

Abstract
Breast cancer is the most frequent malignancy and second leading cause of cancer-related deaths among women. Despite advances in genetic and biochemical analyses, the incidence of breast cancer and its associated mortality remain very high. About 60 – 70% of breast cancers are Estrogen Receptor alpha (ER-α) positive and are dependent on estrogen for growth. Selective estrogen receptor modulators (SERMs) have therefore provided an effective targeted therapy to treat ER-α positive breast cancer patients. Unfortunately, development of resistance to endocrine therapy is frequent and leads to cancer recurrence. Our understanding of molecular mechanisms involved in the development of ER-α positive tumors and their resistance to ER antagonists is currently limited due to lack of experimental models of ER-α positive breast cancer. In most mouse models of breast cancer, the tumors that form are typically ER-negative and independent of estrogen for their growth. However, in recent years more attention has been given to develop mouse models that develop different subtypes of breast cancers, including ER-positive tumors. In this review, we discuss the currently available mouse models that develop ER-α positive mammary tumors and their potential use to elucidate the molecular mechanisms of ER-α positive breast cancer development and endocrine resistance.
Keywords: Breast cancer, estrogen receptor-α, mouse models
INTRODUCTION
Breast cancer is the most common cancer among women and leading cause of cancer-related mortality, worldwide. Globally, breast cancer accounts for >400,000 deaths.[1] The life-time risk of breast cancer among American women continues to remain at an astonishing one in eight, and the breast cancer incidence (26%) and mortality rate (15%) among all cancers in women remain very high. Many factors are believed to contribute to this high burden of breast cancer, including lifestyle, environmental, genetic, and biological factors.[2,3]
Clinical–epidemiological studies have shown a strong correlation between the actions of ovarian steroid hormones, particularly 17β-estradiol, and carcinogenesis in the mammary gland and uterus.[4,5] The biological effects of estrogens are mediated by estrogen receptor alpha (ER-α), a member of the superfamily of nuclear receptors that function as ligand-inducible transcription factors.[6] A related ER, termed ER-β, encoded by a distinct gene, has been identified and may mediate estrogen signaling in some tissues.[7] However, for this review we will focus on ER-α (hereby called ER). About 60 – 70% of human breast cancers are ER-positive and estrogen-dependent,[8] and ER and progesterone receptor (PR) expression is an important indicator of potential responses to hormonal therapy.[9] Based on the molecular classification of breast cancers, ER-positive tumors fall under luminal A (~40%; ERhigh, HER2low) and luminal B subtypes (~20%; ERlow, HER2low), with luminal A subtype having a better outcome.[10–11] Despite higher or lower levels of ER expression, most tumors positive for ER respond to anti-estrogen therapies; however, approximately 30% of the ER-positive tumors fail to respond to anti-estrogen therapies, and thus carry a poorer prognosis.[12] The factors that control ER expression in tumor cells are unknown, and it is not well understood why a small fraction of ER-positive breast cancers do not respond to anti-hormone therapies. A majority of studies in this context have been restricted to xenograft models of a relatively small number of available ER positive cell lines.[13] In addition to drug resistance studies, these xenograft models are also essential for pre-clinical testing of inhibitors of steroid receptor signaling.[14] Thus, genetically engineered mouse (GEM) models that develop ER-positive breast tumors similar to human cancers would be extremely useful to help decipher the mechanisms involved in the pathogenesis of ER-positive breast cancers and to understand the biology of anti-estrogen resistance.
Hormonal regulation of mammary gland development is similar in mice and humans; yet, while 60 – 70% of human tumors are ER positive, most tumors arising in GEM models are ER-negative. Thus, apparently most GEM models do not precisely recapitulate the steroid receptor-dependent signaling events responsible for the formation of ER-positive tumors.[13] Notably, in contrast to apparently hormone-independent mammary tumors that arise in GEM models, chemically induced rat mammary tumors are generally hormone-dependent adenocarcinomas.[15] For this reason, the rat mammary carcinogenesis model has been utilized extensively to investigate hormone-dependent breast cancer and the protective role of pregnancy in breast cancer.[16–18] Despite convincing studies demonstrating hormonal sensitivity of mammary tumors in rats, difficulties with genetic engineering of rat models have thus far precluded their widespread use. On the other hand, ease of genetic manipulations in mice has made GEM models a preferred choice to study breast cancer pathogenesis. In this review, we discuss the pros and cons of various ER-positive mouse models that have been used to delineate steps and pathways in the formation of hormone-dependent mammary adenocarcinomas. How these pathways are linked to ER-dependent cell proliferation is depicted in Figure 1.
Figure 1.
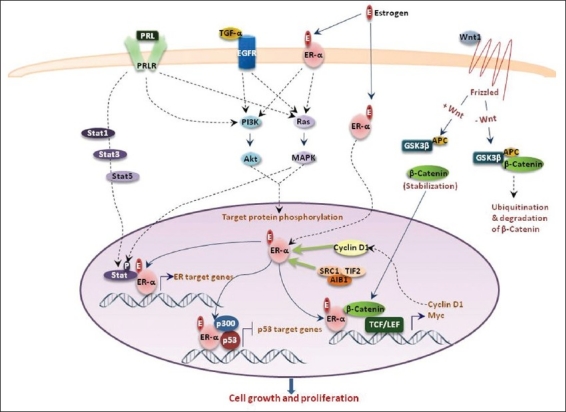
Estrogen functions through multiple pathways. Binding of Estrogen (E) to the Estrogen Receptor-α (ER-α) leads to translocation of the ligand receptor complex to the nucleus, where it affects transcription of an independent set of genes.[95] In addition, ER also exerts its effect on growth and proliferation of cells by binding to and affecting transactivation activity of various growth-related transcription factors (TF). Binding of the Wnt ligand to frizzled receptor leads to stabilization of β-catenin and its subsequent translocation to the nucleus, where it binds to TCF / LEF TF and drives its target genes. β-catenin activity in the nucleus is modulated by ER, leading to enhanced transactivation of the Wnt target genes.[96] Binding of estrogen to the membrane-bound ER activates downstream signaling pathways that include PI3K and Ras-MAPK pathways.[95] Transforming growth factor alpha (TGF-α) binds to its receptor, the epidermal growth factor receptor (EGFR), leading to its activation and a subsequent transcription of proliferative genes, through activation of PI3K and Ras-MAPK signaling.[21] Prolactin (PRL) binds to its trans-membrane cell-surface receptor, the prolactin receptor (PRLR), and triggers a tyrosine kinase-mediated signaling cascade, which leads to the activation of Stat1, Stat3, and Stat5, leading to their translocation to nucleus, where they bind to their target gene promoters.[97] ER binds to Stat target genes and enhances the transcriptional activity of Stats. Additionally, cyclin D1 binds directly to the hormone-binding domain of the estrogen receptor, resulting in an increased binding of the receptor to estrogen response element sequences, and upregulates ER-mediated transcription.[98–99] Similarly, AIB1 in complex with Src1 and TIF2, potentiates transcription of ER-regulated genes.[100] Finally, ER has been shown to bind to p53 on the p53 target gene promoters in breast cancer cells, leading to repression of the p53 transactivation function.[101–102]
Historical perspectives
The earliest ER-positive breast cancer mouse model dates back to 1980; when it was shown that 7,12-dimethylbenzanthracene (DMBA) induced mammary tumors with a high incidence in virgin (69%) as well as hormone-stimulated (81%) female C57BL / 6 x DBA / 2fF1 mice.[19] The investigators characterized these tumors for their histopathology, dependence on ovarian hormones, and expression of mouse mammary tumor virus (MMTV) antigens. Of the tumors examined, nearly half exhibited a significant dependence on the status of the ovarian hormonal axis: one group of tumors showed dependence on ovarian hormones (tumors that grew better in the presence of an intact ovary); another group of tumors were ovary sensitive (tumors that grew better in the absence of the ovary). The propensity of C57BL / 6 x DBA / 2f F1 mice to develop ovary-responsive tumors after chemical carcinogen treatment has led to the extensive use of DMBA-induced mouse models to study ER-positive tumors. As recombinant DNA technology and mouse genome engineering has advanced, a number of transgenic mouse models have been generated (reviewed below).
Transforming growth factor alpha transgenic mouse model
Transforming growth factor alpha (TGF-α) is a member of the epidermal growth factor family that is known to be frequently overexpressed in breast cancers.[20] TGF-α binds to its receptor ErbB1 (EGFR, HER1), and induces its homodimerization or heterodimerization with another member (ErbB2 / HER2 / neu, ErbB3 or ErbB4) of the ErbB receptor family.[21] ErbB1 and ErbB2 are often upregulated in treatment refractory breast cancers and are more often found expressed in ER- or ER+ / PR-tumors.[22–23] TGF-α is a potent mitogen and also induces an increase in the level of its own receptor, ErbB1. TGF-α transgenic mice were independently generated in 1990 by two groups: one group used the zinc-inducible metallothionein promoter to drive TGF-α overexpression, which directs relatively low mammary transgene expression, with expression in several other adult tissues;[24] the second group used the MMTV promoter,[25] which is hormone-responsive and activated by progesterone.[26] Although both groups reported the formation of mammary hyperplasia and adenocarcinomas in transgenic female mice, the hormone receptor expression of adenocarcinomas was not examined in either study. In another study, TGF-α was expressed under the Whey acidic protein (WAP) promoter (also a hormone-responsive promoter) and shown to be exclusively expressed in the mammary gland. In this study, multiparous female transgenic mice developed mammary tumors with 100% incidence but with variable latency; however, the ER status of the tumors was not analyzed.[27] Recent studies utilized a non-hormone responsive neu-related lipocalin (NRL) promoter to direct TGF-α transgene expression to mammary ductal and alveolar cells.[28] NRL-TGF-α transgenic mice developed hyperplasia as well as cystic and solid tumors. ER protein was shown to be present in all lesion types and in the surrounding stromal cells using immunohistochemistry analyses, however, PR protein was detected in normal epithelial cells and in rare cells of the small epithelial hyperplasia, but was absent in stromal or tumor cells. The tumors that formed in this model were hormone-dependent as ovariectomy significantly delayed the tumor development and decreased the tumor incidence. Thus in this mouse model, unregulated TGF-α expression in the mammary gland led to oncogenesis that was dependent on ovarian steroids. The resulting tumors exhibited ER+ / PR- phenotype, reminiscent of similar clinically observed human tumors. Furthermore, when these mice were crossed with p53 heterozygous mice the resulting tumors were ER- / PR-. Taken together, transgenic mice overexpressing TGF-α under the non-hormone-responsive NRL promoter appear to provide a good GEM model to study the formation of ER-positive mammary tumors.
Mouse models of breast cancer driven by alterations of p53
The well-known tumor suppressor protein p53 is a stress responsive transcription factor that regulates the expression of a large number of genes in response to a variety of cellular insults, including oncogene activation and DNA damage. These signals activate p53 primarily through post-translational modifications that result in augmented p53 protein level and transactivation activity.[29] Transcriptional targets of activated p53 mediate various responses to cellular stress, including cell cycle arrest, apoptosis, DNA repair, and differentiation, mechanisms that help repair and / or eliminate cells with damaged genomes, a key to the ability of p53 to suppress cellular transformation.[30] Mutations in p53 are observed in nearly half of all human cancers, including breast carcinomas.[31] As a result, several laboratories have generated mouse mammary tumor models that mimic p53 lesions observed in human cancers, including p53 deletion and mutations. Characterization of a number of these p53-based mouse mammary tumor models suggests their potential suitability to study the biology of ER-positive breast cancers.
One set of models is provided by the alterations induced by p53 deletion in the mouse mammary epithelium. Since rarer and slower mammary tumor formation in p53 null mice[32] is often complicated by lethality from an early-onset of lymphomas and sarcomas,[33–34] investigators have employed transplantation of mammary epithelium from p53 null mice in the mammary gland fat pad of wild-type hosts to form tumors. Others have used conditional deletion of p53 or conditional expression of mutant p53 in the mouse mammary gland. The p53-null mammary transplant into cleared mammary fat pads of wild-type p53 BALB / c hosts allowed long-term analyses of mammary tumor phenotypes and assessment of the effects of hormone stimulation and DMBA treatment on tumor incidence.[35] Tumors arose from the transplanted p53 null epithelium at high rates and these were mostly adenocarcinomas. Interestingly, the incidence of tumors in this system increased dramatically with continuous hormonal stimulation; however, the ER status of tumors was not examined in this study.
Subsequently, the same group characterized the hormone receptor status of a series of serially-transplanted ductal premalignant outgrowth lines derived from p53-null mammary epithelium.[36] Ten out of twelve lines characterized, were ER positive (with > 5% positive cells). A majority of these transplantable lines had retained their estrogen receptor status in ductal outgrowths and in simple ductal hyperplasia observed in these mice; however, tumor formation was not characterized in this study. In a separate study, the same investigators showed that, in comparison to p53 wild-type epithelium, tumorigenesis of p53 null epithelium was strongly enhanced by estrogen, progesterone or their combination.[37] Premalignant outgrowth lines generated from p53 null epithelium in the previous studies were transplanted in mice to investigate tumor formation and hormone dependence of tumors. Although six out of the eight (75%) lines used were ER positive, only three out of the 27 (11%) tumors that formed in mice were hormone-responsive for sustained growth. The p53-null premalignant cells used in the study were highly responsive to the absence or presence of ovarian hormones during premalignant stages, and these cells were ER positive to start with, yet they formed tumors that were frequently ER negative. Interestingly, a recent study has shown that tumors arising in this mouse model are not only ER positive, but also include tumors representing other subtypes of human breast cancers, making it a potentially good mouse model to study breast cancer subtypes.[38]
In a complementary approach, Lin et al. showed that mammary tumors arising in mice in which p53 was conditionally deleted in mammary epithelial cells using the Cre / loxP system, were ER-positive as well as ER-negative.[39] Interestingly specific developmental stages at which p53 deletion was induced determined whether mammary tumors were ER-positive. Deletion of p53 using a constitutively-active WAP-Cre promoter in prepubertal / pubertal mice, but not when MMTV Cre was used for deletion in adult mice, led to the development of ER-positive tumors. Notably, these tumors had frequent genetic alterations in c-myc as well as activation of Her2 / Neu / ErbB2, two well-known breast cancer–related oncogenes. These findings suggest that this model may be useful in examining ER-positive breast cancer progression.
Finally, transgenic mouse model that conditionally expresses the R270H (equivalent to human R273H) p53 mutant allele has been developed.[40] Crossing of these mice with WAP-Cre mice showed that heterozygous p53R270H / +WAP-Cre mice developed spontaneous mammary tumors at high frequency. Treatment of mice with DMBA further accelerated tumor formation. Most of the tumors that formed, either spontaneously or upon DMBA induction, were ER positive and showed gene alterations similar to those implicated in human breast cancer. Thus, this mouse model also appears to be well-suited to study the initiation and progression of ER-positive breast tumors and the responsiveness of mammary gland tumors to chemotherapeutics.
Wnt-1 transgenic mouse model
The prototype member of the Wnt family, Wnt-1, was originally identified as a site of integration of the mouse mammary tumor virus.[41] The Wnt family proteins have now been implicated in multiple aspects of mammary gland development, including mammary morphogenesis and progenitor cell renewal.[42–43] Wnt proteins are made as secreted glycoproteins and they exert their biological effects by binding to membrane receptors, the frizzled and low-density-lipoprotein receptor related proteins. These signaling events lead to stabilization of β-catenin, which then translocates to the nucleus and transactivates different sets of genes depending on the cellular context. The finding of Wnt-1 as a site of MMTV integration prompted the generation of MMTV-Wnt-1 transgenic mouse by Tsukamoto et al., in 1988.[44] In these mice, Wnt-1 was overexpressed from its endogenous promoter linked to MMTV enhancer elements. In this model, the mammary glands of male as well as virgin female mice showed hyperplasia, and formed mammary and salivary adenocarcinomas. However, the ER status of the tumors was not reported. Subsequent studies demonstrated cooperation of the MMTV-Wnt1 transgene with expression of another proto-oncogene, int-2, or with deletion of p53 during oncogenesis, but the hormone receptor status of the tumors formed, remained undefined.[45–46] To address the hormone dependency of tumors formed in MMTV-Wnt1 transgenic mice, these were crossed with ER-α knockout mouse (ERKO).[47] Interestingly, these ERKO / Wnt1 transgenic mice showed a nearly two-fold delay in tumor appearance compared to that observed in Wnt-1 transgenic mice, indicating an important role of hormone stimulation in tumor formation. A subsequent study (Zhang et al., 2005) showed that estrogen positivity in Wnt-1 transgenic mouse mammary tumors is influenced by collaborating oncogenic mutations.[48] The authors documented that Wnt-1 induces ER-positive tumors and this was unaffected by the loss of PTEN or gain of Ras mutations during the evolution of tumors, but concurrent overexpression of Neu or loss of p53 led to the formation of ER-negative tumors. These results suggest that ER expression in mouse models of breast cancer, as in human breast cancer, may be influenced by specific genetic changes that promote cancer progression. These results also suggest potential mechanisms of development of ER-negative tumors from initially ER-positive tumors.
Recently, the effects of p53 genotype on mammary tumor latency, ER expression, and response to tamoxifen in MMTV-Wnt1 transgenic mice were reported.[49] Clinically, p53 genotype has been shown to correlate with the ER status; ER-positive tumors typically have WT p53, whereas ER-negative tumors usually have p53 alterations. The investigators examined MMTV-Wnt1 transgenic animals on WT p53 or heterozygous background for their study. Interestingly, ER-positive tumor formation was delayed in p53 WT mice treated with tamoxifen, whereas, tumors arising in p53 heterozygous mice had reduced levels of ER and tamoxifen treatment had no effect. This study provides an example of how the MMTV-Wnt1 mice may be useful for studying anti-estrogenic responses in tumors and to assess the effects of collaborating mutations on tumor outcome. These mice could provide excellent models to understand hormonal resistance in breast cancers.
Amplified in breast cancer 1 mouse model
Amplified in breast cancer 1 (AIB1), also known as Steroid Receptor Coactivator 3 (SRC-3) or Nuclear Receptor Coactivator 3 (NCoA-3) is a member of the p160 nuclear receptor co-activator family that has emerged as an important oncogene in breast cancer.[50] AIB1 is a transcriptional co-activator that promotes the transcriptional activity of multiple nuclear hormone receptors, such as estrogen receptor, and a number of other transcription factors, including E2F-1, AP-1, NFκB, and STAT6.[51] Overexpression of AIB1 in human breast cancers is linked with poor clinical prognosis.[52] Increased AIB1 levels in conjunction with members of the epidermal growth factor receptor tyrosine kinase family, such as HER2, is associated with resistance to tamoxifen therapy and decreased disease-free survival.[53] Mouse models overexpressing AIB1 have increased our understanding of the role of steroid co-activators in mammary tumorigenesis. It has been recently reported that mice overexpressing AIB1 under the transcriptional control of the MMTV LTR in mammary epithelium led to mammary hyperplasia and development of ER-positive mammary tumors.[54] Notably, the expression of AIB1 transgene in these mice was not specific to the mammary gland and the transgene was detected, as expected, in other tissues such as lung, pituitary, and uterus, and these tissues also showed tumors. Another recent study further confirmed the role of AIB1 in mammary tumorigenesis.[55] Similar to the study noted above, this report also documented mammary hyperplasia in AIB1-overexpressing mice, but tumors were not detected, apparently due to a more moderate level of AIB1 overexpression. The status of ER expression in mammary hyperplasia was not reported in this study. In another report, overexpression of a human AIB1 isoform, AIB1-Δ3, from a cytomegalovirus (CMV) promoter was shown to induce partial oncogenic progression.[56] Notably, even though AIB1-Δ3 isoform is a more potent transcriptional co-activator than the full-length AIB1, the transgenic mice only developed ductal hypertrophy of the mammary gland, along with increased proliferation of mammary epithelial cells, but did not develop mammary tumors. Interestingly, the ductal hypertrophy was not associated with any changes in the expression pattern of ER-α. Thus, the mouse model developed by these authors[54] appears to be suitable for examining ER-positive breast tumor progression in mice.
Estrogen Receptor-α transgenic mouse model
ER transgenic mouse models were reported as early as in 1994 to study ER's role in reproductive processes.[57] These mice that had ER overexpressed in various tissues at low levels under mouse metallothionein-I (MT) promoter, showed reproductive abnormalities, but did not form tumors. A subsequent analysis of the MT-ER females reported that the transgenic animals were more susceptible to uterine tumors after neonatal exposure to diethylstilbestrol, a potent synthetic estrogen, than the wild-type littermate controls.[58] Furthermore, researchers achieved tetracycline-dependent conditional expression of ER in mice by generating TetO-ER mice and crossing them with MMTV-tTA (tetracycline transactivator) (tet-off gene regulation) mice, whereby, transgene expression could be stalled by administrating doxycycline to the double transgenic mice.[59] This study also focused on the effect of ER overexpression on normal reproductive system and did not report any tumor formation in the double transgenic mice. Given the role of ER-α in breast cancer pathogenesis, investigators then introduced the TetO-ER transgene (described above) into the conditional Simian Virus 40 T antigen (TAg) mouse model (MMTV-tTA / TetO-TAg).[60] The mice developed mammary adenocarcinomas with a mean latency of 11 months and exhibited histological features similar to those of human breast cancers. Steroid-binding studies conducted on adenocarcinoma lysates demonstrated binding to estradiol. Tumor explant studies in the presence and absence of estradiol in ovariectomized athymic nude mice revealed that the growth of mammary tumors was stimulated by estrogen. In addition, the presence of ER altered the tumor spectrum in other MMTV-targeted tissues in the tTA / TAg female mice. Experiments utilizing this mouse model have shown that both ER-positive and ER-negative mammary adenocarcinomas develop when deregulated ER is combined with BRCA1 loss, and p53 haploinsuffiency[61] and Stat5a contributed to ER initiated preneoplasia, but loss of Stat5a did not prevent the development of invasive cancer in this model.[62] It is noteworthy that ER-α is overexpressed in this model, while ER-α has not been found to be overexpressed in breast cancers. Instead, merely the presence or absence of ER-α appears to be important in imparting responsiveness to a hormonal therapeutic regimen. Nonetheless, this mouse model provides an important tool to study hormone-responsive growth of mammary tumors.
Prolactin transgenic mouse model
Prolactin (PRL) is a peptide hormone normally secreted by the anterior pituitary lactotroph cells. In primates, however, other cell types including normal and neoplastic human mammary epithelium also synthesize PRL, which permits autocrine / paracrine actions within the mammary gland, independent of pituitary prolactin.[63] Prolactin has a well established role in stimulating mammary growth and differentiation during puberty and lactation.[64] Prolactin binds to its trans-membrane cell-surface receptor, the prolactin receptor (PRLR), and triggers a tyrosine kinase-mediated signaling cascade.[65] Further studies have implicated components of this pathway in the progression of breast cancer.[63] Up to 98% of human mammary tumors reportedly express PRLR,[66–68] with higher levels of expression in neoplastic tissue compared to the adjacent normal tissue.[69] Rose-Hellekant et al. generated mice that overexpress prolactin within mammary epithelial cells under the control of the hormonally-nonresponsive NRL promoter.[70] These mice developed invasive ER-positive and ER-negative mammary neoplasms. The tumors were of various histological subtypes, with papillary adenocarcinomas and adenosquamous neoplasms being the most frequent. Interestingly, significant variations in the percentage of ER-positive tumors were seen in different transgenic lines used; with 50% ER-positive tumors in one line, but only 13% ER-positive mammary tumors in a second line. Although this variability is a concern, these mice provide a good model to examine the biology of hormone-dependent breast cancer. Further studies by the same investigators showed co-expression of prolactin with the EGFR ligand and the mammary oncogene, TGF-α, dramatically reduced the latency of pre-neoplastic lesions.[71] The pre-neoplastic lesions common in prolactin transgenic mice were epithelial hyperplasia with high rate of adenocarcinoma formation, whereas, in TGF-α transgenic mice the most common pre-neoplastic lesions were macrocysts with a rare incidence of adenosis. Interestingly, macrocysts were the most common pre-neoplastic lesions in the double transgenic mice, followed by epithelial hyperplasia and adenosis. ER-α expression varied widely among individual cysts, and within the cells lining the macrocysts in the double transgenic mice. Very recently, the same group demonstrated that prolactin can promote diverse carcinomas in these transgenic mice, varying in histological subtypes, ER / PR expression, and signaling cascades activated.[72] Many of the tumors arising in this model resembled human luminal breast cancers based on comparison with the molecular signature associated with ER-positive luminal subtype of human breast cancers. As the NRL promoter is not altered by estrogen, the investigators used the ER-selective antagonist, Faslodex, to assess the effect of anti-estrogen therapy on ER-positive tumors. The tumors did not respond well to the therapy indicating drug resistance. Overall, the NRL–PRL mice provide a good experimental model to examine the pathogenesis, progression, and treatment responsiveness of luminal subtype of breast cancer.
Cyclin D1 transgenic mouse model
Cyclin D1 is induced upon mitogenic signaling during G1 phase and plays a pivotal role in the regulation of progression from G1 to S phase of the cell cycle by allosterically regulating cyclin-dependent kinases (CDK) 4 and 6.[73] These active kinase complexes translocate to the nucleus and phosphorylate substrates including the retinoblastoma gene product, pRb, thus relieving pRb's inhibitory function on S phase entry by triggering E2F-dependent transcription of genes required for S phase entry.[74–76] Various mechanisms control this rate-limiting step in cell cycle progression, including cyclin D1 abundance.[76] Thus, expressional or functional deregulation of cyclin D1 gene is a probable contributor to the loss of normal cell cycle control during carcinogenesis. Several studies have shown that cyclin D1 deregulation is observed in various cancers, including those of the breast, esophagus, bladder, and lung.[77–80] In breast cancer cells, over expression of cyclin D1 has been linked with the development of endocrine resistance.[81–82] Researchers have demonstrated that cyclin D1 is essential for the development of mouse mammary cancers induced by c-neu and v-Ha-ras, but not those induced by c-myc or Wnt-1.[83] Mammary adenocarcinoma formation in transgenic mice overexpressing cyclin D1 under MMTV promoter has been reported, however, the observed mean age of tumor onset was ~ 18 months, suggesting that cyclin D1, on its own, is a weak oncogene and may require cooperating oncogenes for efficient carcinogenesis.[84] Experimental models support the idea that mutations that interfere with the nuclear exclusion of cyclin D1 / CDK4 during S-phase trigger neoplastic conversion. Indeed overexpression of a mutant cyclin D1 (D1T286A) that is defective in phosphorylation-mediated nuclear export and subsequent proteolysis, triggers B cell lymphoma in a mouse model of mantle cell lymphoma.[85] In 2008, researchers directed overexpression of D1T286A in the mammary gland by introducing transgene expression under the control of MMTV promoter.[86] These mice developed mammary adenocarcinomas at increased rates and shorter latency, compared to mice overexpressing wild-type cyclin D1 (MMTV-D1). The mean age of tumor onset in MMTV-D1 and MMTV-D1T286A mice was reported to be 19.9 months and 15.8 months, respectively. Interestingly, 50% of the tumors that formed in MMTV-D1T286A were ER-positive, whereas, MMTV-D1 mice formed about 37% ER-positive tumors. Considering that aberrations in cyclin D1 proto-oncogene are common in breast cancers and almost half of the mammary tumors formed in cyclin D1 transgenic mice are ER-positive, these mice provide a relevant GEM model to study human breast cancer.
Myristoylated AKT1 transgenic mouse model
Akt (also known as protein kinase B) was first discovered as an oncogene that affected both proliferation and survival pathways.[87] AKT1 / PKB is a serine / threonine protein kinase that regulates biological processes such as proliferation, apoptosis, and growth in a variety of cell types. Activation of AKT is initiated by translocation to the membrane by binding to phosphatidylinositol-3, 4, 5-trisphosphate produced by phosphoinositide 3-kinase (PI3K) activation in response to cell surface receptor stimulation.[88–89] The PI3K-Akt pathway is one of the most frequently deregulated pathways in cancer due to frequent genetic alterations seen in various enzymes of the pathway and their effectors such as activating mutations and amplification of the PI3K catalytic subunit, PTEN loss, AKT mutations, and receptor tyrosine kinase amplification.[90] Importantly, AKT activity has been linked to both proliferative and anti-apoptotic effects of ER in breast tumor cells, both in an estrogen-dependent and -independent manner.[91–92] Recently, researchers overexpressed a myristoylated form of Akt1 in the mouse mammary epithelium under the control of MMTV LTR; myristoylation makes the protein active, by constitutively localizing it to the plasma membrane.[93] These mice did not form spontaneous tumors, but did so upon DMBA treatment. Histologically, the tumors formed in response to DMBA alone were sarcomas; while DMBA treated AKT transgenic mice formed adenocarcinomas or adenosquamous tumors. Interestingly, 100% of DMBA-induced mammary tumors and benign lesions in these mice expressed ER-α, suggesting that ER deregulation might be due to activation of Akt. The propensity of essentially all tumors formed in these mice to be ER-positive makes the DMBA / constitutive AKT1 model a potentially relevant mouse model to study ER-positive breast cancer.
Mouse model for Luminal subtype of breast cancer
Kumar et al. reported a model of spontaneous ER-positive mammary tumors using a heterozygous NIH nude mouse strain.[94] By performing sibling mating, the investigators derived a colony of heterozygous breeding females that showed ER-positive tumors around the age of six months. Histologically, these tumors showed invasive nodular masses of pleomorphic tubular neoplastic epithelial cells that exhibited invasion in the surrounding stroma, adjacent dermis, and the subcutaneous tissue. Significantly, the investigators observed metastasis through hematogenous and regional lymph nodes into liver, lungs, spleen, heart, and dermal lymphatics. In comparison to the normal mammary gland, the tumors showed high expression of ER along with a moderate-to-high expression of proliferating cell nuclear antigen, moderate expression of vimentin and Cytokeratin 19, and low expression of p53. These mice exhibit the phenotype of ER-positive luminal epithelial-like human breast cancer, making it an attractive model relevant to a particular subtype of breast cancer.
Concluding remarks
A vast majority of human breast cancers are ER-positive and are treated with anti-estrogenic therapies. Therefore, developing GEM models of ER-positive breast cancer is critical for examining the molecular characteristics and progression of ER-positive tumorigenesis, to understand mechanisms of hormone resistance in vivo, and to develop and test novel chemotherapeutic agents in pre-clinical trials. Although it has been difficult to thoroughly recapitulate the characteristics of human ER-positive tumors in mice, several mouse models have been engineered over the years that exhibit a reasonable resemblance to aspects of human ER-positive breast cancers. Many of the mouse models are promising and should collectively provide a good platform to study the various aspects of ER-dependent breast tumor biology. Several studies have shown that the promoters that drive the transgene expression in the mammary gland apparently play an important role. For example, studies of hormone resistance of ER-positive tumors are ideally investigated using a mouse model driven by a steroid hormone non-responsive promoter, such as the NRL promoter; however, mouse models that use hormone responsive promoters such as MMTV or WAP have also proven to be useful in studying hormone-dependent tumorigenesis and hormone resistance in tumors. Furthermore, the specific transgenes used to generate the models [Figure 1] are important deciding factors in choosing the appropriate mouse model, depending on the goals of a particular study. Mouse models employing genetic alterations that are commonly seen in human breast cancer and are known to regulate ER expression [Figure 1] would be ideal for studies of hormone dependency of tumors. Development of GEMs that faithfully mimic in vivo ER-positive breast cancers is an important goal of breast cancer biologists.
AUTHOR'S PROFILE
Mr. Shakur Mohibi, Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, Nebraska
Dr. Sameer Mirza, Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, Nebraska
Dr. Hamid Band, Departments of Genetics, Cell Biology and Anatomy; Biochemistry and Molecular Biology; Pathology and Microbiology; and Pharmacology and Experimental Neuroscience, College of Medicine, and the Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, Nebraska
Dr. Vimla Band, Department of Genetics, Cell Biology and Anatomy, College of Medicine, and the Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, Nebraska
ACKNOWLEDGMENTS
We apologize to many of our colleagues whose original work could not be cited due to space constraints. Work in our laboratories is supported by the NIH grants CA96844 and CA144027 to VB and CA87986, CA99163, CA105489, CA116552, and NCI 5U01CA151806-02 to HB; and Department of Defense grants W81XWH-07-1-0351 and W81XWH-11-1-0171 to VB; and the NCI Core Support Grant to the UNMC-Eppley Cancer Center.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Cummings SR, Tice JA, Bauer S, Browner WS, Cuzick J, Ziv E, et al. Prevention of breast cancer in postmenopausal women: Approaches to estimating and reducing risk. J Natl Cancer Inst. 2009;101:384–98. doi: 10.1093/jnci/djp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McTiernan A, Porter P, Potter JD. Breast cancer prevention in countries with diverse resources. Cancer. 2008;113(8 Suppl):2325–30. doi: 10.1002/cncr.23829. [DOI] [PubMed] [Google Scholar]
- 4.Pike MC, Spicer DV, Dahmoush L, Press MF. Estrogens, progestogens, normal breast cell proliferation, and breast cancer risk. Epidemiol Rev. 1993;15:17–35. doi: 10.1093/oxfordjournals.epirev.a036102. [DOI] [PubMed] [Google Scholar]
- 5.Boyd J. Estrogen as a carcinogen: The genetics and molecular biology of human endometrial carcinoma. Prog Clin Biol Res. 1996;394:151–73. [PubMed] [Google Scholar]
- 6.Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid / thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–86. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 7.Mosselman S, Polman J, Dijkema R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 8.Masood S. Estrogen and progesterone receptors in cytology: a comprehensive review. Diagn Cytopathol. 1992;8:475–91. doi: 10.1002/dc.2840080508. [DOI] [PubMed] [Google Scholar]
- 9.Trichopoulos D, MacMahon B, Cole P. Menopause and breast cancer risk. J Natl Cancer Inst. 1972;48:605–13. [PubMed] [Google Scholar]
- 10.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 11.Malhotra GK, Zhao X, Band H, Band V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther. 2010;10:955–60. doi: 10.4161/cbt.10.10.13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allred DC, Brown P, Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004;6:240–5. doi: 10.1186/bcr938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner KU. Models of breast cancer: Quo vadis, animal modeling? Breast Cancer Res. 2004;6:31–8. doi: 10.1186/bcr723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem / progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15:193–7. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Russo J, Russo IH. Atlas and histologic classification of tumors of the rat mammary gland. J Mammary Gland Biol Neoplasia. 2000;5:187–200. doi: 10.1023/a:1026443305758. [DOI] [PubMed] [Google Scholar]
- 16.Russo J, Balogh GA, Heulings R, Mailo DA, Moral R, Russo PA, et al. Molecular basis of pregnancy-induced breast cancer protection. Eur J Cancer Prev. 2006;15:306–42. doi: 10.1097/00008469-200608000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Russo J, Balogh G, Mailo D, Russo PA, Heulings R, Russo IH. The genomic signature of breast cancer prevention. Recent Results Cancer Res. 2007;174:131–50. doi: 10.1007/978-3-540-37696-5_12. [DOI] [PubMed] [Google Scholar]
- 18.Blakely CM, Stoddard AJ, Belka GK, Dugan KD, Notarfrancesco KL, Moody SE, et al. Hormone-induced protection against mammary tumorigenesis is conserved in multiple rat strains and identifies a core gene expression signature induced by pregnancy. Cancer Res. 2006;66:6421–31. doi: 10.1158/0008-5472.CAN-05-4235. [DOI] [PubMed] [Google Scholar]
- 19.Medina D, Butel JS, Socher SH, Miller FL. Mammary tumorigenesis in 7,12-dimethybenzanthracene-treated C57BL × DBA / 2f F1 mice. Cancer Res. 1980;40:368–73. [PubMed] [Google Scholar]
- 20.Rudland PS, Fernig DG, Smith JA. Growth factors and their receptors in neoplastic mammary glands. Biomed Pharmacother. 1995;49:389–99. doi: 10.1016/0753-3322(96)82676-x. [DOI] [PubMed] [Google Scholar]
- 21.Roepstorff K, Grovdal L, Grandal M, Lerdrup M, van Deurs B. Endocytic downregulation of ErbB receptors: Mechanisms and relevance in cancer. Histochem Cell Biol. 2008;129:563–78. doi: 10.1007/s00418-008-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- 23.Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, et al. Epidermal growth factor receptor / HER2 / insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer. 2005;12(Suppl 1):S99–111. doi: 10.1677/erc.1.01005. [DOI] [PubMed] [Google Scholar]
- 24.Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–46. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- 25.Matsui Y, Halter SA, Holt JT, Hogan BL, Coffey RJ. Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell. 1990;61:1147–55. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 26.Otten AD, Sanders MM, McKnight GS. The MMTV LTR promoter is induced by progesterone and dihydrotestosterone but not by estrogen. Mol Endocrinol. 1988;2:143–7. doi: 10.1210/mend-2-2-143. [DOI] [PubMed] [Google Scholar]
- 27.Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995;55:3915–27. [PubMed] [Google Scholar]
- 28.Rose-Hellekant TA, Schroeder MD, Brockman JL, Zhdankin O, Bolstad R, Chen KS, et al. Estrogen receptor-positive mammary tumorigenesis in TGFalpha transgenic mice progresses with progesterone receptor loss. Oncogene. 2007;26:5238–46. doi: 10.1038/sj.onc.1210340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 30.Oren M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003;10:431–42. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 31.Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, Hainaut P. New approaches to understanding p53 gene tumor mutation spectra. Mutat Res. 1999;431:199–209. doi: 10.1016/s0027-5107(99)00162-1. [DOI] [PubMed] [Google Scholar]
- 32.Harvey M, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225–9. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- 33.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 34.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 35.Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, et al. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–8. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 36.Medina D, Kittrell FS, Shepard A, Stephens LC, Jiang C, Lu J, et al. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–3. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- 37.Medina D, Kittrell FS, Shepard A, Contreras A, Rosen JM, Lydon J. Hormone dependence in premalignant mammary progression. Cancer Res. 2003;63:1067–72. [PubMed] [Google Scholar]
- 38.Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, et al. Breast Cancer Special Feature: Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin SC, Lee KF, Nikitin AY, Hilsenbeck SG, Cardiff RD, Li A, et al. Somatic mutation of p53 leads to estrogen receptor alpha-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res. 2004;64:3525–32. doi: 10.1158/0008-5472.CAN-03-3524. [DOI] [PubMed] [Google Scholar]
- 40.Wijnhoven SW, Zwart E, Speksnijder EN, Beems RB, Olive KP, Tuveson DA, et al. Mice expressing a mammary gland-specific R270H mutation in the p53 tumor suppressor gene mimic human breast cancer development. Cancer Res. 2005;65:8166–73. doi: 10.1158/0008-5472.CAN-05-1650. [DOI] [PubMed] [Google Scholar]
- 41.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 42.Alonso L, Fuchs E. Stem cells in the skin: Waste not, Wnt not. Genes Dev. 2003;17:1189–200. doi: 10.1101/gad.1086903. [DOI] [PubMed] [Google Scholar]
- 43.Chu EY, Hens J, Andl T, Kairo A, Yamaguchi TP, Brisken C, et al. Canonical WNT signaling promotes mammary placode development and is essential for initiation of mammary gland morphogenesis. Development. 2004;131:4819–29. doi: 10.1242/dev.01347. [DOI] [PubMed] [Google Scholar]
- 44.Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55:619–25. doi: 10.1016/0092-8674(88)90220-6. [DOI] [PubMed] [Google Scholar]
- 45.Kwan H, Pecenka V, Tsukamoto A, Parslow TG, Guzman R, Lin TP, et al. Transgenes expressing the Wnt-1 and int-2 proto-oncogenes cooperate during mammary carcinogenesis in doubly transgenic mice. Mol Cell Biol. 1992;12:147–54. doi: 10.1128/mcb.12.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, et al. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 1995;9:882–95. doi: 10.1101/gad.9.7.882. [DOI] [PubMed] [Google Scholar]
- 47.Bocchinfuso WP, Hively WP, Couse JF, Varmus HE, Korach KS. A mouse mammary tumor virus-Wnt-1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor-alpha. Cancer Res. 1999;59:1869–76. [PubMed] [Google Scholar]
- 48.Zhang X, Podsypanina K, Huang S, Mohsin SK, Chamness GC, Hatsell S, et al. Estrogen receptor positivity in mammary tumors of Wnt-1 transgenic mice is influenced by collaborating oncogenic mutations. Oncogene. 2005;24:4220–31. doi: 10.1038/sj.onc.1208597. [DOI] [PubMed] [Google Scholar]
- 49.Fuchs-Young R, Shirley SH, Lambertz I, Colby JK, Tian J, Johnston D, et al. P53 genotype as a determinant of ER expression and tamoxifen response in the MMTV-Wnt-1 model of mammary carcinogenesis. Breast Cancer Res Treat. 2011;130:399–408. doi: 10.1007/s10549-010-1308-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–8. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 51.Lahusen T, Henke RT, Kagan BL, Wellstein A, Riegel AT. The role and regulation of the nuclear receptor co-activator AIB1 in breast cancer. Breast Cancer Res Treat. 2009;116:225–37. doi: 10.1007/s10549-009-0405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao C, Yasui K, Lee CJ, Kurioka H, Hosokawa Y, Oka T, et al. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer. 2003;98:18–23. doi: 10.1002/cncr.11482. [DOI] [PubMed] [Google Scholar]
- 53.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2 / neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–61. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 54.Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, et al. High tumor incidence and activation of the PI3K / AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–74. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 55.Avivar A, Garcia-Macias MC, Ascaso E, Herrera G, O’Connor JE, de Mora JF. Moderate overexpression of AIB1 triggers pre-neoplastic changes in mammary epithelium. FEBS Lett. 2006;580:5222–6. doi: 10.1016/j.febslet.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 56.Tilli MT, Reiter R, Oh AS, Henke RT, McDonnell K, Gallicano GI, et al. Overexpression of an N-terminally truncated isoform of the nuclear receptor coactivator amplified in breast cancer 1 leads to altered proliferation of mammary epithelial cells in transgenic mice. Mol Endocrinol. 2005;19:644–56. doi: 10.1210/me.2004-0106. [DOI] [PubMed] [Google Scholar]
- 57.Davis VL, Couse JF, Goulding EH, Power SG, Eddy EM, Korach KS. Aberrant reproductive phenotypes evident in transgenic mice expressing the wild-type mouse estrogen receptor. Endocrinology. 1994;135:379–86. doi: 10.1210/endo.135.1.8013372. [DOI] [PubMed] [Google Scholar]
- 58.Couse JF, Davis VL, Hanson RB, Jefferson WN, McLachlan JA, Bullock BC, et al. Accelerated onset of uterine tumors in transgenic mice with aberrant expression of the estrogen receptor after neonatal exposure to diethylstilbestrol. Mol Carcinog. 1997;19:236–42. doi: 10.1002/(sici)1098-2744(199708)19:4<236::aid-mc4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 59.Hruska KS, Tilli MT, Ren S, Cotarla I, Kwong T, Li M, et al. Conditional over-expression of estrogen receptor alpha in a transgenic mouse model. Transgenic Res. 2002;11:361–72. doi: 10.1023/a:1016376100186. [DOI] [PubMed] [Google Scholar]
- 60.Tilli MT, Frech MS, Steed ME, Hruska KS, Johnson MD, Flaws JA, et al. Introduction of estrogen receptor-alpha into the tTA / TAg conditional mouse model precipitates the development of estrogen-responsive mammary adenocarcinoma. Am J Pathol. 2003;163:1713–19. doi: 10.1016/s0002-9440(10)63529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones LP, Tilli MT, Assefnia S, Torre K, Halama ED, Parrish A, et al. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene. 2008;27:794–802. doi: 10.1038/sj.onc.1210674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miermont AM, Parrish AR, Furth PA. Role of ERalpha in the differential response of Stat5a loss in susceptibility to mammary preneoplasia and DMBA-induced carcinogenesis. Carcinogenesis. 2010;31:1124–31. doi: 10.1093/carcin/bgq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Binart N, Ormandy CJ, Kelly PA. Mammary gland development and the prolactin receptor. Adv Exp Med Biol. 2000;480:85–92. doi: 10.1007/0-306-46832-8_10. [DOI] [PubMed] [Google Scholar]
- 65.Goffin V, Kelly PA. The prolactin / growth hormone receptor family: Structure / function relationships. J Mammary Gland Biol Neoplasia. 1997;2:7–17. doi: 10.1023/a:1026313211704. [DOI] [PubMed] [Google Scholar]
- 66.Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology. 1997;138:5555–60. doi: 10.1210/endo.138.12.5605. [DOI] [PubMed] [Google Scholar]
- 67.Ormandy CJ, Hall RE, Manning DL, Robertson JF, Blamey RW, Kelly PA, et al. Coexpression and cross-regulation of the prolactin receptor and sex steroid hormone receptors in breast cancer. J Clin Endocrinol Metab. 1997;82:3692–9. doi: 10.1210/jcem.82.11.4361. [DOI] [PubMed] [Google Scholar]
- 68.Mertani HC, Garcia-Caballero T, Lambert A, Gerard F, Palayer C, Boutin JM, et al. Cellular expression of growth hormone and prolactin receptors in human breast disorders. Int J Cancer. 1998;79:202–11. doi: 10.1002/(sici)1097-0215(19980417)79:2<202::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 69.Touraine P, Martini JF, Zafrani B, Durand JC, Labaille F, Malet C, et al. Increased expression of prolactin receptor gene assessed by quantitative polymerase chain reaction in human breast tumors versus normal breast tissues. J Clin Endocrinol Metab. 1998;83:667–74. doi: 10.1210/jcem.83.2.4564. [DOI] [PubMed] [Google Scholar]
- 70.Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene. 2003;22:4664–74. doi: 10.1038/sj.onc.1206619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arendt LM, Rose-Hellekant TA, Sandgren EP, Schuler LA. Prolactin potentiates transforming growth factor alpha induction of mammary neoplasia in transgenic mice. Am J Pathol. 2006;168:1365–74. doi: 10.2353/ajpath.2006.050861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arendt LM, Rugowski DE, Grafwallner-Huseth TA, Garcia-Barchino MJ, Rui H, Schuler LA. Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer. Breast Cancer Res. 2011;13:R11. doi: 10.1186/bcr2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–65. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 74.Hatakeyama M, Brill JA, Fink GR, Weinberg RA. Collaboration of G1 cyclins in the functional inactivation of the retinoblastoma protein. Genes Dev. 1994;8:1759–71. doi: 10.1101/gad.8.15.1759. [DOI] [PubMed] [Google Scholar]
- 75.Calbo J, Parreno M, Sotillo E, Yong T, Mazo A, Garriga J, et al. G1 cyclin / cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J Biol Chem. 2002;277:50263–74. doi: 10.1074/jbc.M209181200. [DOI] [PubMed] [Google Scholar]
- 76.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 77.Musgrove EA. Cyclins: Roles in mitogenic signaling and oncogenic transformation. Growth Factors. 2006;24:13–9. doi: 10.1080/08977190500361812. [DOI] [PubMed] [Google Scholar]
- 78.Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD, et al. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med. 1995;1:1257–60. doi: 10.1038/nm1295-1257. [DOI] [PubMed] [Google Scholar]
- 79.Sutherland RL, Musgrove EA. Cyclin D1 and mammary carcinoma: New insights from transgenic mouse models. Breast Cancer Res. 2002;4:14–7. doi: 10.1186/bcr411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim JK, Diehl JA. Nuclear cyclin D1: An oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–6. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002;62:6916–23. [PubMed] [Google Scholar]
- 82.Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999;5:2069–76. [PubMed] [Google Scholar]
- 83.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 84.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–71. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 85.Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA. Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene. 2006;25:998–1007. doi: 10.1038/sj.onc.1209147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008;27:1231–42. doi: 10.1038/sj.onc.1210738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84:5034–7. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B / Akt. FEBS Lett. 2003;546:108–12. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 89.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–42. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 90.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–11. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- 92.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase / AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–24. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 93.Blanco-Aparicio C, Perez-Gallego L, Pequeno B, Leal JF, Renner O, Carnero A. Mice expressing myrAKT1 in the mammary gland develop carcinogen-induced ER-positive mammary tumors that mimic human breast cancer. Carcinogenesis. 2007;28:584–94. doi: 10.1093/carcin/bgl190. [DOI] [PubMed] [Google Scholar]
- 94.Kumar MJ, Ponvijay KS, Nandhini R, Nagarajan RS, Jose J, Srinivas G, et al. A mouse model for luminal epithelial like ER positive subtype of human breast cancer. BMC Cancer. 2007;7:180. doi: 10.1186/1471-2407-7-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song RX, Santen RJ. Membrane initiated estrogen signaling in breast cancer. Biol Reprod. 2006;75:9–16. doi: 10.1095/biolreprod.105.050070. [DOI] [PubMed] [Google Scholar]
- 96.Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt / beta-catenin / Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005;26:898–915. doi: 10.1210/er.2003-0034. [DOI] [PubMed] [Google Scholar]
- 97.Bachelot A, Binart N. Reproductive role of prolactin. Reproduction. 2007;133:361–9. doi: 10.1530/REP-06-0299. [DOI] [PubMed] [Google Scholar]
- 98.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–15. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 99.Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997;17:5338–47. doi: 10.1128/mcb.17.9.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–41. [PubMed] [Google Scholar]
- 101.Liu W, Ip MM, Podgorsak MB, Das GM. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: A novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;115:43–50. doi: 10.1007/s10549-008-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U S A. 2010;107:15081–6. doi: 10.1073/pnas.1009575107. [DOI] [PMC free article] [PubMed] [Google Scholar]