

Abstract
We recently discovered and reported a series of N-alkyl-isatin acylhydrazone derivatives that are potent cannabinoid receptor 2 (CB2) agonists. In an effort to improve the druglike properties of these compounds and to better understand and improve the treatment of neuropathic pain, we designed and synthesized a new series of 2,3-dihydro-1-benzofuran derivatives bearing an asymmetric carbon atom that behave as potent selective CB2 agonists. We used a multidisciplinary medicinal chemistry approach with binding mode prediction through ligand-steered modeling. Enantiomer separation and configuration assignment were carried out for the racemic mixture for the most selective compound, MDA7 (compound 18). It appeared that the S enantiomer, compound MDA104 (compound 33), was the active enantiomer. Compounds MDA42 (compound 19) and MDA39 (compound 30) were the most potent at CB2. MDA42 was tested in a model of neuropathic pain and exhibited activity in the same range as that of MDA7. Preliminary ADMET studies for MDA7 were performed and did not reveal any problems.
Keywords: agonists, benzofuran derivatives, cannabinoid receptor 2, ligand-steered modelingm, receptors
Introduction
The endogenous cannabinoid system is a complex system consisting of two cannabinoid receptors (cannabinoid receptors 1 [CB1] and 2 [CB2]), seven endogenous (endocannabinoid) ligands,[1] and several proteins responsible for the regulation of endocannabinoid metabolic pathways, such as monoacylglycerol lipase and fatty acid amide hydrolase.[2] Modulation of this system has significant consequences in terms of immunomodulation. CB2 stimulation suppresses microglial cell activation and neuroinflammation,[3, 4] which may explain why CB2 is an emergent target for the treatment of neuropathic pain.[5–8] On the other hand, use of CB2 inverse agonists has been studied for the treatment of allergic contact dermatitis in animal models.[9] Increases in endocannabinoid concentrations seem to be a protective mechanism aimed at counteracting pain and inflammation.[10]
The two cannabinoid receptors that have been characterized and cloned, CB1 and CB2,[11, 12] have significant differences in terms of localization and function. CB1 is found predominantly in the brain[13] and is linked to cognitive impairment and psychoactivity.[14] These adverse effects preclude the development and use of CB1 agonists as therapeutics. CB2 is expressed mainly on immune tissues—the spleen, tonsils, monocytes, and B and T lymphocytes.[12,15] CB2 mRNA and/or protein levels are increased during different inflammatory conditions.[3, 4] Modulation of CB2 appears not to cause central side effects.[6, 7]
Neuropathic pain is caused by lesions in the central or peripheral nervous system and is characterized by hyperalgesia, reduced nociceptive thresholds such that normally innocuous stimuli cause pain, and allodynia (touch-evoked pain). Neuropathic pain is triggered by conditions such as diabetic neuropathy, AIDS-related neuropathy, postherpetic neuralgia, degenerative spinal disease, chemotherapy, radiotherapy, complex regional pain syndrome, phantom limb pain, trigeminal neuralgia, and multiple sclerosis. Neuropathic pain is refractory to traditional analgesics. The current typical treatments for neuropathic pain—opioids, gabapentin, and amitriptyline—are effective in fewer than 30 % of patients.[16–20] Neuropathic pain negatively affects patients’ physical, emotional, and social quality of life.[21] Recently, however, CB2 has emerged as a new target for the treatment of neuropathic pain.[7, 8,22–24] Upregulation of CB2 mRNA and proteins in the dorsal root ganglia and spinal cord is also found in animals after spinal nerve ligation[23,25] or nerve injury.[26–28]
Recently, the potential utility of CB2 agonists as treatments for neuropathic pain has received much attention. Several CB2 selective agonists have been described,[5,6, 29–37] and in the last 2 years, more than 150 patents have been granted on CB2 modulators.
Some of these compounds (Figure 1) have been widely used in in vivo animal models of neuropathic pain. Compound 1 better drug-like profile. A novel series based on a 3,3-disubstituted-2,3-dihydro-1-benzofuran ring was designed to increase bioavailability compared with the isatin series. On the basis of a comparison of the benzofuran and isatin structures, we assumed that the benzofuran scaffold might mimic the isatin scaffold (Figure 2). In Figure 2, ring B in the benzofuran series is superimposable with ring B in the isatin series formed by the hydrazone and the indolone through an internal hydrogen bond. In this case, the two five-membered rings (ring A) from the benzofuran and isatin structures fit. The absence of ring C in the benzofuran series is expected to decrease lipophilicity of the benzofuran series compared with the isatin series. This benzofuran series was synthesized using a colloidal palladium nano-particle-catalyzed tandem cyclization/cross-coupling reaction. We previously described the detailed pharmacological profile of one of these benzofuran compounds, compound 18.[7] This compound reversed neuropathic pain in spinal nerve ligation and paclitaxel-induced neuropathy models in rats without affecting locomotor behavior, and the effects of this compound were selectively antagonized by a CB2 receptor antagonist but not a CB1 receptor antagonist.[7] In this report, we describe the design, synthesis, and structure-activity (Figure 1) has been described as a protean agonist.[38,39] Compound 2, a well-characterized selective CB2 agonist, showed efficacy in models of inflammatory, postoperative, neuropathic, and osteoarthritic pain.[6] Compound 3 was chosen as a clinical candidate for the treatment of inflammatory pain.[30] Recently, the pharmacokinetics and safety in humans of compound 4, a nonselective CB1/CB2 agonist with limited brain penetration acting on the peripheral nervous system,[40] have been reported on the basis of results of phase I studies.[41] We recently described compound 5, which has a profile similar to that of compound 4 and which showed potent antiallodynic effects in a rat model of neuropathic pain but did not affect rat locomotor activity.[42] Systemic administration or intrathecal administration of compound 6[43] reduced both evoked and spontaneous wide-dynamic-range neuronal activity in neuropathic but not sham-treated rats. The effects in neuropathic rats were blocked by pre-administration of a CB2, but not a CB1, receptor antagonist.[44]
Figure 1.

Structures of CB2-selective modulators 1–6, a nonselective CB agonist 7, and a CB1-selective antagonist 8: 1 (AM1241),[105] 2 (A-796260),[106] 3 (GW842166X),[30] 4 (CRA13),[40] 5 (MDA19),[42] 6 (A-83633),[43] 7 (CP55,940),[107] 8 (AM251).[108]
Figure 2.
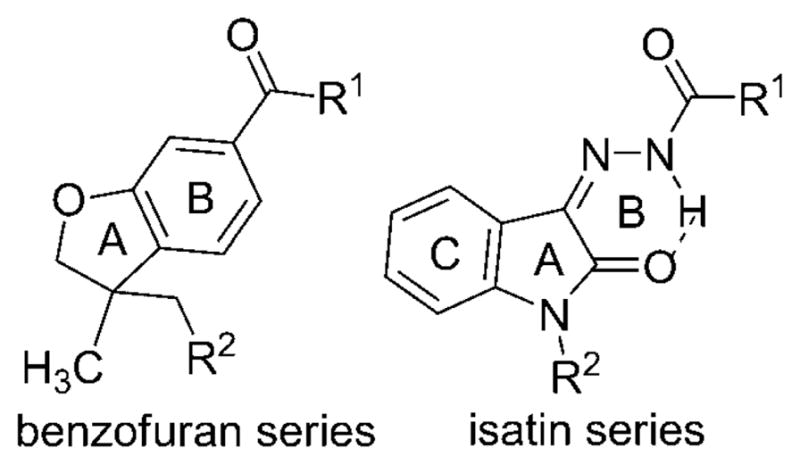
Comparison of the 3,3-disubstituted-2,3-dihydro-1-benzofuran scaffold with the isatin scaffold.
We recently described a series of N-alkyl isatin acylhydrazone derivatives that are potent CB2-selective agonists[42] and a series of CB2-selective inverse agonists.[45] The agonists suffer from poor water solubility, and in our efforts to better understand and improve the treatment of neuropathic pain, we sought to design and synthesize a series of agonists with a relationships (SARs) of this series of benzofuran compounds and the biological activities of both enantiomers of compound 18. Structural modeling studies based on ligand-steered modeling of the agonist: CB2 complex binding site are also presented, supporting a structural rationalization of SAR data.
Methods
Chemistry
The syntheses are outlined in Schemes 1–3. Compounds 10 a (Scheme 1) and 10 b (Scheme 2) were obtained from the corresponding phenols[46,47] by coupling with 3-bromo-2-methylpropene using potassium carbonate in methyl ethyl ketone. The resulting ethers were submitted to a palladium-catalyzed tandem cyclization/Suzuki-coupling reaction to afford benzofuran 11 a, 11 b, and 11 c.[48,49] After saponification, the resulting carboxylic acids (12 a to 12 c) were coupled with amines using HATU, DiPEA to afford compounds 13 and 14 and 16 to 24. To study the impact of substitution at carbone 3, the palladium-catalyzed tandem cyclization/Suzuki-coupling reaction was done in the last step (Scheme 3). Carboxylic acid 25 was obtained from 10 b by saponification using sodium hydroxide. Then, 25 was coupled with cyclohexylamine or piperidine using HATU, DiPEA to afford compounds 26 a and 26 b. Compounds 26 a and 26 b were submitted to palladium-catalyzed tandem cyclization/Suzuki-coupling reaction to afford compounds respectively 27 and compounds 28 to 32. This synthetic pathway (Scheme 3) was not used for compounds 19 and 20, as their retention factors were close to the retention factors of, respectively, compounds 26 b and 26 a.
Scheme 1.

Reagents and conditions: a) K2CO3, methyl ethyl ketone, 3-bromo-2-methylpropene; b) K2CO3, [Pd(OAc)2], nBu4NCl, DMF, PhB(OH)2; c) NaOH, EtOH, THF, H2O; d) HATU, DiPEA, DMF, CH2Cl2, 2-iodoaniline for 13 or cyclohexylamine for 14 or piperidine for 15.
Scheme 3.

Reagents and conditions: a) NaOH, EtOH, THF, H2O; b) HATU, DiPEA, DMF, CH2Cl2, cyclohexylamine for 26 a or piperidine for 26 b; c) K2CO3, [Pd(OAc)2], nBu4NCl, DMF, 1-naphthylboronic acid for 27 and 28 or 2-naphthylboronic acid for 29 or 4-chlorophenylboronic acid for 30 or 4-methoxyboronic acid for 31 or 4-pyridylboronic acid for 32.
Scheme 2.

Reagents and conditions: a) K2CO3, methyl ethyl ketone, 3-bromo-2-methylpropene; b) K2CO3, [Pd(OAc)2], nBu4NCl, DMF, PhB(OH)2 for 11 b or 1-penten-1-ylboronic acid for 11 c; c) NaOH, EtOH, THF, H2O; d) HATU, DiPEA, DMF, CH2Cl2, 2-iodoaniline for 16 or cyclohexylamine for 17 and 20 or piperidine for 18 and 19 or neopentylamine for 21 or morpholine for 22 or N-tert-butylmethylamine for 23 or 4-(aminomethyl)tetrahydropyran for 24.
Enantiomeric separation
Seven hundred fifty milligrams of the racemic mixture of compound 18 was processed using a Cyclobond DMP column and a mixture of acetonitrile, acetic acid, and triethylamine as mobile phase at a temperature of 10°C. Purity analysis indicated that compound 33 has an enantiomeric purity of 97.3 % while compound 34 has an enantiomeric purity of 97.5 %.
Crystallographic analysis and assignment of absolute configuration
The structure of compound 33 was confirmed by X-ray diffraction. Compound 33 yielded crystals of suitable quality for X-ray diffraction by slow evaporation of an ethyl acetate/heptane solution (Figure 3). The crystal structure was determined using X-ray data collected at 90 K, with Cu Kα radiation (λ=1.54178 Å) on a Bruker Kappa Apex-II diffractometer. Crystals are mono-clinic, space group P21 with Z = 2. All H atoms were visible in difference maps and were placed in calculated positions in the refinement, with a torsional parameter refined for the methyl group, leading to R =0.029, Rw =0.075 for 228 refined parameters and 3026 independent reflections having θmax = 68.8°. The absolute configuration was determined, on the basis of resonant scattering from light atoms only, to be (S) at C14, using 1266 Bijvoet pairs. The Flack parameter[50] has a value of x =0.07(19), and the Hooft parameter[51] has a value of y = 0.03(8), corresponding to a probability of 1.000 that the reported absolute configuration is correct. The CIF has been deposited at the Cambridge Crystallographic Data Centre, CCDC 725803.
Figure 3.

Compound 33 with atom labeling shown.
Ligand-steered modeling of the agonist: CB2 complex binding site
It should be emphasized that there is no crystal structure of an active-state class-A G-protein-coupled receptor (GPCR). Thus, structural modeling studies were performed on the benzofuran derivatives to identify a putative binding mode of this class of CB2 agonists. The crude homology model of the active-state CB2 receptor developed in this study was based on a multitemplate approach[52–54] using (1) the recently described crystallized structures of the β2-adrenergic and ligand-free opsin class-A GPCRs, to take advantage of several features of the purported active state of class-A GPCRs that are not completely present in either crystal structure, and (2) the information obtained from several computational studies that have proposed the active state models of CB2 and other class-A GPCRs.[55–65]
The CB2, β2-adrenergic (PDB code 2RH1), and ligand-free opsin (PDB code 3CAP) sequences were aligned on the basis of existing information on conserved residues within class-A GPCRs.[66] CB2 lacks the conserved proline in helix 5, so the next highly conserved residue, tyrosine, was used for the alignment as described by Xie et al.[67] The nomenclature of Ballesteros, Weinstein, and Stuart is used, whereby the most conserved residue in helix X is labeled X.50.[68] The resulting sequence alignment was used as an input to MODELLER 9v4[69] to develop a crude CB2 model. The N- and C-terminus residues (amino acids 1–26 and 316–360) of CB2, the T4 L residues connecting helix 5 and 6 of β2, helices 5–7 of β2, and helices 1–4 of ligand-free opsin were omitted. Mutagenesis and other CB2 modeling studies suggest the possibility of a disulfide bond between residues Cys 174 and Cys 179 in the E2-loop, which was included in our homology model.[70,71] On the basis of information in the literature,[62] helix 3 of CB2 was rotated counterclockwise (as seen from the extracellular side) using the GPCR Helix Manipulator script available under Maestro from Schrçdinger LLC.[72] This was followed by a restraint-minimization procedure to relieve the structural strain stemming from the replacement of nonconserved residues in the homology modeling process while the pocket was kept intact.
The ligand-steered modeling method has already been described in full.[45,73, 74] Briefly, starting from the crude model developed through a multitemplate approach, a known agonist was seeded into the pocket, and a structural ensemble of 200 structures was generated by randomizing the position and orientation of the ligand, followed by a multistep energy minimization in which the van der Waals interaction was gradually switched from soft to full interaction, as performed in other cases.[73, 75–77] The ligand and receptor were held flexible in this stage without any restraints. The structures in the ensemble were ranked using crude binding energy estimation, and then 40 structures were subjected to a full flexible-ligand-flexible-side chain Monte Carlo-based global energy optimization. The top-ranking structures were then visually inspected for ligand:receptor interactions, and complex candidates were selected.
Pharmacology
Cannabinoid-receptor-mediated functional activity
Functional activity was evaluated using γ-[35S]GTP assays in Chinese hamster ovarian cell membrane extracts expressing recombinant human CB1 (hCB1) or human CB2 (hCB2). The assay relies on the binding of γ-[35S]GTP a radiolabeled nonhydrolyzable GTP analogue, to the G protein upon binding of an agonist of the GPCR. In this system, agonists stimulate γ-[35S]GTP binding, whereas antagonists have no effect and inverse agonists decrease γ-[35S]GTP basal binding. CB1 and CB2 assay data are presented as the mean of two determinations. Assay reproducibility was monitored by the use of a reference compound, compound 7. For replicate determinations, the maximum variability tolerated in the test was of ± 20 % around the average of the replicates. Efficacies (Emax) for CB1 and CB2 are expressed as a percentage of the efficacy of compound 7.
Binding assays
Compounds 18, 19, 20, 29, and 30 were screened in a competitive binding experiment using membranes of Chinese hamster ovarian K1 cells selectively expressing hCB2 at different concentrations in duplicate.[78] The competitive binding experiment was performed in 96-well plates (Masterblock) containing binding buffer (50 mm Tris, pH 7.4, 2.5 mm EDTA, 0.5 % protease-free bovine serum albumin), recombinant membrane extracts (0.25 μg protein/well), and 1 nm [3H]7 (PerkinElmer, NEX-1051, 161 Ci/mmol, diluted in binding buffer). Nonspecific binding was determined in the presence of 10 μm compound 7 (Tocris Bioscience). The sample was incubated in a final volume of 0.1 mL for 60 min at 30°C and then filtered on a GF/B UniFilter microplate (PerkinElmer, catalogue number 6005177) pre-soaked in 0.5 % polyethyleneimine for 2 h at room temperature. Filters were washed six times with 4 mL of cold binding buffer (50 mm Tris, pH 7.4, 2.5 mm EDTA, 0.5 % protease-free bovine serum albumin), and the amount of bound [3H]7 was determined by liquid scintillation counting. Median inhibitory concentration (IC50) values were determined by nonlinear regression using the one-site competition equation. The inhibition constant (Ki) values were calculated using the Cheng-Prus-off equation (Ki =IC50/(1+(L/KD)), where L = concentration of radioligand in the assay and KD = affinity of the radioligand for the receptor.
In vivo evaluation
Animals
Adult male Sprague Dawley rats (Harlan Sprague Dawley) weighing 120–150 gm were used in experimental procedures approved by the Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center. Animals were housed three per cage on a 12-hour-light/12-hour-dark cycle with water and food pellets available ad libitum.
Paclitaxel-induced neuropathy model
Groups of rats (n =5–7) received either vehicle (10 % Cremophor EL in saline) or 1.0 mg kg−1 of paclitaxel daily by intraperitoneal injection for 4 consecutive days for a final cumulative dose of 4 mg kg−1;[7, 79] the injection volume was 1 mL kg−1. Baseline responses to mechanical stimulation of the hind paw (see next paragraph) were established on day 0 and continued daily until the development of neuropathy was confirmed.
Assessment of mechanical withdrawal thresholds
For assessment of antiallodynic effect, rats were placed in a compartment with a wire mesh bottom and allowed to acclimate for a minimum of 30 min before testing. Mechanical sensitivity was assessed using a series of Von Frey filaments with logarithmic incremental stiffness (0.41, 0.70, 1.20, 2.00, 3.63, 5.50, 8.50, and 15.1 g) (Stoelting, Wood Dale, IL), as previously described,[80] and 50 % probability withdrawal thresholds were calculated with the up-down method.[81] In brief, beginning with the 2.00 g probe, filaments were applied one by one to the plantar surface of a hind paw for 6–8 s. If no withdrawal response was observed, the next stiffer filament was applied; if there was a withdrawal response, the next less stiff filament was applied. Six consecutive responses after the first change in the response were used to calculate the withdrawal threshold (in grams). When response thresholds fell outside the range of detection, 15.00 g was assigned for absence of response to all tested fibers, and 0.25 g was assigned for withdrawal response to all tested fibers. The percentage maximal possible effect was calculated as ([postdrug threshold−baseline threshold]/[cutoff threshold (15 g)−baseline threshold])× 100.
Data analysis
Statistical analyses were carried out using BMDP 2007 (Statistical Solutions, Saugus, MA). Data were analyzed using one-way analysis of variance (ANOVA) or t test, where appropriate. If findings on ANOVA were significant, Tukey–Kramer post hoc analysis was used for multiple group comparison. Area under the curve was calculated using the trapezoidal rule. The results are presented as mean ±standard error of the mean and were considered significant at P <0.05.
ADMET
Compound 18 was tested for the following: aqueous solubility (PBS, pH 7.4) at 2.0E−04 m at a chromatographic wave length of detection of 230 nm,[82] plasma protein binding with %recovery (human) at 1.0E−05m.,[83] A-B permeability at 1.0E−05 m.,[84] and B-A permeability at 1.0E−05m.[85] Ames test[86] and automated whole-cell patch-clamp technique (Qpatch 16 by Sophion Biosciences) were used to record outward potassium currents (hERG) from single cells.[87]
Results and Discussion
Ligand-steered modeling of the agonist–CB2 complex
Because there is no published crystal structure of the active state of CB2, we aimed to structurally characterize the agonist:CB2 complex to identify residues involved in ligand recognition and thus explain SAR data for the benzofuran class of agonists. Recently, ligand-steered homology modeling, in which known ligands are explicitly used to shape and optimize the binding site through a docking-based stochastic global energy minimization procedure, was developed and tested in melanin-concentrating hormone receptor 1, a class-A GPCR target.[73] The ligand and receptor are considered flexible throughout the modeling process, which ensures proper coverage of the interaction energy landscape. This method was recently successfully applied in two different applications in the drug discovery process: high-throughput docking-based discovery of novel chemotype inhibitors for melanin-concentrating hormone receptor 1[73] and structural characterization of inverse agonists binding to CB2.[45] Ligand-steered modeling of the binding site is especially useful when there is only limited and inconclusive structural information available about protein:ligand interactions. To accomplish our aim, we implemented this methodology to build a multitemplate homology model of CB2 in a putative agonist-bound conformation, based on available information about similar active-state models of class-A GPCRs[58–61,63–65,88] and crystal structures of the β2-adrenergic[55–57] and ligand-free opsin receptors.[89]
Multi-template approach to develop an initial CB2 homology model
It is well known that the quality and accuracy of homology models depends on the choice of the template protein structure and its sequence alignment with the target sequence.[74,90,91] However, a single template structure may not represent a physiologically relevant conformation, and if this is the case, the result is an inaccurate homology model. The use of two or more homologous template structures, termed multitemplate homology modeling, has recently been successfully used to improve the accuracy of structural models.[52–54] In the case of CB2 and other class-A GPCRs, rotational movement of helix 3 and/or helix 6, translational motion of helix 5 towards helix 6, and outward tilting of helix 6 coupled with structural changes, like breakage of “ionic lock” between R3.50 and D/E6.30 residues, seem to characterize the active state, thus complicating the modeling.[58–65,88] Though the crystal structure of the ligand-free opsin[89] explains the movements of helices 5 and 6 and the breakage of ionic lock relevant for agonist-induced conformations, it fails to explain the displacement of extracellular loop 2 away from the transmembrane core, as seen in the crystal structure of human β2-adrenergic receptor, which may play an important role in capturing diffusible ligands by providing the ligands access from the extracellular environment into the transmembrane core of GPCRs.[64,89,92, 93] This displacement of extracellular loop 2 away from the transmembrane core is difficult to explain on the basis of the ligand-free structure of opsin. However, it should be noted that the β2-adrenergic receptor was crystallized with inverse agonist carazolol;[55–57] hence, it may not be suited to model an active-state structure by itself. Therefore, considering that neither crystal structure is suitable as is to model an active-state GPCR, and on the basis of the experimental and modeling evidence reported above, we incorporated several features from both crystal structures (helices 1–4 and extracellular loop 2 from β2-adrenergic crystal structure, helices 5–7 from ligand-free opsin structure and manual rotation of helix 3), thus developing an initial multitemplate model of active-state CB2. Thus, this initial model includes several well-known structural features of the agonist-bound conformations of class-A GPCRs as described earlier.
Structural characterization of the agonist-bound CB2 binding site in agreement with experimental evidence
The current experimental and computational modeling studies indicate that hydrogen bonds, π-π stacking, and/or van der Waals interactions with residues of helices 3–7 account for most of the known ligand:CB2 interactions.[60,62, 72,94–97] Prior modeling, SAR, and mutagenesis studies have suggested the presence of an aromatic pocket within helices 3, 5–7 surrounded by residues Y5.39, F5.46, W5.43, and W6.48, and hydrogen-bond interactions with S3.31, T3.35, Y5.39, and N7.45.[60,62, 94–98] Mutagenesis studies seem to indicate that interactions with S3.31 and F5.46 are crucial to improve selectivity as compared with CB1.[95] However, ligand:receptor interactions vary with ligand chemotypes, and there is no total agreement on the binding site and mode of CB2 agonist compounds. From the set of compounds displaying CB2 agonist activity (Table 1), we selected compound 33 to model the binding site with the ligand-steered method, analogous to what has been done with GPCRs and other receptors.[45,73, 75,76,99–102] Because of the sensitivity of hCB2 half-maximal effective concentration (EC50) values (Table 1) to C3 substitutions, we hypothesized that the R1 group was facing towards the hydrophobic pocket located in the interior of the transmembrane domain. Hence, in our initial model, the R1 group was oriented towards that pocket. Given the similarities between the isatin scaffold and the benzofuran scaffold, it was assumed that as in the case of isatin series, the lack of carbonyl group results in loss of hCB2 functional activity for benzofuran-scaffold-based agonist compounds. The R2 moiety (Table 1) was orientated such that the carbonyl group could possibly make hydrogen bond interactions with S3.31 or Y5.39 as suggested in earlier studies described above. These two conformations were seeded into the pocket and used as the starting structures for the ligand-steered modeling of the binding site. An initial ensemble of 200 structures for each conformation was generated by randomizing the position and orientation of the ligand, followed by a flexible-ligand:flexible-receptor docking procedure to select the most promising complexes (see Methods for a description of this methodology). From these, two representative structures were chosen, the binding sites were visually inspected, and the final complex was retained in which the R1 group was oriented toward the lipophilic pocket as described earlier and the carbonyl was positioned close enough to any residue side chain to form a hydrogen bond. As described earlier, since we hypothesized that the absence of the carbonyl group resulted in lack of affinity for benzofuran compounds, we assumed that absence of hydrogen-bond interaction with the carbonyl possibly was related to a wrong pose.
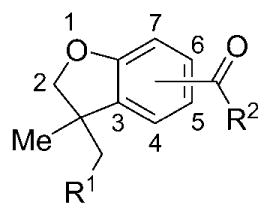
Table 1.
Potency (EC50) and maximal stimulation (Emax)s of hCB1 and hCB2 by compounds 13–32.[a]
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R2 position | γ-[35S]GTP hCB1 | γ-[35S]GTP hCB2 | ||
| EC50 [nm ± SEM] | Emax [%][b] | EC50 [nm ± SEM] | Emax [%][b] | ||||
| 13 | phenyl | N-(2-iodophenyl) | 5 | >10 000 | ND[c] | >10 000 | ND |
| 14 | phenyl | N-cyclohexyl | 5 | >10 000 | ND | 406 ± 1.4 | 47.1 |
| 15 | phenyl | 1-piperidyl | 5 | >10 000 | ND | >10 000 | ND |
| 16 | phenyl | N-(2-iodophenyl) | 6 | >10 000 | ND | >10 000 | ND |
| 17 | phenyl | N-cyclohexyl | 6 | >10 000 | ND | 478 ± 1.3 | 54.3 |
| 18 | phenyl | 1-piperidyl | 6 | >10 000 | ND | 128 ± 32 | 88.3 |
| 33 | S enantiomer (1) of 18 | 6 | >10 000 | ND | 108.02 | 86 | |
| 34 | R enantiomer (2) of 18 | 6 | >10 000 | ND | 960.89 | 43.3 | |
| 19 |
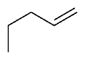
|
1-piperidyl | 6 | 856 ± 1.2 | 60 | 48.9± 1.4 | 97.1 |
| 20 |
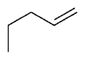
|
N-cyclohexyl | 6 | >10 000 | ND | 839 ± 5.5 | 105 |
| 21 | phenyl |
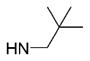
|
6 | >10 000 | ND | 246 ± 1.3 | 48.7 |
| 22 | phenyl | morpholine | 6 | >10 000 | ND | 5583 ± 4.4 | 93 |
| 23 | phenyl |
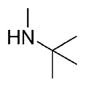
|
6 | 2580 ± 1.5 | ND | 95.3±1.8 | 91 |
| 24 | phenyl |
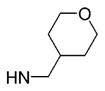
|
6 | >10 000 | ND | 659 ±1.4 | 49 |
| 27 | 1-naphthyl | N-cyclohexyl | 6 | >10 000 | ND | >10 000 | ND |
| 28 | 1-naphthyl | 1-piperidyl | 6 | >10 000 | ND | >10 000 | ND |
| 29 | 2-naphthyl | 1-piperidyl | 6 | >10 000 | ND | 875 ± 1.4 | 112 |
| 30 | 4-chlorohenyl | 1-piperidyl | 6 | 1363 ± 1.4 | 86.9 | 56.2± 1.2 | 102 |
| 31 | 4-methoxyphenyl | 1-piperidyl | 6 | >10 000 | ND | 234.68 | 77.1 |
| 32 | 4-pyridine | 1-piperidyl | 6 | >10 000 | ND | 2801.44 | 62.5 |
CB1 and CB2 assay data are presented as the mean of two determinations; reproducibility was monitored by the use of compound 7 as reference. For replicate determinations, the maximum variability tolerated in the test was ±20 % around the average of the replicates.
Efficacies for CB1 or CB2 are expressed as percentage relative to the efficacy of compound 7.
ND =not determined (plateau was not reached at a dose of 10 μm).
Correlation between ligand binding to the CB2 structural model and SAR data
Given the limited and inconclusive experimental evidence available to validate our model, we evaluated the accuracy of the model by assessing its ability to explain the SAR data of our compounds. In our representative model with compound 33 (Figure 4), the carbonyl group is located at 1.8 Å from the side chain of S3.31, suggesting a possible ligand-receptor interaction via hydrogen bond, as described in earlier studies62. Mutagenesis of S3.31 with glycine (S3.31G) and/or F5.46 with valine (F5.46 V) in CB2 resulted in loss of affinity/activity for several CB2 agonists, which confirmed the role of these residues in developing CB2-selective agonists.[62,95, 96,98] The R1 group (Table 1) is properly located in the aromatic pocket to form van der Waals interactions with residues F5.46 and W6.48. The R2 group (1-piperidyl for compound 33) is oriented towards F7.35. These observations suggest the overall binding mode and possible ligand receptor interactions of the benzofuran series of agonist compounds. It is important to note the difference in functional activity with different R1 moieties facing the aromatic domain. Introduction of chlorine (compound 30) at the phenyl ring facing the aromatic pocket results in increased hCB2 functional activity compared with compound 33, while introduction of polar moieties such as methoxyphenyl (compound 31) or 4-pyridine (compound 32) in the aromatic pocket causes unfavorable interactions with F5.46 and W6.48, resulting in loss of CB2 functional activity. Decreasing negative electrostatic potential of phenyl ring by introduction of a chlorine (compound 30) might result in an increase of the pi-stacking interaction with the electron-rich W6.48. Alkenyl moiety (compound 19) seems to be well tolerated in the aromatic pocket. Introduction of bulky moiety such as 1-naphthyl at the R1 position with 1-piperidyl at R2 (compound 28) causes steric clashes of the naphthyl group with side chains in TM3, which results in complete loss of hCB2 functional activity. For compound 29: the 2-naphthyl group is oriented deeper into the aromatic pocket, which abolishes the pi-stacking interaction with W6.48 and possibly the hydrogen bond interaction with S3.31, which explains its loss of activity.
Figure 4.

A) CB2 complexed with compound 33 (yellow carbon atoms). Transmembrane regions (TM) are shown as white ribbons; the image was prepared using PyMOL (http://www.pymol.org). B) A putative hydrogen bond between S3.31 and compound 33 (yellow carbon atoms) is represented as red dashes. The aromatic pocket is enclosed by residues F5.46 and W6.48 (green carbon atoms).
Combination of a cyclohexyl ring at R2 with a 1-naphthyl ring at R1 causes steric clashes with residues in transmembrane 3 and F7.35 and disruption of the hydrogen bond with S3.31, explaining the complete loss of CB2 functional activity for compound 27. As the aromatic pocket formed by residues F5.46 and W6.48 was well suited to tolerate either an alkene or a phenyl ring, we decided to explore the receptor-ligand interactions for R2, keeping either an alkene or a phenyl group for R1. Replacing the piperidine ring of compound 19 by a cyclohexylamine ring (20) results in direct steric clashes with F7.35 and decrease of CB2 functional activity. Moving the carboxamide moiety from position 6 to position 5 (compounds 13–15) results in loss of hydrogen-bond interaction with S3.31, explaining dramatic loss of CB2 functional activity of these compounds. The same disruption of the hydrogen bond with S3.31 by steric clashes between F7.35 and the bulky iodoaniline ring might explain the loss of activity for compound 16. However, some activity is regained when the iodoaniline ring is replaced by a cyclohexyl group (compound 17). On the other hand, neopentylamine moiety (compound 21) or N-tert-butylmethylamine moiety (compound 23) exhibited the same range of CB2 potency as the piperidine analog 18. It should be noted that compound 21 showed an efficacy of 48 %. As the vicinity of the R2 moiety is lipophilic, the lower lipophilicity of the neopentylamine nitrogen (compound 21) compared to the trisubsituted piperidine nitrogen (compound 18) or N-tert-butylmethylamine (compound 23) might explain the decrease of CB2 efficacy. Introduction of a morpholine ring in compound 22 disrupts the aromatic interactions with F7.35 and possibly hydrogen bond with S3.31, explaining the loss of CB2 functional activity. However, replacing the morpholine ring by a methyltetrahydropyran (compound 24) restores some CB2 functional activity. The additional flexibility of the methyltetrahydropyran ring compared to the morpholine group might result in exposing compound 24 to the solvent. For compound 34, the R en-antiomer of compound 33, accommodation of the phenyl group in the aromatic binding pocket results in a flipped benzofuran core, which may explain the decreased activity.
Some compounds were selected for binding studies at hCB1 and hCB2 (Table 2). Compounds 20 allow us to rule out any potential CB2 antagonist activity. Binding results were in agreement with the corresponding hCB2 functional activities. Both compound 20 and compound 30 exhibited CB2 affinity in agreement with the corresponding CB2 functional activities. Compound 18 would be expected to show the same range of affinity for CB2 as compounds 19 and 30 according to the hCB2 functional activities. However, the CB2 affinity of compound 18 was four times lower, which might be explained by a different positioning of the radiolabeled ligand in the receptor compared with compound 18.
Table 2.
Radioligand competitive binding assay data.
| Ligand | Ki [nm][a] | |
|---|---|---|
| hCB1 | hCB2 | |
| 18 | >10 000 | 422 ± 123 |
| 19 | ND[b] | 83.6 ± 23.8 |
| 20 | >1000 | >1000 |
| 29 | 430 ± 39 | 490 ± 45 |
| 30 | ND | 123 ± 16.5 |
| 7 | 3.4 | 1.8 ± 1.1 |
| 8 | 1.16 ± 0.01 | ND |
Values represent the mean ±SEM.
ND = not determined.
Effects of compounds 19 and 1 on tactile allodynia in a paclitaxel-induced neuropathic pain model
Tactile allodynia developed in 100 % of rats 10 days after the start of paclitaxel administration. In paclitaxel-treated rats, intraperitoneal administration of compound 19 suppressed mechanical allodynia (Figure 5) in a dose-dependent manner. The %MPE (maximum peak effect) for reversing mechanical allodynia for the 15 mg kg−1 compound 19 dose was significantly different (P <0.05) from that of the vehicle.
Figure 5.

Effects of A) compound 1 and B) compound 19 administered by intraperitoneal injection on tactile allodynia in a paclitaxel-induced neuropathic pain model in rats (n =5–6 per group). C) Compound 19 dose-dependently attenuated tactile allodynia in this model, as evidenced by an increase in the percent MPE withdrawal threshold area under the curve (AUC); P <0.05 between the vehicle (V) and compound 19 at 15 mg kg−1 (ANOVA followed by Tukey–Kramer test for multiple group comparison). Compound 1 did not affect the paw withdrawal threshold. The effects of compound 1, a CB2 ligand,[103] at 1, 3, and 5 mg kg−1 were not significantly different from the effects induced by the vehicle.
The effects of 1 mg kg−1, 3 mg kg−1, and 5 mg kg−1 of compound 1, a CB2 ligand,[103] were not significantly different from the effects induced by the vehicle (Figure 5). The antinociceptive effects of compound 1 have been shown to involve the μ-opioid receptor system and β-endorphin and to be blocked by administration of the opioid receptor antagonist naloxone or antiserum to β-endorphin.[7, 104]
ADMET
Because the complete pharmacological profile was completed for compound 18,[7] we performed ADME-Tox assessment for compound 18. No main issues appeared in the preliminary compound-18 profile (Table 3). Aqueous solubility and plasma protein binding are in the range of those of many marketed drugs. The partition coefficient (Log D, n-octanol/PBS, pH 7.4) was not determined because compound 18 was below the limit of quantitation in the aqueous phase. Intestinal permeability is compatible with oral administration with no efflux. The Ames test was performed to determine whether compound 18 was mutagenic. No genetic toxicity up to 10 μm was detected (Table 3).[86] Compound 18 did not show any cardiac toxicity.
Table 3.
ADMET profile for compound 18.
| Parameter | Value |
|---|---|
| Aqueous solubility (PBS, pH 7.4) at 2.0 × 10−4 m: | 31.8 ± 1.8 μm |
| Protein bound (human): | 82.8 ± 11.1 % |
| Protein recovery (human): | 95.6 ± 13.6 % |
| A–B permeability (TC7, pH 6.5/7.4) [10−6 cm s−1]: | 52.2 ± 1.7 |
| A–B permeability (% recovery): | 63.5 ± 6.5 % |
| B–A permeability (TC7, pH 6.5/7.4) [10−6 cm s−1]: | 16.2 ± 0.2 |
| B–A permeability (% recovery): | 85.5 ± 0.5 % |
| Ames test (strain TA98): | <0 |
| Ames test (strain TA98 +S9): | <0 |
| K+ channel hERG automated patch-clamp (cardiac toxicity): | inactive at 1 μm |
Conclusions
In summary, we have discovered a novel series of CB2 agonists that are potent and selective. A multidisciplinary approach was carried out with the aim of improving understanding of the interaction between our new series of compounds and the cannabinoid receptor CB2. Organic synthesis, enantiomer separation, configurational assignment, and in vitro biological testing were carried out and validated our CB2 homology model. Compound 33 and compound 34, the two enantiomers of compound 18, were tested at CB2 and CB1. The S enantiomer, compound 33, is responsible for compound 18 CB2 activity, and the R enantiomer, compound 34, exhibited weak CB2 functional activity. The discrimination between the two enantiomers for CB2 functional activity was predicted by our homology model. Compound 18 exhibited the best selectivity at CB2 compared to CB1. Compound 19 and compound 30 were the most potent at CB2. Compound 19 was active in a model of neuropathic pain in the same range as compound 18. Preliminary ADME-Tox studies for compound 18 were performed and did not show any issues. Thus, we have identified a novel series of compounds from which it should be possible to develop new drugs for the treatment of neuropathic pain. Pharmacokinetic and metabolism studies are ongoing for compound 18.
Experimental Section
All chemicals were purchased from Sigma–Aldrich or Acros. Thin-layer chromatographic analyses were performed on Sigma–Aldrich 60 F254 TLC plates. Column chromatography was performed with silica gel (230–400 mesh). 1H and 13C NMR spectra were recorded on Bruker 300 and 500 MHz DPX NMR spectrometers, respectively. Chemical shifts (δ, in ppm) are reported relative to either residual dimethyl sulfoxide (DMSO; 3.35 ppm) or CHCl3 (7.24 ppm) as internal standards. Signals are abbreviated as follows: br=broad, s= singlet, d =doublet, t =triplet, q=quadruplet, m=multiplet. Coupling constants (J) are expressed in Hertz. When needed, the reactions were performed under a positive pressure of dry N2 gas.
3-Iodo-4-(2-methylallyloxy)benzoic acid methyl ester (10 a)
Finely powdered K2CO3 (1.49 g, 10.78 mmol) was added to a solution of methyl 4-hydroxy-3-iodobenzoate (1.5 g, 5.4 mmol) in anhydrous methyl ethyl ketone (60 mL) followed by 3-bromo-2-methylpropene (0.81 mL, 1.1 g, 8.15 mmol). The reaction mixture was heated at 70°C for 4 h. The mixture was diluted with EtOAc and filtered. The filtrate was washed with H2O and dried over MgSO4. Evaporation of the solvent and of the remaining bromopropene under vacuum afforded 1.77 g (98 %) of pure 10a as a yellow oil. 1H NMR (300 MHz, CDCl3): δ = 8.46 (d, J = 1.8 Hz, 1 H), 7.98 (dd, J = 8.7 Hz, J =1.8 Hz, 1 H), 6.80 (d, J =8.7 Hz, 1 H), 5.19 (d, J =1.2 Hz, 1 H), 5.04 (d, J =1.2 Hz, 1 H), 4.54 (s, 2H), 3.09 (s, 3H), 1.88 (d, J = 1.2 Hz, 3H); 13C NMR (500 MHz, CDCl3): δ=165.49 (C=O), 160.64 (C), 140.99 (CH), 139.52 (C), 131.44 (CH), 113.39 (CH2), 111.09 (CH), 90.50 (C), 72.71 (CH2), 52.10 (CH3), 19.41 (CH3).
4-Iodo-3-(2-methylallyloxy)benzoic acid methyl ester (10 b)
Methyl 3-hydroxy-4-iodobenzoate (1.5 g, 5.4 mmol) was submitted to the same procedure described above for the preparation of 10 a. Evaporation of the solvent and of the remaining bromopropene under vacuum afforded 1.4 g (78 %) of pure 10 b as a yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.86 (d, J = 8.1 Hz, 1 H), 7.42 (d, J =1.7 Hz, 1H), 7.36 (dd, J =8.1 Hz, 1.8 Hz, 1 H), 5.23 (s, 1H), 5.04 (s, 1 H), 4.54 (s, 2 H), 3.91 (s, 3 H), 1.88 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 166.58 (C=O), 157.24 (C), 139.78 (C), 139.52 (CH), 131.49 (C), 123.36 (CH), 113.25 (CH2), 112.43 (CH), 93.22 (C), 72.68 (CH2), 52.31 (CH3), 19.50 (CH3).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid methyl ester (11 a)
K2CO3 (379 mg, 2.74 mmol), tetra-n-butylammonium chloride (380 mg, 1.37 mmol), and PhB(OH)2 (200 mg, 1.64 mmol) were added to a solution of 10a (455 mg, 1.37 mmol) in DMF (15 mL). The resulting mixture was heated at 115°C, and [Pd(OAc)2] (25.6 mg, 0.136 mmol) dissolved in DMF (5 mL) was added. The resulting mixture was stirred for 3 h at 115 °C, cooled to room temperature, filtered over silica, washed with H2O, dried over MgSO4, and concentrated under vacuum. Column chromatography (silica gel, heptane/CH2Cl2 4:6) afforded 202 mg (52%) of the title compound as a light brown oil. 1H NMR (300 MHz, CDCl3): δ = 7.89 (dd, J =8.4, 1.8, 1H), 7.72 (d, J =1.8 Hz, 1 H), 7.23 (m, 3H), 6.98 (m, 2H), 6.74 (d, J =8.4 Hz, 1H), 4.60 (d, J =8.9 Hz, 1 H), 4.13 (d, J = 8.9 Hz, 1 H), 3.89 (s, 3 H), 2.93 (d, J =13.4 Hz, 1H), 2.87 (d, J = 13.4 Hz, 1 H), 1.40 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ = 167.00 (C= O), 163.75 (C), 137.06 (C), 135.39 (C), 131.25 (CH), 130.22 (CH), 128.08 (CH), 126.67 (CH), 125.23 (CH), 122.70 (C), 109.45 (CH), 82.56 (CH2), 51.83 (CH3), 46.66 (CH2), 45.93 (C), 25.04 (CH3).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid methyl ester (11 b)
Compound 10b (455 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a. Column chromatography (silica gel, heptane/CH2Cl2 4:6) afforded 368 mg (95 %) of the title compound as a light brown oil that crystallized; mp: 52°C; 1H NMR (500 MHz, CDCl3): δ = 7.59 (dd, J =7.7 Hz, 1.2 Hz, 1 H), 7.39 (d, J =1.0 Hz, 1H), 7.22 (m, 3H), 6.96 (m, 3 H), 4.54 (d, J =8.7 Hz, 1 H), 4.11 (d, J =8.7 Hz, 1 H), 3.89 (s, 3 H), 2.88 (q, J =13.3, 2H), 1.37 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 167.06 (C=O), 159.73 (C), 140.30 (C), 137.01 (C), 130.53 (C), 130.34 (CH), 128.05 (CH), 126.68 (CH), 123.19 (CH), 122.41 (CH), 110.64 (CH), 82.33 (CH2), 52.13 (CH3), 46.43 (CH2), 46.39 (C), 24.46 (CH3).
3-((E)-Hex-2-enyl)-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid methyl ester (11 c)
Compound 10 b (65 mg, 0.2 mmol) was submitted to the same procedure described above for the preparation of 11 a using 1-penten-1-ylboronic acid (26 mg, 0.23 mmol). Column chromatography (silica gel, heptane/CH2Cl2 6:4) afforded 290 mg (48%) of the title compound as a light yellow oil. 1H NMR (500 MHz, CDCl3): δ = 7.60 (dd, J = 7.7 Hz, 1.4 Hz, 1H), 7.40 (d, J = 1.1 Hz, 1H), 7.11 (d, J =7.7 Hz, 1 H), 5.41–5.47 (m, 1 H), 5.31–5.21 (m, 1 H), 4.43 (d, J =8.6 Hz, 1 H), 4.16 (d, J =8.6 Hz, 1H), 3.89 (s, 3 H), 2.35–2.23 (m, 2H), 1.94 (q, J =7.1, 2H), 1.33 (m, 6 H), 0.85 (t, J =7.4, 3 H); 13C NMR (500 MHz, CDCl3): δ=167.04 (C=O), 159.73 (C), 140.74 (C), 135.03 (CH), 130.35 (C), 124.68 (CH), 122.66 (CH), 122.49 (CH), 110.46 (CH), 82.28 (CH2), 52.08 (CH3), 45.50 (C), 43.65 (CH2), 34.66 (CH2), 25.01 (CH3), 22.54 (CH2), 13.57 (CH3).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid (12 a)
A mixture of compound 10 a (125 mg, 0.44 mmol), NaOH (120 mg, 3 mmol), EtOH (6 mL), and H2O (1 mL) in THF (6 mL) was stirred for 12 h at room temperature. The reaction medium was acidified by adding a solution of 1.2m HCl and was extracted with EtOAc. The organic phase was washed with H2O, dried (Na2SO4), and concentrated in a rotary evaporator. The product was obtained as a light brown oil (116 mg, 97 %); 1H NMR (300 MHz, CDCl3): δ = 11.43 (s, 1 H), 7.89 (dd, J =8.4 Hz, 1.8 Hz, 1 H), 7.69 (d, J =1.6 Hz, 1 H), 7.11–7.14 (m, 3 H), 6.87 (dd, J =6.4 Hz, 2.9 Hz, 2 H), 6.67 (d, J = 8.4 Hz, 1 H), 4.51 (d, J =8.9 Hz, 1 H), 4.05 (d, J =8.9 Hz, 1 H), 2.83 (d, J =13.4 Hz, 1H), 2.77 (d, J =13.3 Hz, 1 H), 1.30 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ = 172.34 (C=O), 164.61 (C), 136.99 (C), 134.45(C), 132.25 (CH), 130.28 (CH), 128.15 (CH), 126.77 (CH), 125.99 (CH), 121.85 (C), 109.69 (CH), 82.81 (CH2), 46.69 (CH2), 45.91 (C), 25.04 (CH3).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid (12 b)
Compound 11 b (300 mg, 1.06 mmol) was submitted to the same procedure described above for the preparation of 12 a. The product was obtained as a white solid (300 mg, 100 %); mp: 165 °C; 1H NMR (500 MHz, CDCl3): δ=7.67 (dd, J =7.7 Hz, 1.3 Hz, 1 H), 7.46 (d, J =1.0 Hz, 1 H), 7.22–7.25 (m, 3 H), 7.02–6.94 (m, 3 H), 4.57 (d, J =8.7 Hz, 1H), 4.14 (d, J =8.7 Hz, 1H), 2.91 (q, J =13.4 Hz, 2 H), 2.87 (q, J =13.4 Hz, 2H), 1.39 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ = 171.61 (C=O), 159.79 (C), 141.33 (C), 136.92 (C), 130.34 (CH), 129.61 (C), 128.08 (CH), 126.73 (CH), 123.31 (CH), 123.11 (CH), 111.14 (CH), 82.37 (CH2), 46.45 (C), 46.41 (CH2), 24.43 (CH3).
3-((E)-Hex-2-enyl)-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid (12 c)
Compound 11 c (200 mg, 0.73 mmol) was submitted to the same procedure described above for the preparation of 12 a. The product was obtained as a colorless oil (155 mg, 82%) that crystallized. mp: 172–173°C; 1H NMR (500 MHz, CDCl3): δ=7.70 (dd, J =7.7 Hz, 1.2 Hz, 1 H), 7.49 (d, J =0.9 Hz, 1 H), 7.17 (d, J = 7.7 Hz, 1H), 5.48 (dt, J =13.9 Hz, 6.8 Hz, 1 H), 5.30 (dt, J =14.9 Hz, 7.3 Hz, 1 H), 4.48 (d, J =8.6 Hz, 1 H), 4.20 (d, J =8.6 Hz, 1 H), 2.39–2.27 (m, 2 H), 1.98 (dd, J =14.1, 7.1 Hz, 2H), 1.42–1.31 (m, 6 H), 0.88 (t, J =7.4 Hz, 3H); 13C NMR (500 MHz, CDCl3): δ=170.99 (C=O), 159.79 (C), 141.77 (C), 135.15 (CH), 129.31 (C), 124.58 (CH), 123.21 (CH), 122.81 (CH), 110.96 (CH), 100.00 (C), 82.32 (CH2), 45.57 (C), 43.63 (CH2), 34.67 (CH2), 25.00 (CH3), 22.54 (CH2), 13.58 (CH3).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid-o-iodoanilide (13)
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU; 94 mg, 0.25 mmol) and a solution of N,N-diisopropylethylamine (DiPEA; 44 mg, 59 μL, 0.34 mmol) in DMF (1 mL) were added to a stirred suspension of compound 12a (60 mg, 0.225 mmol) and 2-iodoaniline (54 mg, 0.25 mmol) in CH2Cl2 (2 mL) and DMF (1 mL). The reaction mixture was stirred at room temperature for 18 h. The reaction medium was acidified by adding a solution of 1.2 m HCl and extracted with EtOAc. The organic phase was washed with H2O, dried (MgSO4), and concentrated to give the amide, which was purified by flash chromatography (EtOAc/heptane 4:6) to afford 10 mg (10%) of a light brown oil. 1H NMR (300 MHz, CDCl3): δ = 8.75 (dd, J = 4.4 Hz, 1.3 Hz, 1 H), 8.47 (dd, J =8.4 Hz, 1.4 Hz, 1H), 8.17 (dd, J =8.6 Hz, 2.0 Hz, 1H), 7.90 (d, J =1.9 Hz, 1H), 7.47 (dd, J =8.4 Hz, 4.5 Hz, 1H), 7.28–7.22 (m, 4H), 7.01 (dd, J =7.4 Hz, 1.7 Hz, 2 H), 6.89 (d, J = 8.5 Hz, 1 H), 4.70 (d, J =9.1 Hz, 1 H), 4.24 (d, J =9.1 Hz, 1 H), 2.98 (d, J =13.3 Hz, 1H), 2.91 (d, J =13.3 Hz, 2H), 1.45 (s, 3H).
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid cyclohexylamide (14)
Compound 12 a (60 mg, 0.225 mmol) was submitted to the same procedure described above for the preparation of 13 using cyclohexylamine (25 mg, 29 μL, 0.25 mmol). The organic phase was washed with H2O, dried (MgSO4), and concentrated to give the amide as a white solid. The solid was washed with a mixture of heptane and CH2Cl2/heptanes (9:1) to afford 38 mg (48 %) of a white solid. 1H NMR (300 MHz, CDCl3): δ = 7.56 (dd, J = 8.3, 1.9, 1H), 7.24 (dd, J =5.0 Hz, 1.9 Hz, 4H), 6.98 (dd, J =6.5 Hz, 2.8 Hz, 2 H), 6.75 (d, J =8.3 Hz, 1 H), 5.72 (d, J =7.4 Hz, 1 H), 4.57 (d, J =8.9 Hz, 1H), 4.14 (d, J =8.8 Hz, 1 H), 4.03–3.86 (m, 1 H), 2.93 (d, J =13.3 Hz, 1H), 2.86 (d, J =13.2 Hz, 1 H), 2.00–2.04 (m, 1 H), 1.83–1.53 (m, 4H), 1.52–1.32 (m, 5 H), 1.32–1.11 (m, 3H); 13C NMR (500 MHz, CDCl3): δ = 166.39 (C=O), 162.24 (C), 137.21 (C), 135.12 (C), 130.44 (CH), 127.96 (CH), 127.77 (CH), 127.41 (C), 126.68 (CH), 122.79 (CH), 109.34 (CH), 82.83 (CH2), 48.59 (CH), 46.64 (C), 46.06 (CH2), 33.34 (CH2), 25.64 (CH2), 24.95 (CH2), 24.68 (CH3); HRMS (ES+) calcd for C23H27NO2 [M+H]+ m/z: 350.2120, found: 350.2095.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid piperidine amide (15)
Compound 12 a (60 mg, 0.225 mmol) was submitted to the same procedure described above for the preparation of 13 using piperidine (21 mg, 25 μL, 0.25 mmol). The organic phase was washed with H2O, dried (MgSO4), and concentrated to give the amide, which was purified by flash chromatography (EtOAc/heptane 3:7) to afford 54 mg (71.5%) of the desired amide as a colorless oil. 1H NMR (300 MHz, CDCl3): δ = 7.25–7.16 (m, 4 H), 7.03 (d, J =1.7, 1H), 6.99 (dd, J =8.2 Hz, 1.7 Hz, 2 H), 6.73 (d, J = 8.2 Hz, 1 H), 4.53 (t, J =8.4 Hz, 1 H), 4.10 (d, J =8.4 Hz, 1 H), 3.53 (br s, 4H), 2.92 (d, J =13.3 Hz, 1 H), 2.86 (d, J =13.3 Hz, 1 H), 1.75–1.48 (m, 6 H), 1.37 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ = 170.77 (C=O), 160.80 (C), 137.37 (C), 135.14 (C), 130.44 (CH), 128.68 (C), 128.16 (CH), 128.02 (CH), 126.72 (CH), 123.05 (CH), 109.42 (CH), 82.58 (CH2), 46.68 (CH2), 46.32 (C), 29.84 (CH2), 24.81 (CH2), 24.72 (CH3); HRMS (ES +) calcd for C22H25NO2 [M+H]+ m/z: 336.1964, found: 336.1932.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid (2-iodophenyl)amide (16)
Compound 12b (80 mg, 0.3 mmol) was submitted to the same procedure described above for the preparation of 13 using 2-iodoaniline (72 mg, 0.33 mmol). The crude amide was purified by flash chromatography (EtOAc/heptane 4:6) to afford 22 mg of the desired amide as a pale yellow solid (16%). 1H NMR (300 MHz, CDCl3): δ = 8.74 (dd, J = 4.5 Hz, 1.4 Hz, 1 H), 8.46 (dd, J =8.4 Hz, 1.4 Hz, 1H), 7.85 (dd, J =7.8 Hz, 1.5 Hz, 1H), 7.61 (d, J =1.3 Hz, 1H), 7.46 (dd, J =8.4 Hz, 4.5 Hz, 1H), 7.29–7.26 (m, 2 H), 7.07 (d, J =7.8 Hz, 1H), 6.99 (dd, J =6.4 Hz, 3.0 Hz, 2H), 4.62 (d, J = 8.9 Hz, 1H), 4.21 (d, J =8.9 Hz, 1 H), 2.98 (d, J =13.3 Hz, 1 H), 2.92 (d, J =13.4 Hz, 1 H), 1.44 (s, 3H); 13C NMR (500 MHz, CDCl3): δ= 165.41 (C=O), 160.38 (C), 139.73 (C), 138.99 (C), 138.52 (CH), 137.12 (C), 135.27 (C), 130.54 (CH), 129.57 (CH), 128.27 (CH), 126.91 (CH), 126.17 (CH), 124.01 (CH), 121.96 (CH), 119.93, 108.64 (CH), 90.39 (C-I), 82.72 (CH2), 46.63(C), 46.56 (CH2), 24.74 (CH3); HRMS (ES +) calcd for C23H20INO2 [M+H] + m/z: 470.0617, found: 470.0638.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid cyclohexylamide (17)
Compound 12 b (80 mg, 0.3 mmol) was submitted to the same procedure described above for the preparation of 13 using cyclohexylamine (33 mg, 38 μL, 0.33 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 2:8) to afford 70 mg (67 %) of a white solid; mp: 119–121 °C; 1H NMR (300 MHz, CDCl3): δ=7.28–7.20 (m, 6 H), 7.11 (d, J =1.4 Hz, 1H), 6.98 (dd, J =6.4 Hz, 3.1 Hz, 2 H), 6.92 (d, J =7.7 Hz, 1H), 5.87 (d, J = 6.8 Hz, 1 H), 4.54 (d, J =8.8 Hz, 1 H), 4.11 (d, J =8.8 Hz, 1H), 4.03–3.88 (m, 1 H), 2.91 (d, J =13.3 Hz, 1H), 2.86 (d, J =13.3 Hz, 1 H), 2.02 (m, 2 H), 1.82–1.60 (m, 3 H), 1.52–1.32 (m, 5H), 1.31–1.13 (m, 3 H); 13C NMR (500 MHz, CDCl3): δ = 166.48 (C=O), 159.88 (C), 138.29 (C), 137.08 (C), 135.76 (C), 130.36 (CH), 128.03 (CH), 126.62 (CH), 123.39 (CH), 119.31 (CH), 108.17 (CH), 82.46 (CH2), 48.64 (CH), 46.43 (CH2), 46.26 (C), 33.23 (CH2), 25.59 (CH2), 24.90 (CH2), 24.59 (CH3); HRMS (ES+) calcd for C23H27NO2 [M+H] + m/z: 350.2120, found: 350.2090.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid piperidine amide (18)
Compound 12 b (80 mg, 0.3 mmol) was submitted to the same procedure described above for the preparation of 13 using piperidine (28 mg, 33 μL, 0.33 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 4:6) to afford 50 mg (50%) of a white solid; mp: 108°C; 1H NMR (500 MHz, CDCl3): δ = 7.25–7.20 (m, 3H), 7.00 (dd, J = 7.0 Hz, 1.2 Hz, 2H), 6.94 (d, J =7.5 Hz, 1 H), 6.89 (dd, J =7.5 Hz, 1.2 Hz, 1 H), 6.75 (d, J = 0.7 Hz, 1H), 4.53 (d, J =8.7 Hz, 1H), 4.09 (d, J =8.7 Hz, 1H), 3.41 (br s, 2 H), 3.69 (br s, 2 H), 2.90 (d, J =13.3 Hz, 1H), 2.86 (d, J = 13.3 Hz, 1H), 1.71–1.46 (m, 6H), 1.36 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 170.18 (C=O), 159.47 (C), 137.26 (C), 136.76 (C), 136.20 (C), 130.36 (CH), 127.99 (CH), 126.58 (CH), 123.42 (CH), 119.08 (CH), 108.20 (CH), 82.28 (CH2), 46.56 (CH2), 46.22 (C), 24.64 (CH2), 24.56 (CH3); HRMS (ES +) calcd for C22H25NO2 [M+H]+ m/z: 336.1964, found: 336.1964.
3-((E)-Hex-2-enyl)-3-methyl-2,3-dihydrobenzofuran-6-yl]piperidin-1-ylmethanone (19)
Compound 12c (70 mg, 0.27 mmol) was submitted to the same procedure described above for the preparation of 13 using piperidine (25 mg, 0.29 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 3:7) to afford 69 mg (78%) of a colorless oil. 1H NMR (300 MHz, CDCl3): δ = 7.07 (d, J =7.5 Hz, 1 H), 6.89 (dd, J =7.5 Hz, 1.4 Hz, 1 H), 6.77 (d, J = 1.0 Hz, 1H), 5.50–5.41 (dt, J =13.8 Hz, 6.8 Hz, 1 H), 5.35–5.24 (m, 1 H), 4.41 (d, J =8.6 Hz, 1 H), 4.13 (d, J =8.6 Hz, 1H), 3.68 (s, 2 H), 3.35 (s, 2 H), 2.28 (d, J =7.1 Hz, 2 H), 1.96 (dd, J =14.0 Hz, 7.1 Hz, 2 H), 1.76–1.58 (m, 6 H), 1.42–1.23 (m, 4H), 0.86 (t, J =7.3 Hz, 3 H); 13C NMR (500 MHz, CDCl3): δ = 170.27 (C=O), 159.48 (C), 136.62 (C), 136.52 (C), 134.80 (CH), 124.96 (CH), 122.99 (CH), 119.14 (CH), 108.06 (CH), 82.28 (CH2), 45.31 (C), 43.70 (CH2), 34.67 (CH2), 25.16 (CH3), 24.64 (CH2), 22.58 (CH2), 13.61 (CH3); HRMS (ES +) calcd for C21H29NO2 [M+H]+ m/z: 328.2277, found: 328.2247.
3-((E)-Hex-2-enyl)-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid cyclohexylamide (20)
Compound 12 c (70 mg, 0.27 mmol) was submitted to the same procedure described above for the preparation of 13 using cyclohexylamine (30 mg, 0.3 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 2:8) to afford 83 mg (90%) of a colorless oil. 1H NMR (300 MHz, CDCl3): δ=7.27 (dd, J =7.7 Hz, 1.3 Hz, 1 H), 7.12 (d, J =1.3 Hz, 1H), 7.09 (d, J =7.7 Hz, 1H), 5.86 (br d, J =8.5 Hz, 1 H), 5.51–5.39 (m, 1 H), 5.33–5.20 (m, 1 H), 4.42 (d, J =8.7 Hz, 1H), 4.15 (d, J =8.7 Hz, 1H), 4.04–3.87 (m, 1 H), 2.34–2.23 (m, 2H), 2.10–1.89 (m, 4H), 1.81–1.69 (m, 4 H), 1.52–1.13 (m, 9H), 0.85 (t, J =7.3 Hz, 3H); 13C NMR (500 MHz, CDCl3): δ = 165.06 (C=O), 158.36 (C), 137.23 (C), 134.08 (C), 133.41 (CH), 123.25 (CH), 121.35 (CH), 117.83 (CH), 106.49 (CH), 80.83 (CH2), 47.06 (CH3), 43.83 (CH2), 42.13 (C), 33.14 (CH2), 31.70 (CH2), 28.17 (CH2), 24.05 (CH2), 23.60 (CH3), 23.35 (CH2), 21.02 (CH2), 12.07 (CH3); HRMS (ES +) calcd for C22H31NO2 [M+H]+ m/z: 342.2433, found: 342.2463.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-5-carboxylic acid (2,2-dimethylpropyl)amide (21)
Compound 12 b (60 mg, 0.225 mmol) was submitted to the same procedure described above for the preparation of 13 using neopentylamine (29 mg, 0.33 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 2:8) to afford 53 mg (60 %) of a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 7.29 (dd, J = 7.7 Hz, 1.3 Hz, 1H), 7.25–7.22 (m, 3 H), 7.13 (d, J =1.1 Hz, 1 H), 6.99 (dd, J =7.0 Hz, 2.0 Hz, 2H), 6.94 (d, J =7.7 Hz, 1H), 6.08 (s, 1H), 4.55 (d, J =8.7 Hz, 1H), 4.12 (d, J = 8.7 Hz, 1H), 3.26 (d, J =6.3 Hz, 2 H), 2.91 (d, J =13.4 Hz, 1 H), 2.87 (d, J =13.3 Hz, 1H), 1.37 (s, 3H), 0.98 (s, 9 H); 13C NMR (500 MHz, CDCl3): δ = 167.56 (C=O), 159.91 (C), 138.43 (C), 137.06 (C), 135.70 (C), 130.37 (CH), 128.04 (CH), 126.65 (CH), 123.50 (CH), 119.36 (CH), 108.11(CH), 82.48 (CH2), 51.00 (CH2), 46.42 (CH2), 46.28 (C), 32.15 (C), 27.30 (CH3), 24.58 (CH3); HRMS (ES +) calcd for C22H27NO2 [M+H]+ m/z: 338.2120, found: 338.2148.
(3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-yl)morpholinomethanone (22)
Compound 12 b (70 mg, 0.26 mmol) was submitted to the same procedure described above for the preparation of 13 using morpholine (48 mg, 0.56 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 4:6) to afford 56 mg (64 %) of a colorless oil. 1H NMR (300 MHz, CDCl3): δ = 7.22–7.24 (m, 2 H), 7.00–7.02 (m, 2H), 6.96 (d, J =7.5 Hz, 1 H), 6.90 (dd, J =7.6 Hz, 1.3, 1H), 6.76 (d, J =0.9 Hz, 1H), 4.54 (d, J =8.8 Hz, 0 H), 4.10 (d, J = 8.8 Hz, 0 H), 3.59 (br m, 4H), 2.91 (d, J =13.4 Hz, 1 H), 2.85 (d, J = 13.4 Hz, 1 H), 1.36 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ = 170.34 (C= O), 159.58 (C), 137.13 (C), 136.85 (C), 135.50 (C), 130.34 (CH), 128.01 (CH), 126.63 (CH), 123.61 (CH), 119.38 (CH), 108.44 (CH), 82.35 (CH2), 66.92 (CH2), 46.51 (C), 46.24, 24.56(CH3); HRMS (ES +) calcd for C21H23NO3 [M+H] + m/z: 338.1756, found: 338.1763.
3-Benzyl-N-tert-butyl-N,3-dimethyl-2,3-dihydrobenzofuran-6-carboxamide (23)
Compound 12 b (70 mg, 0.26 mmol) was submitted to the same procedure described above for the preparation of 13 using N-tert-butylmethylamine (29 mg, 0.29 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 2:8) to afford 69 mg (76%) of an off-white oil; mp: 71°C; 1H NMR (300 MHz, CDCl3): δ = 7.22–7.23 (m, 3 H), 6.99 (dd, J = 6.4 Hz, 3.0, 2 H), 6.94 (dd, J =7.6 Hz, 1.3 Hz, 1H), 6.89 (d, J =7.5 Hz, 1H), 6.78 (d, J =1.1 Hz, 1H), 4.52 (d, J =8.8 Hz, 0 H), 4.08 (d, J =8.8 Hz, 0 H), 2.94–2.81 (m, 5H), 1.49 (s, 9H), 1.35 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ = 172.98 (C=O), 159.42 (C), 139.44 (C), 137.26 (C), 136.21 (C), 130.35 (CH), 127.95 (CH), 126.53 (CH), 123.25 (CH), 119.63 (CH), 108.63 (CH), 82.25 (CH2), 56.42 (C), 46.53 (CH2), 46.18 (C), 35.34 (CH3), 27.73 (CH3), 24.50 (CH3); HRMS (ES +) calcd for C22H27NO2 [M+H]+ m/z: 338.2120, found: 338.2147.
3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid (tetrahydropyran-4-ylmethyl)amide (24)
Compound 12 b (70 mg, 0.26 mmol) was submitted to the same procedure described above for the preparation of 13 using 4-(aminomethyl)tetrahydropyran (46 mg, 0.4 mmol). Crude amide was purified by flash chromatography (EtOAc/heptane 5:5) to afford 41 mg (43%) of a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 7.28 (dd, J = 7.7, 1.4, 1H), 7.23–7.24 (m, 3H), 7.12 (d, J =1.2, 1 H), 6.97 (dd, J =6.9 Hz, 2.4 Hz, 2H), 6.93 (d, J =7.7 Hz, 1H), 6.26 (br s, 1H), 4.54 (d, J =8.7 Hz, 1H), 4.12 (d, J =8.7 Hz, 1 H), 3.98 (dd, J =11.3 Hz, 3.2 Hz, 2H), 3.42–3.31 (m, 4 H), 2.91 (d, J =13.3 Hz, 1 H), 2.86 (d, J =13.3 Hz, 1 H), 1.95–1.82 (m, 1H), 1.66 (d, J =11.6 Hz, 3 H), 1.48–1.27 (m, 5 H); 13C NMR (500 MHz, CDCl3): δ = 167.60 (C=O), 159.95 (C), 138.61 (C), 137.03 (C), 135.20 (C), 130.36 (CH), 128.05 (CH), 126.66 (CH), 123.50 (CH), 119.37 (CH), 108.12 (CH), 82.47 (CH2), 67.62 (CH2), 46.43 (CH2), 46.29 (C), 45.69 (CH2), 35.39 (CH), 30.70 (CH2), 24.57 (CH3); HRMS (ES +) calcd for C23H27NO3 [M+H]+ m/z: 366.2069, found: 366.2065.
4-Iodo-3-(2-methylallyloxy)benzoic acid (25)
Compound 10 b (2 g, 6 mmol) was submitted to the same procedure described above for the preparation of 12 a. The product was obtained as a colorless oil (1.8 g, 94%). 1H NMR (300 MHz, CDCl3): δ = 7.91 (d, J = 7.9 Hz, 1 H), 7.46 (m, 2 H), 5.23 (s, 1 H), 5.06 (s, 1H), 4.57 (s, 2H), 1.89 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ=171.43 (C=O), 157.34 (C), 139.75 (CH), 139.69 (C), 130.51 (C), 123.99 (CH), 113.39 (CH2), 112.73 (CH), 94.62 (C), 72.73 (CH2), 19.49 (CH3).
N-Cyclohexyl-4-iodo-3-(2-methylallyloxy)benzamide (26 a)
HATU (449 mg, 1.18 mmol) and then DiPEA (229 mg, 1.77 mmol, 0.31 mL) were added to a stirred suspension of 25 (350 mg 1.1 mmol) and cyclohexylamine (117 mg, 1.18 mmol, 0.134 mL) in CH2Cl2 (5 mL) and DMF (5 mL). The reaction mixture was stirred at room temperature for 18 h. The reaction medium was acidified by the addition of a solution of HCl (3.7 %) and extracted with CH2Cl2. The organic phase was washed three times with H2O, dried (MgSO4), and concentrated to give the crude amide. Column chromatography (silica gel, heptane/EtOAc 7:3) afforded the title compound as a white solid (418 mg, 95 %); mp: 124–126 °C; 1H NMR (500 MHz, CDCl3): δ=7.80 (d, J =8.0 Hz, 1H), 7.29 (d, J =1.7 Hz, 1H), 6.94 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 5.93 (d, J =7.4 Hz, 1 H), 5.22 (s, 1 H), 5.04 (s, 1 H), 4.55 (s, 2 H), 4.02–3.90 (m, 1H), 2.03 (dd, J =12.4 Hz, 3.1 Hz, 2 H), 1.87 (s, 3H), 1.80–1.71 (m, 2H), 1.70–1.54 (m, 5H), 1.49–1.36 (m, 2 H), 1.20–1.27 (m, 4H); 13C NMR (500 MHz, CDCl3): δ = 165.87, (C= O) 157.55 (C), 139.86 (C), 139.24 (CH), 136.53 (C), 119.50 (CH), 113.16 (CH2), 111.20 (CH), 90.48 (C), 72.65 (CH2), 48.92 (CH), 33.15 (CH2), 25.54 (CH2), 24.88 (CH2), 19.48 (CH3).
[4-Iodo-3-(2-methylallyloxy)phenyl]-(1-piperidyl)methanone (26 b)
Compound 25 (1.4 g, 4.4 mmol) was submitted to the same procedure described above for the preparation of 26 a. The resulting oil was purified by column chromatography (silica gel, CH2Cl2). After concentration, 500 mg (30%) of a white solid was obtained; mp: 43 °C; 1H NMR (500 MHz, CDCl3): δ = 7.79 (d, J = 7.9 Hz, 1 H), 6.83 (d, J =1.6 Hz, 1 H), 6.71 (dd, J =7.9 Hz, 1.7 Hz, 1 H), 5.19 (s, 1H), 5.02 (s, 1H), 4.50 (s, 2 H), 3.69 (br s, 2 H), 3.33 (brs, 2H), 1.87 (s, 3 H), 1.68 (br s, 4 H), 1.51 (br s, 2H); 13C NMR (500 MHz, CDCl3): δ = 169.27 (C =O), 157.28 (C), 139.86 (C), 139.37 (CH), 137.81(C), 120.57 (CH), 113.13 (CH2), 110.78 (CH), 87.77 (C), 72.64 (CH2), 24.56 (CH2), 19.46 (CH3).
3-Methyl-3-naphthalen-1-ylmethyl-2,3-dihydrobenzofuran-6-carboxylic acid cyclohexylamide (27)
Compound 26 a (547 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a using 1-naphthylboronic acid (282 mg, 1.64 mmol). Column chromatography (silica gel, heptane/EtOAc 8:2) afforded 368 mg (43 %) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3): δ = 7.88–7.79 (m, 2 H), 7.75 (d, J = 8.2 Hz, 1 H), 7.47–7.31 (m, 3 H), 7.12–7.17 (m, 3 H), 6.84 (d, J = 8.0 Hz, 1 H), 5.85 (d, J =7.8 Hz, 1 H), 4.57 (d, J =8.8 Hz, 1 H), 4.09 (d, J =8.8 Hz, 1 H), 3.96 (tdt, J =12.2 Hz, 8.3 Hz, 4.0 Hz, 1 H), 3.44 (d, J = 13.9 Hz, 1H), 3.33 (d, J =13.9 Hz, 1H), 2.02 (d, J =13.1 Hz, 2 H), 1.82–1.61 (m, 3 H), 1.53–1.35 (m, 5H), 1.32–1.13 (m, 3 H); 13C NMR (500 MHz, CDCl3): δ = 166.47 (C=O), 159.78 (C), 138.62 (C), 135.81 (C), 133.80 (C), 133.45 (C), 133.00 (C), 128.81 (CH), 128.72 (CH), 127.47 (CH), 125.75 (CH), 125.36 (CH), 124.97 (CH), 124.05 (CH), 123.42 (CH), 119.26 (CH), 108.33 (CH), 82.71 (CH2), 48.60 (CH), 47.07 (CH2), 41.40 (C), 33.22 (CH2), 25.57 (CH2), 24.87 (CH2), 24.47(CH3); HRMS (ES +) calcd for C27H29NO2 [M+H]+ m/z: 400.2277, found: 400.2292.
(3-Methyl-3-naphthalen-1-ylmethyl-2,3-dihydrobenzofuran-6-yl)-piperidin-1-ylmethanone (28)
Compound 26 b (528 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a using 1-naphthylboronic acid (282 mg, 1.64 mmol). Column chromatography (silica gel, heptane/EtOAc 7:3) afforded 227 mg (43%) of the title compound as a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 7.81 (dd, J = 8.1 Hz, 3.7 Hz, 2H), 7.75 (d, J =8.2 Hz, 1H), 7.43–7.32 (m, 3H), 7.20 (d, J =6.9 Hz, 1H), 6.80 (s, 1 H), 6.77–6.71 (m, 2H), 4.58 (d, J =8.7 Hz, 1 H), 4.09 (d, J = 8.7 Hz, 1H), 3.68 (br s, 2 H), 3.47 (d, J =13.9 Hz, 1H), 3.31 (m, 3H), 1.64–1.47 (m, 6 H), 1.43 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 24.32 (CH3), 24.64 (CH2), 41.60 (CH2), 46.94 (C), 82.84 (CH2), 108.33 (CH), 119.02 (CH), 123.56 (CH), 124.19 (CH), 124.98 (CH), 125.27 (CH), 125.61 (CH), 127.46 (CH), 128.69 (CH), 128.77 (CH), 133.06 (C), 133.68 (C), 133.80 (C), 136.29 (C), 136.76 (C), 159.47 (C), 170.15 (C= O); HRMS (ES +) calcd for C26H27NO2 [M+H] + m/z: 386.2120, found: 386.2126.
(3-Methyl-3-naphthalen-2-ylmethyl-2,3-dihydrobenzofuran-6-yl)-piperidin-1-ylmethanone (29)
Compound 26 b (528 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a using 2-naphthylboronic acid (282 mg, 1.64 mmol). Column chromatography (silica gel, heptane/EtOAc 7:3) afforded 116 mg (22%) of the title compound as a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 7.83–7.77 (m, 1 H), 7.72 (dd, J = 12.1 Hz, 6.1 Hz, 2H), 7.48–7.41 (m, 3H), 7.12 (dd, J =8.4 Hz, 1.4 Hz, 1 H), 6.95 (d, J =7.5 Hz, 1 H), 6.89 (dd, J =7.5 Hz, 1.2 Hz, 1H), 6.75 (d, J =0.7 Hz, 1H), 4.61 (d, J =8.7 Hz, 1 H), 4.12 (d, J =8.7 Hz, 1 H), 3.69 (s, 2 H), 3.33 (s, 2H), 3.06 (d, J =13.4 Hz, 1H), 3.03 (d, J = 13.4 Hz, 1H), 1.58–1.67 (m, 6H), 1.41 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 170.18 (C=O), 159.50 (C), 136.82 (C), 136.14 (C), 134.88 (C), 133.18 (C), 132.21 (C), 128.94 (CH), 128.79 (CH), 127.58 (CH), 127.57 (CH), 127.41 (CH), 125.99 (CH), 125.56 (CH), 123.51 (CH), 119.08 (CH), 108.24 (CH), 82.35 (CH2), 46.75 (CH2), 46.47 (C), 24.64 (CH2), 24.59(CH3); HRMS (ES +) calcd for C26H27NO2 [M+H] + m/z: 386.2120, found: 386.2121.
[3-(4-Chlorobenzyl)-3-methyl-2,3-dihydrobenzofuran-6-yl]piperidin-1-ylmethanone (30)
Compound 26 b (528 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a using 4-chlorophenylboronic acid (256 mg, 1.64 mmol). Column chromatography (silica gel, heptane/EtOAc 6:4) afforded 187 mg (37 %) of the title compound as a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 7.19 (d, J = 8.3 Hz, 2 H), 6.90 (dd, J =6.1 Hz, 2.1 Hz, 4H), 6.74 (s, 1H), 4.48 (d, J =8.8 Hz, 1H), 4.11 (d, J =8.8 Hz, 1 H), 3.69 (br s, 2 H), 3.33 (br s, 2 H), 2.85 (d, J =13.5 Hz, 1 H), 2.82 (d, J =13.5 Hz, 1H), 1.66 (m, 6H), 1.36 (s, 3 H); 13C NMR (500 MHz, CDCl3): δ = 170.08(C=O), 159.47(C), 136.92(C), 135.67(C), 135.58(C), 132.58(C), 131.60 (CH), 128.10 (CH), 123.44 (CH), 119.13 (CH), 108.28 (CH), 82.23 (CH), 46.17 (C), 46.01 (C), 24.62 (CH2), 24.37 (CH2); HRMS (ES +) calcd for C22H24ClNO2 [M+H] + m/z: 370.1574, found: 370.1596.
[3-[(4-Methoxyphenyl)methyl]-3-methyl-2,3-dihydrobenzofuran-6-yl]-(1-piperidyl)methanone (31)
Compound 26b (275 mg, 0.71 mmol) was submitted to the same procedure described above for the preparation of 11 a using 4-methoxyphenylboronic acid (130 mg, 0.85 mmol). Column chromatography (silica gel, heptane/EtOAc 6:4) afforded 133 mg (51 %) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3): δ = 6.94–6.85 (m, 4 H), 6.76 (dt, J =4.8 Hz, 2.8 Hz, 2 H), 4.49 (t, J =9.3 Hz, 1H), 4.08 (d, J =8.7 Hz, 1 H), 3.73–3.61 (m, 2 H), 3.33 (s, 2 H), 2.83 (d, J =13.5 Hz, 1 H), 2.78 (d, J =13.5 Hz, 1 H), 1.75–1.44 (m, 6 H), 1.33 (s, 3H); 13C NMR (500 MHz, CDCl3): δ = 170.66 (C=O), 159.87 (C), 158.73 (C), 136.99 (C), 136.68 (C), 131.70 (C), 129.70 (CH), 123.89 (CH), 119.46 (CH), 113.76 (CH), 108.53 (CH), 82.69 (CH2), 55.60 (CH3), 46.68 (C), 46.10 (CH2), 25.02 (CH2), 24.91 (CH3); HRMS (ES +) calcd for C23H27NO3 [M+H]+ m/z: 366.2069, found: 336.2075.
[3-Methyl-3-(4-pyridylmethyl)-2,3-dihydrobenzofuran-6-yl]-(1-piperidyl)methanone (32)
Compound 26 b (528 mg, 1.37 mmol) was submitted to the same procedure described above for the preparation of 11 a using 4-pyridineboronic acid (201 mg, 1.64 mmol). Column chromatography (silica gel, CH2Cl2/EtOH 9:1) afforded 133 mg (15%) of the title compound as an off-white solid; mp: 115°C; 1H NMR (300 MHz, CDCl3): δ = 8.45 (d, J = 5.0 Hz, 2 H), 6.99–6.83 (m, 5 H), 6.75 (s, 1H), 4.47 (d, J =8.9 Hz, 1H), 4.12 (d, J =8.9 Hz, 1H), 3.69 (br s, 2H), 3.33 (br s, 2H), 22.89 (d, J =13.1 Hz, 1 H), 2.86 (d, J =13.1 Hz, 1 H), 1.52–1.68 (m, 6 H), 1.40 (s, 3H); 13C NMR (500 MHz, CDCl3): δ=170.09 (C=O), 159.61 (C), 149.56 (CH), 146.29 (C), 137.37 (C), 135.21 (C), 125.73 (CH), 123.48 (CH), 119.41 (CH), 108.62 (CH), 82.30 (CH2), 46.19 (C), 46.14 (CH2), 24.76 (CH2), 24.58 (CH3); HRMS (ES +) calcd for C21H24N2O2 [M+H]+ m/z: 337.1916, found: 337.1902.
(S)-3-Benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid piperidine amide (33) and (R)-3-benzyl-3-methyl-2,3-dihydrobenzofuran-6-carboxylic acid piperidine amide (34)
Optimal analytical conditions for separation of the two enantiomers from racemic mixture 18 were determined using the following conditions: CYCLOBOND DMP column (Supelco Cat. No. 20724AST) 25 cm × 4.6 mm i.d., 5 μm, mobile phase: CH3CN/AcOH/TEA 100:0.01:0.05, T =10°C, flow rate: 0.3 mL min−1, detection: UV (λ=288 nm), injection volume: 10 μL, sample: 4.0 mg mL−1 of compound 18 in solution (CH3CN/AcOH/TEA 100:0.01:0.05); tR for compound 33: 15.73 min; tR for compound 34: 16.63 min.
A loading study revealed that up to 40 μg (10 μL of a 4.0 mgmL−1 solution in CH3CN/AcOH/TEA 100:0.01:0.05) could be loaded on an analytical CYCLOBOND DMP column while maintaining good resolution and selectivity. After translation of analytical loading conditions to the preparative scale, optimal conditions were obtained with the following parameters: CYCLOBOND DMP column (Supelco Cat. No. 20744AST) 25 cm × 21.2 mm i.d., 5 μm, mobile phase: CH3CN/AcOH/TEA 100:0.01:0.05, T = 10 °C, flow rate: 6.4 mL min−1, detection: UV (λ=288 nm), injection volume: 80 μL, sample: 4.0 mg mL−1 of compound 18 in a solution of CH3CN/AcOH/TEA 100:0.01:0.05. Fractions for compounds 33 and 34 were collected separately. Fractions for each peak were concentrated to dryness by rotary evaporation at 40°C under vacuum. Fractions were then resuspended in CH3CN and re-concentrated (3× in 25 mL CH3CN) to remove residual moisture. Hexane was added to each flask to precipitate each enantiomer in the form of an off-white solid. The solid off-white powder resulting from each peak was scraped from the round-bottom flask, transferred to a vial, and left to air-dry overnight on a hot plate at low heat (30 °C) to remove residual solvent. An enantiomeric purity analysis was then performed on each final product. Enantiomeric purity of compound 33 fell within acceptable limits (>95% ee); however, this was not the case with compound 34. To ensure that sufficient material from compound 34 was produced, separation of additional racemic compound was re-initiated. The fractions from peak 2 were then collected, and sample recovery steps 2 and 3 were repeated. Because separations thus far produced peaks outside the acceptable limits of enantiomeric purity, the semi-pure compound 34 was re-purified on the preparatory system to remove residual compound 33 impurities. Sample recovery steps 2–4 were then repeated to obtain material with acceptable enantiomeric purity. Approximately 750 mg of racemic material was processed. Purity analysis indicated that the final solid material of compound 33 (tR = 16.84 min) has an enantiomeric purity of 97.3 % while the final solid material of compound 34 (tR = 18.38 min) has an enantiomeric purity of 97.5 %. A total of 109.4 mg of compound 33 was recovered; a total of 112.5 mg of compound 34 was recovered.
Supplementary Material
Acknowledgments
This work was supported in part by NIH Grant P30 M64023. We thank Dr. Kumar Kaluarachchi for performing NMR studies. The NMR facility at the M. D. Anderson Cancer Center is supported by Cancer Center Support Grant CA16672 awarded by the National Cancer Institute. We thank Ms. Beverly Parker for performing LC–MS and HRMS analyses.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.200900226: details of ADMET studies, further characterization data (HPLC–MS) for compounds 13–32, as well as crystallographic data for compound 33.
References
- 1.McPartland JM. Brain Res Rev. 2004;45:18–29. doi: 10.1016/j.brainresrev.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. J Neuroinflammation. 2005;2:29–41. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romero-Sandoval A, Eisenach JC. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- 5.Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, Mark L, Pearson MS, Miller W, Shan S, Rabadi L, Rotshteyn Y, Chaffer SM, Turchin PI, Elsemore DA, Toth M, Koetzner L, Whiteside GT. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, Grayson GK, Zhu CZ, Pai M, Chandran P, Salyers AK, Wensink EJ, Honore P, Sullivan JP, Dart MJ, Meyer MD. Br J Pharmacol. 2008;153:390–401. doi: 10.1038/sj.bjp.0707568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naguib M, Diaz F, Xu J, Astruc-Diaz F, Craig S, Vivas-Mejia P, Brown DL. Br J Pharmacol. 2008;155:1104–1116. doi: 10.1038/bjp.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guindon J, Hohmann AG. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karsak M, Gaffal E, Date R, Wang-Eckhardt L, Rehnelt J, Petrosino S, Starowicz K, Steuder R, Schlicker E, Cravatt B, Mechoulam R, Buettner R, Werner S, Di Marzo V, Tuting T, Zimmer A. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- 10.Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 12.Munro S, Thomas KL, Abu-Shaar M. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 13.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ameri A. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 15.Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A. Proc Natl Acad Sci USA. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warms CA, Turner JA, Marshall HM, Cardenas DD. Clin J Pain. 2002;18:154–163. doi: 10.1097/00002508-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Davis MP. Support Care Cancer. 2007;15:363–372. doi: 10.1007/s00520-006-0156-0. [DOI] [PubMed] [Google Scholar]
- 18.Khaliq W, Alam S, Puri N. Cochrane Database Syst Rev. 2007;2:CD004846. doi: 10.1002/14651858.CD004846.pub2. [DOI] [PubMed] [Google Scholar]
- 19.Tassone DM, Boyce E, Guyer J, Nuzum D. Clin Ther. 2007;29:26–48. doi: 10.1016/j.clinthera.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Gilron I, Coderre TJ. Expert Opin Emerging Drugs. 2007;12:113–126. doi: 10.1517/14728214.12.1.113. [DOI] [PubMed] [Google Scholar]
- 21.Jensen MP, Chodroff MJ, Dworkin RH. Neurology. 2007;68:1178–1182. doi: 10.1212/01.wnl.0000259085.61898.9e. [DOI] [PubMed] [Google Scholar]
- 22.Cox ML, Haller VL, Welch SP. Eur J Pharmacol. 2007;570:50–56. doi: 10.1016/j.ejphar.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, Reggiani A. Eur J Neurosci. 2006;23:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- 24.Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP., Jr Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 25.Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J. Neuroscience. 2005;135:235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O’Donnell D. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- 27.Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. J Neurosci Res. 2006;83:1310–1322. doi: 10.1002/jnr.20821. [DOI] [PubMed] [Google Scholar]
- 29.Page D, Yang H, Brown W, Walpole C, Fleurent M, Fyfe M, Gaudreault F, St-Onge S. Bioorg Med Chem Lett. 2007;17:6183–6187. doi: 10.1016/j.bmcl.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Giblin GMP, O’Shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, Rawlings DA, Goldsmith P, Brown AJ, Haslam CP, Clayton NM, Wilson AW, Chessell IP, Wittington AR, Green R. J Med Chem. 2007;50:2597–2600. doi: 10.1021/jm061195+. [DOI] [PubMed] [Google Scholar]
- 31.Huffman JW. Mini Rev Med Chem. 2005;5:641–649. doi: 10.2174/1389557054368844. [DOI] [PubMed] [Google Scholar]
- 32.Huffman JW, Yu S, Showalter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH. J Med Chem. 1996;39:3875–3877. doi: 10.1021/jm960394y. [DOI] [PubMed] [Google Scholar]
- 33.Murineddu G, Lazzari P, Ruiu S, Sanna A, Loriga G, Manca I, Falzoi M, Dessi C, Curzu MM, Chelucci G, Pani L, Pinna GA. J Med Chem. 2006;49:7502–7512. doi: 10.1021/jm060920d. [DOI] [PubMed] [Google Scholar]
- 34.Manera C, Benetti V, Castelli MP, Cavallini T, Lazzarotti S, Pibiri F, Saccomanni G, Tuccinardi T, Vannacci A, Martinelli A, Ferrarini PL. J Med Chem. 2006;49:5947–5957. doi: 10.1021/jm0603466. [DOI] [PubMed] [Google Scholar]
- 35.Salo OMH, Raitio KH, Savinainen JR, Nevalainen T, Lahtela-Kakkonen M, Laitinen JT, J3rvinen T, Poso A. J Med Chem. 2005;48:7166–7171. doi: 10.1021/jm050565b. [DOI] [PubMed] [Google Scholar]
- 36.Ohta H, Ishizaka T, Yoshinaga M, Morita A, Tomishima Y, Toda Y, Saito S. Bioorg Med Chem Lett. 2007;17:5133–5135. doi: 10.1016/j.bmcl.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Ohta H, Ishizaka T, Tatsuzuki M, Yoshinaga M, Iida I, Tomishima Y, Toda Y, Saito S. Bioorg Med Chem Lett. 2007;17:6299–6304. doi: 10.1016/j.bmcl.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Brink CB. Trends Pharmacol Sci. 2002;23:454. doi: 10.1016/s0165-6147(02)02079-5. [DOI] [PubMed] [Google Scholar]
- 39.Kenakin T. Trends Pharmacol Sci. 1995;16:256–258. doi: 10.1016/s0165-6147(00)89037-9. [DOI] [PubMed] [Google Scholar]
- 40.Dziadulewicz EK, Bevan SJ, Brain CT, Coote PR, Culshaw AJ, Davis AJ, Edwards LJ, Fisher AJ, Fox AJ, Gentry C, Groarke A, Hart TW, Huber W, James IF, Kesingland A, LaVecchia L, Loong Y, Lyothier I, McNair K, O’Farrell C, Peacock M, Portmann R, Schopfer U, Yaqoob M, Zadrobilek J. J Med Chem. 2007;50:3851–3856. doi: 10.1021/jm070317a. [DOI] [PubMed] [Google Scholar]
- 41.Gardin A, Kucher K, Kiese B, Appel-Dingemanse S. Drug Metab Dispos. 2009;37:827–833. doi: 10.1124/dmd.108.024000. [DOI] [PubMed] [Google Scholar]
- 42.Diaz P, Xu JJ, Astruc-Diaz F, Pan H-M, Brown DL, Naguib M. J Med Chem. 2008;51:4932–4947. doi: 10.1021/jm8002203. [DOI] [PubMed] [Google Scholar]
- 43.Yao BB, Hsieh G, Daza AV, Fan Y, Grayson GK, Garrison TR, El Kouhen O, Hooker BA, Pai M, Wensink EJ, Salyers AK, Chandran P, Zhu CZ, Zhong C, Ryther K, Gallagher ME, Chin C-L, Tovcimak AE, Hradil VP, Fox GB, Dart MJ, Honore P, Meyer MD. J Pharmacol Exp Ther. 2009;328:141–151. doi: 10.1124/jpet.108.145011. [DOI] [PubMed] [Google Scholar]
- 44.McGaraughty S, Chu KL, Dart MJ, Yao BB, Meyer MD. Neuroscience. 2009;158:1652–1661. doi: 10.1016/j.neuroscience.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 45.Diaz P, Phatak SS, Xu J, Astruc-Diaz F, Cavasotto CN, Naguib M. J Med Chem. 2009;52:433–444. doi: 10.1021/jm801353p. [DOI] [PubMed] [Google Scholar]
- 46.Edgar KJ, Falling SN. J Org Chem. 1990;55:5287–5291. [Google Scholar]
- 47.Diaz P, Gendre F, Stella L, Charpentier B. Tetrahedron. 1998;54:4579–4590. [Google Scholar]
- 48.Szlosek-Pinaud M, Diaz P, Martinez J, Lamaty F. Tetrahedron Lett. 2003;44:8657–8659. [Google Scholar]
- 49.Szlosek-Pinaud M, Diaz P, Martinez J, Lamaty F. Tetrahedron. 2007;63:3340–3349. [Google Scholar]
- 50.Flack HD. Acta Crystallogr. 1983;A39:876–881. [Google Scholar]
- 51.Hooft RWW, Straver LH, Spek AL. J Appl Crystallogr. 2008;41:96–103. doi: 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng J. BMC Struct Biol. 2008;8:18–30. doi: 10.1186/1472-6807-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hazai E, Bikadi Z. J Struct Biol. 2008;162:63–74. doi: 10.1016/j.jsb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 54.Larsson P, Wallner B, Lindahl E, Elofsson A. Protein Sci. 2008;17:990–1002. doi: 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 57.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 58.Bhattacharya S, Hall SE, Li H, Vaidehi N. Biophys J. 2008;94:2027–2042. doi: 10.1529/biophysj.107.117648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niv MY, Skrabanek L, Filizola M, Weinstein H. J Comput-Aided Mol Des. 2006;20:437–448. doi: 10.1007/s10822-006-9061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Chem Biol. 2008;15:1207–1219. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tikhonova IG, Best RB, Engel S, Gershengorn MC, Hummer G, Costanzi S. J Am Chem Soc. 2008;130:10141–10149. doi: 10.1021/ja0765520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuccinardi T, Ferrarini PL, Manera C, Ortore G, Saccomanni G, Martinelli A. J Med Chem. 2006;49:984–994. doi: 10.1021/jm050875u. [DOI] [PubMed] [Google Scholar]
- 63.Vilardaga JP. Nat Chem Biol. 2006;2:395–396. doi: 10.1038/nchembio0806-395. [DOI] [PubMed] [Google Scholar]
- 64.Weis WI, Kobilka BK. Curr Opin Struct Biol. 2008;18:734–740. doi: 10.1016/j.sbi.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobilka BK. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mirzadegan T, Benko G, Filipek S, Palczewski K. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie XQ, Chen JZ, Billings EM. Proteins Struct Funct Bioinf. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 68.Ballesteros JA, Weinstein H, Stuart CS. Methods in Neurosciences. Vol. 25. Academic Press; 1995. pp. 366–428. [Google Scholar]
- 69.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 70.Gouldson P, Calandra B, Legoux P, Kerneis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Eur J Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 71.Montero C, Campillo NE, Goya P, Paez JA. Eur J Med Chem. 2005;40:75–83. doi: 10.1016/j.ejmech.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Tao Q, McAllister SD, Andreassi J, Nowell KW, Cabral GA, Hurst DP, Bachtel K, Ekman MC, Reggio PH, Abood ME. Mol Pharmacol. 1999;55:605–613. [PubMed] [Google Scholar]
- 73.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O’Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 74.Cavasotto CN, Phatak SS. Drug Discovery Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 75.Cavasotto CN, Kovacs JA, Abagyan RA. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 76.Cavasotto CN, Liu G, James SY, Hobbs PD, Peterson VJ, Bhattacharya AA, Kolluri SK, Zhang XK, Leid M, Abagyan R, Liddington RC, Dawson MI. J Med Chem. 2004;47:4360–4372. doi: 10.1021/jm030651g. [DOI] [PubMed] [Google Scholar]
- 77.Kovacs JA, Cavasotto CN, Abagyan R. J Comput Theo Nanosci. 2005;2:354–361. [Google Scholar]
- 78.Mukherjee S, Adams M, Whiteaker K, Daza A, Kage K, Cassar S, Meyer M, Yao BB. Eur J Pharmacol. 2004;505:1–9. doi: 10.1016/j.ejphar.2004.09.058. [DOI] [PubMed] [Google Scholar]
- 79.Polomano RC, Mannes AJ, Clark US, Bennett GJ. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 80.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 81.Dixon W. J Am Stat Assoc. 1965;60:967–978. [Google Scholar]
- 82.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 83.Banker MJ, Clark TH, Williams JA. J Pharm Sci. 2003;92:967–974. doi: 10.1002/jps.10332. [DOI] [PubMed] [Google Scholar]
- 84.Gres MC, Julian B, Bourrie M, Meunier V, Roques C, Berger M, Boulenc X, Berger Y, Fabre G. Pharm Res. 1998;15:726–733. doi: 10.1023/a:1011919003030. [DOI] [PubMed] [Google Scholar]
- 85.Hunter J, Jepson MA, Tsuruo T, Simmons NL, Hirst BH. J Biol Chem. 1993;268:14991–14997. [PubMed] [Google Scholar]
- 86.Maron DM, Ames BN. Mutat Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 87.Mathes C. Expert Opin Ther Targets. 2006;10:319–327. doi: 10.1517/14728222.10.2.319. [DOI] [PubMed] [Google Scholar]
- 88.Gouldson PR, Kidley NJ, Bywater RP, Psaroudakis G, Brooks HD, Diaz C, Shire D, Reynolds CA. Proteins Struct Funct Bioinf. 2004;56:67–84. doi: 10.1002/prot.20108. [DOI] [PubMed] [Google Scholar]
- 89.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 90.Fernandez-Fuentes N, Rai BK, Madrid-Aliste CJ, Fajardo JE, Fiser A. Bioinformatics. 2007;23:2558–2565. doi: 10.1093/bioinformatics/btm377. [DOI] [PubMed] [Google Scholar]
- 91.Read RJ, Chavali G. Proteins Struct Funct Bioinf. 2007;69:27–37. doi: 10.1002/prot.21662. [DOI] [PubMed] [Google Scholar]
- 92.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M. Nat Struct Mol Biol. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kobilka B, Schertler GF. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 94.Chin CN, Murphy JW, Huffman JW, Kendall DA. J Pharmacol Exp Ther. 1999;291:837–844. [PubMed] [Google Scholar]
- 95.Ashton JC, Wright JL, McPartland JM, Tyndall JD. Curr Med Chem. 2008;15:1428–1443. doi: 10.2174/092986708784567716. [DOI] [PubMed] [Google Scholar]
- 96.Song ZH, Slowey CA, Hurst DP, Reggio PH. Mol Pharmacol. 1999;56:834–840. [PubMed] [Google Scholar]
- 97.McAllister SD, Tao Q, Barnett-Norris J, Buehner K, Hurst DP, Guarnieri F, Reggio PH, Nowell Harmon KW, Cabral GA, Abood ME. Biochem Pharmacol. 2002;63:2121–2136. doi: 10.1016/s0006-2952(02)01031-6. [DOI] [PubMed] [Google Scholar]
- 98.Poso A, Huffman JW. Br J Pharmacol. 2008;153:335–346. doi: 10.1038/sj.bjp.0707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cavasotto CN, Abagyan RA. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 100.Cavasotto CN, Orry AJ, Abagyan RA. Proteins Struct Funct Bioinf. 2003;51:423–433. doi: 10.1002/prot.10362. [DOI] [PubMed] [Google Scholar]
- 101.Monti MC, Casapullo A, Cavasotto CN, Napolitano A, Riccio R. ChemBioChem. 2007;8:1585–1591. doi: 10.1002/cbic.200700217. [DOI] [PubMed] [Google Scholar]
- 102.Monti MC, Casapullo A, Cavasotto CN, Tosco A, Dal Piaz F, Ziemys A, Margarucci L, Riccio R. Chemistry. 2009;15:1155–1163. doi: 10.1002/chem.200801512. [DOI] [PubMed] [Google Scholar]
- 103.Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lai J, Porreca F, Makriyannis A, Malan TP., Jr Proc Natl Acad Sci USA. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Davar G, Makriyannis A, Vanderah TW, Mata HP, Malan TP., Jr Proc Natl Acad Sci USA. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Malan TP, Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, Porreca F, Makriyannis A. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 106.Pace JM, Tietje KR, Dart MJ, Meyer MD. 3-Cycloalkylcarbonylindoles as Cannabinoid Receptor Ligands and Their Preparation, Pharmaceutical Compositions and Use for Treatment of Pain. 2006069196. Patent WO. 2006
- 107.Johnson MR, Melvin LS. Cannabinoids as Therapeutic Agents. CRC Press; Boca Raton: 1986. [Google Scholar]
- 108.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.