defects (Frank et al., 2002; Vitelli et al., 2002), and Fgf8 modifies the vascular phenotypes resulting from Tbx1 haploinsufficiency (Vitelli et al., 2006) Furthermore, loss of Crkl function, another gene in the 22q11 deletion that contributes the human phenotypes, disrupts phosphorylation of FRS2α downstream of FGF8/FGFR interactions (Guris et al., 2001; Moon et al., 2006). Our new findings implicate FRS2α-mediated signaling as a molecular mechanism underlying the cardiovascular features in these mouse models and possibly in affected humans. Moreover, our data uncover branchpoints in the downstream effector pathways that mediate distinct aspects of FGF-signaling.
FRS2α-mediated signals secondarily regulate EMT and NCC contribution during cardiac OFT cushion formation
The process of endocardial EMT generates a subset of the OFT cushion mesenchymal cells and is regulated both by autonomous signals within the endocardium, and by paracrine signals from the myocardium (Armstrong and Bischoff, 2004). We have shown that expression of Bmp4, which is required in the myocardium for EMT (JFM, manuscript in preparation) and NCC invasion (Liu et al., 2004), is decreased in response to Frs2α ablation in the SHF. We further demonstrate that Frs2α function is essential for suppression of VEGF-A expression in the myocardium. Myocardial NFATc2/3/4 promotes EMT by suppressing VEGF-A expression (Chang et al., 2004). Furthermore, the transcriptional activity of NFATc2/3/4 can be regulated by MAP kinase and PI3K/AKT pathways downstream of the FGF signaling axis (Macian, 2005). Indeed, ablation of Frs2α in mouse embryonic fibroblasts blocked NFAT transcriptional activity in response to FGF stimulation (Fig. 4F). In the accompanying report, Park and colleagues show that soluble factors from wildtype OFT can rescue EMT defects in ex vivo cultured Fgf8 mutant OFTs.
Nkx2.5Cre is homogenously expressed in the OFT endocardium, whereas Mef2ccre is mosaically expressed in this domain. However, we ruled out a requirement for Frs2α in the endocardium during EMT because ablating Frs2α with Tie2Cre did not disrupt EMT. Together, our data suggest that the FGF signaling axis promotes endocardial EMT by promoting Bmp4 expression and suppressing myocardial VEGF-A production via an FRS2α-MAP kinase-NFATc pathway.
Although Nkx2.5cre is not expressed in NCCs, ablation of Fgfr1/Fgfr2 or Frs2α with Nkx2.5cre reduced the NCC contribution to the OFT cushions. Notably, ablation of Frs2α in NCCs with Wnt1-Cre did not cause OFT defects (data not shown). Park and colleagues also demonstrate that ablation of Fgf receptors or overexpression of the FGFR antagonist Sprouty2 in the SHF, but not in NCC, also suppresses NCC invasion of the OFT cushions (accompanying report). Interestingly, suppression of the FGFR signaling axis, either by ablation of Fgf8, Fgfr, or Frs2α, or by overexpression of Sprouty2 (herein, and accompanying report), reduced Bmp expression in the SHF and PE, which has been shown to be critical for NCC to contribute to the OFT cushion (Liu et al., 2004). Consistent with the expression pattern of Nkx2.5Cre and Mef2ccre, Frs2αcn/Nkx embryos had reduced Bmp4 expression in both SM and PE, while Frs2αcn/Mef only had reduced Bmp4 expression in the SHF (Fig. 6A, panels j and l). Given the fact that Mef2ccre is homogenously expressed in the SHF (Ai et al., 2007), the results suggest that early BMP4 signaling from both PE and SHF are required for NCC contribution to the OFT cushion. Together, these results indicate that the FGF8-FGFR/FRS2α signaling axis in the SM and/or PE indirectly regulates NCC contribution to the OFT cushion via the BMP signaling axis.
In summary, here we report that an FRS2α-mediated FGF signaling pathway in the SHF and PE controls OFT extension and alignment by promoting expansion of OFT precursors in the SHF, and also controls OFT septation by regulating OFT cushion formation through promoting the EMT of the cushion endocardium and NCC recruitment. Our findings provide a mechanism as to how the FGF signaling axis regulates OFT morphogenesis.
Supplementary Material
Table 2.
Survival analyses of Frs2αcn/Mef embryos and postnatal pups
| Age | Total | Frs2αcn/Mef | Expected |
|---|---|---|---|
| E8.5-E17.5 | 265 | 62 | 66 |
| Postnatal | 44 | 4 | 11 |
Acknowledgments
We thank Dr. David M. Ornitz for the Fgfr2-floxed mice, Dr. Juha Partanen for the Fgfr1-floxed mice, Dr. Jeffery D. Molkentin for the NFAT reporter, Dr. Wallace L. McKeehan, Ms. Young Xu and Mary Cole for critical reading of the manuscript, and Kerstin McKeehan for excellent technical support. The work was supported by Public Health Service Grants NIH-CA96824, AHA0655077Y from The American Heart Association, and DAMD17-03-0014 from the U.S. Department of Defense.
Abbreviations footnote
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- FRS2α
FGFR substrate 2α
- DORV
double outlet right ventricle
- OA
overriding aorta
- OFT
outflow tract
- TA
truncus arteriosus
- FSH
first heart field
- SHF
second heart field
- NCCs
neural crest cells
- PE
pharyngeal endoderm
- SM
splanchnic mesoderm
- VSD
ventricular septal defect
References
- Aggarwal VS, Liao J, Bondarev A, Schimmang T, Lewandoski M, Locker J, Shanske A, Campione M, Morrow BE. Dissection of Tbx1 and Fgf interactions in mouse models of 22q11DS suggests functional redundancy. Hum Mol Genet. 2006;15:3219–28. doi: 10.1093/hmg/ddl399. [DOI] [PubMed] [Google Scholar]
- Ai D, Fu X, Wang J, Lu MF, Chen L, Baldini A, Klein WH, Martin JF. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc Natl Acad Sci U S A. 2007;104:9319–24. doi: 10.1073/pnas.0701212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai D, Liu W, Ma L, Dong F, Lu MF, Wang D, Verzi MP, Cai C, Gage PJ, Evans S, et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev Biol. 2006;296:437–49. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–70. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery AW, Figueroa C, Vojtek AB. UNC-51-like kinase regulation of fibroblast growth factor receptor substrate 2/3. Cell Signal. 2007;19:177–84. doi: 10.1016/j.cellsig.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–89. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–63. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Choi KY, Kim HJ, Lee MH, Kwon TG, Nah HD, Furuichi T, Komori T, Nam SH, Kim YJ, Kim HJ, et al. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev Dyn. 2005;233:115–21. doi: 10.1002/dvdy.20323. [DOI] [PubMed] [Google Scholar]
- Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, Morrisey EE. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest. 2007;117:1794–804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–6. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–63. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- Enciso JM, Gratzinger D, Camenisch TD, Canosa S, Pinter E, Madri JA. Elevated glucose inhibits VEGF-A-mediated endocardial cushion formation: modulation by PECAM-1 and MMP-2. J Cell Biol. 2003;160:605–15. doi: 10.1083/jcb.200209014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli D, Dominguez JN, Zaffran S, Munk A, Brown NA, Buckingham ME. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development. 2008;135:1157–67. doi: 10.1242/dev.014563. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Manova K, Tanaka S, Murohashi M, Hadari Y, Lee A, Hamada Y, Hiroe T, Ito M, Kurihara T, et al. The docking protein FRS2alpha is an essential component of multiple fibroblast growth factor responses during early mouse development. Mol Cell Biol. 2005;25:4105–16. doi: 10.1128/MCB.25.10.4105-4116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–29. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A. 2001;98:8578–83. doi: 10.1073/pnas.161259898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch RV, Soriano P. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development. 2006;133:663–73. doi: 10.1242/dev.02242. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Zhang P, Stadt HA, Sato AK, Li YX, Burch J, Creazzo TL, Kirby ML. Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx. Dev Biol. 2006;295:486–97. doi: 10.1016/j.ydbio.2006.02.052. [DOI] [PubMed] [Google Scholar]
- Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–45. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–40. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- Lamers WH, Moorman AF. Cardiac septation: a late contribution of the embryonic primary myocardium to heart morphogenesis. Circ Res. 2002;91:93–103. doi: 10.1161/01.res.0000027135.63141.89. [DOI] [PubMed] [Google Scholar]
- Larsson H, Klint P, Landgren E, Claesson-Welsh L. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain- containing adaptor protein Crk. J Biol Chem. 1999;274:25726–34. doi: 10.1074/jbc.274.36.25726. [DOI] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–85. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Lin L, Cui L, Zhou W, Dufort D, Zhang X, Cai CL, Bu L, Yang L, Martin J, Kemler R, et al. Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc Natl Acad Sci U S A. 2007a;104:9313–8. doi: 10.1073/pnas.0700923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Zhang J, Zhang Y, Wang F. Generation of an Frs2alpha conditional null allele. Genesis. 2007b;45:554–9. doi: 10.1002/dvg.20327. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci U S A. 2004;101:4489–94. doi: 10.1073/pnas.0308466101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–11. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Marguerie A, Bajolle F, Zaffran S, Brown NA, Dickson C, Buckingham ME, Kelly RG. Congenital heart defects in Fgfr2-IIIb and Fgf10 mutant mice. Cardiovasc Res. 2006;71:50–60. doi: 10.1016/j.cardiores.2006.03.021. [DOI] [PubMed] [Google Scholar]
- McDougall K, Kubu C, Verdi JM, Meakin SO. Developmental expression patterns of the signaling adapters FRS-2 and FRS-3 during early embryogenesis. Mech Dev. 2001;103:145–8. doi: 10.1016/s0925-4773(01)00337-9. [DOI] [PubMed] [Google Scholar]
- McKeehan WL, Wang F, Kan M. The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog Nucleic Acid Res Mol Biol. 1998;59:135–76. doi: 10.1016/s0079-6603(08)61031-4. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Dionne CA, Li W, Li N, Spivak T, Honegger AM, Jaye M, Schlessinger J. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature. 1992;358:681–4. doi: 10.1038/358681a0. [DOI] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell. 2006;10:71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–80. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Ong SH, Goh KC, Lim YP, Low BC, Klint P, Claesson-Welsh L, Cao X, Tan YH, Guy GR. Suc1-associated neurotrophic factor target (SNT) protein is a major FGF- stimulated tyrosine phosphorylated 90-kDa protein which binds to the SH2 domain of GRB2. Biochem Biophys Res Commun. 1996;225:1021–6. doi: 10.1006/bbrc.1996.1288. [DOI] [PubMed] [Google Scholar]
- Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol. 2000;20:979–89. doi: 10.1128/mcb.20.3.979-989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays. 2000;22:108–12. doi: 10.1002/(SICI)1521-1878(200002)22:2<108::AID-BIES2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–33. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, Williams LT. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358:678–81. doi: 10.1038/358678a0. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, et al. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–48. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–18. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Partanen J. Fibroblast growth factor signalling and regional specification of the pharyngeal ectoderm. Int J Dev Biol. 2005;49:797–805. doi: 10.1387/ijdb.051976nt. [DOI] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. Embo J. 2003;22:1811–23. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–45. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Vincentz JW, McWhirter JR, Murre C, Baldini A, Furuta Y. Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis. 2005;41:192–201. doi: 10.1002/gene.20114. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002;129:4605–11. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Zhang Z, Huynh T, Sobotka A, Mupo A, Baldini A. Fgf8 expression in the Tbx1 domain causes skeletal abnormalities and modifies the aortic arch but not the outflow tract phenotype of Tbx1 mutants. Dev Biol. 2006;295:559–70. doi: 10.1016/j.ydbio.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Hatch EP, Karabagli H, Karabagli P, Schoenwolf GC, Mansour SL. Expression of mouse fibroblast growth factor and fibroblast growth factor receptor genes during early inner ear development. Dev Dyn. 2003;228:267–72. doi: 10.1002/dvdy.10362. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yoshino I, Shimazaki T, Murohashi M, Hevner RF, Lax I, Okano H, Shibuya M, Schlessinger J, Gotoh N. Essential role of Shp2-binding sites on FRS2alpha for corticogenesis and for FGF2-dependent proliferation of neural progenitor cells. Proc Natl Acad Sci U S A. 2005;102:15983–8. doi: 10.1073/pnas.0507961102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–74. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McKeehan K, Lin Y, Zhang J, Wang F. FGFR1 tyrosine phosphorylation regulates binding of FRS2alpha but not FRS2beta to the receptor. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.