

Abstract
BACKGROUND:
Recent findings suggest that production of pro-inflammatory cytokines, such as Tumour Necrosis Factor-alpha (TNF-α), is increased in the brain tissue of patients suffering late-onset Alzheimer's disease (LOAD) and play an important role in the pathogenesis of this disease. Several epidemiological studies also suggest that patients taking anti-inflammatory drugs have a decreased risk of developing AD. TNF-α is an important pro inflammatory cytokine that is unregulated in Alzheimer's patients. Functional polymorphisms in tumor necrosis factor alpha (TNF-α) can affect immune response, inflammation, tissue injury and possibly the susceptibility to Alzheimer disease (AD).
METHODS:
We used the polymorphic DNA markers (-308G/A) and (-863C/A) to study the association of TNF-α gene mutations with Late-onset Alzheimer's disease (LOAD) and the relation between clinical features and genotypes in affected individuals. A total of 160 patient samples and 163 healthy controls from west northern Iran (Eastern Azerbaijan) were genotyped for the two polymorphisms by the PCR-RFLP method and genotype frequencies were statistically determined.
RESULTS:
Our data showed significant difference in TNF-α-308 G/A genotype and pro inflammatory cytokine allele frequencies between the Alzheimer disease patients and healthy subjects. Contrary to that, no significant difference was observed in TNF-α-863 C/A genotype and allele frequencies between these two groups.
CONCLUSIONS:
TNF-α-308 G/A gene polymorphism could affect cerebral inflammatory response and the risk of late-onset Alzheimer disease but -863 C/A polymorphism does not influence the risk of this disease and this possible association between TNF-α -308G/A and -863C/A gene polymorphisms have to be further elucidated in larger case control studies.
KEYWORDS: Alzheimer, TNF- α, Polymorphism, PCR-RFLP, β-Amyloid, -308G/A, -863C/A
Alzheimer is the most common form of dementia and is an incurable, degenerative and terminal disease. It is mostly diagnosed in people over 65 year of age, although the less-prevalent early-onset form can occur bellow 65 years.1,2,3 Alzheimer is clinically characterized by a progressive loss of memory and cognitive functions in later life.4
The main causes of AD are not well known.5 Presence of diffuse and neurotic plaque localized to specific regions of brain is a histopathological feature of Alzheimer's disease.6 The main constituent of these plaques is β-amyloid. β-amyloid peptide is derived through proteolytic processing of the soluble precursor protein or Amyloid precursor proteins (APPs).7 APPs are critical to neuron growth, survival and post injury repair.8,9 Cytokines have been detected to influence the production of APPs and ß-amyloid.10,11.
Previous studies suggest that immune mechanisms play a fundamental role in the pathogenesis of AD12,13 . Postmortem studies of the brain in AD patients have revealed the presence of a broad spectrum of immune and inflammatory mediators such as complement proteins, Human leukocyte antigens HLA and proinflammatory cytokines including Interleukin (IL)-1a, 1B, 6, TNF-α in association with the characteristic AD lesions.12,13
TNF-α is an important pro inflammatory cytokine that is unregulated in Alzheimer's patients. This cytokine plays an important role in pro inflammatory responses of immune system including regulation and catabolism.14 TNF is also implicated in activating the nuclear-factor kappa B (NF-KB), a protein which activates the secretion of Apolipoprotein E (APOE).15 Furthermore TNF-α and APOE lead to excessive amyloid formation in the brain before the symptoms of AD arise.16 TNF-α increases the level of damage by free radicals through up regulation of cyclooxygense 2 and in combination with interferon γ it can elevate the production of betaamyloid and inhibits the secretion of APP.15 Finally Tarkowski and colleagues showed increased level of TNF-α in CSF liquid in AD.17
The expression of TNF-α is regulated at transcriptional and post transcriptional levels.18 The TNF-α coding gene is located on chromosome 6P21.3, within the region of HLA class III complex19,20 and compared to other cytokine genes, it has large number of polymorphisms, and the majority of them are located within the 5’ untranslated region.21
Nucleotide variations in genes encoding proteins such as TNF-α may influence their transcriptional and biological activities. Polymorphisms in the promoter of TNF alpha gene have been reported to affect the transcription rate and the release of this protein and thus might influence the risk of AD.22
According to the previous studies, promoter region of TNF alpha encoding gene has several polymorphic regions, amongst them the polymorphisms located on -30bp and -836bp have been shown to be associated with an increased and decreased transcriptional activity of the gene, respectively.23 A growing body of evidence indicates that these polymorphisms may affect the susceptibility to different diseases.24 The molecular mechanisms linking TNF-α to Alzheimer remains elusive and we need to understand if variant levels of TNF-α can really affect the probability of Alzheimer's disease.
Although an increasing number of LOAD associated genes are being diagnosed, replication studies specifically within different ethnic populations are important for confirmation of the previous results. Because no similar study had been done on Azeri Turk population of Iran and the results of previous studies had shown the association of polymorphisms within promoter region of TNF-α gene with LOAD, we performed the present study to determine if -308 and -863 polymorphisms contribute to the risk for LOAD in our study population.
Methods
This study included 160 LOAD patients and 163 healthy controls. Alzheimer subjects were diagnosed by expert clinicians according to the mini-mental state examination (MMSE) criteria.25 The sporadic form of the disease was ensured. No affected individuals were present in first degree relatives of subjects and the age of onset was above 65 years. The Control group included 163 healthy individuals with the same ethnicity to subject group which were randomly selected from a distinguished lab. Blood specimens were collected in sterile tubes containing EDTA, and the DNA was extracted using the salting out method. The TNF-α -308G/A and -863C/a genotypes were determined by PCR-RFLP. The -308G/A PCR reactions were prepared in a total volume of 25 μl, containing 0.1 micrograms of genomic DNA, 0.01 μg each of forward (5’-GCA ATA GGT TTT GAG GGC CAT G- 3’)’ and reverse: (5’ -GGGACA CAC AAG CAT CAA GGA T -3’) primers, 2.5 μg of 10xPCR buffer (670mM Tris-HC1 pH 8.8, 160mM (NH4)2SC4, 0.1%Tween-20), dNTP mix (10mM each), 50 mM MgC12, Taq DNA polymerase (5000u/ml). After denaturation of template DNA at 94°C for 5 minutes, 35 cycles of PCR reactions were performed by denaturation at 94°C for 1 minutes, annealing at 56.4°C for 1 min and extension at 72°C for 1 min. The PCR products were digested using 2unit/μl of NC0I restriction enzyme in a total volume of 25μl, containing 8μl of PCR product in supplied buffer. The mixture was incubated at 65°C for 12-16 hours. The digested PCR product was fractionated on 8% Polyacrylamid gel and visualized after staining by AgNo3.
TNF-α -863C/A genotypes were determined according to the previously described PCR and RFLP conditions with slight modification. The PCR reaction was prepared in a final volume of 25μl, containing 0.1 micrograms of genomic DNA, 0.01 μg each of forward (5’ -GGC TCT GAG GAA TGG GTT AC-3’) and reverse (5’ -CTA CAT GGC CCT VGTC TTC GTT ACG 3’) primers, 2.5μl of 10xPCR buffer (670mMTris-HCl pH8.8, 160mM (NH4)2S04, 0.1%Tween-20), dNTP mix (10mM each), 50 mM MgC12, Taq DNA polymerase (5000u/ml). The cycling conditions were as follows: initial denaturation at 94°C for 4 min followed by 31 cycles of 94°C for 1 min, 55°C for 30 sec, 72°C for 45 sec and final extention at 72°C for 5 min. The PCR products were digested using 2unit/μl of Tail restriction enzyme in a total volume of 25μl, containing 5μl of PCR product in supplied buffer. The mixture was incubated at 65°C for 12-24hours. The digested PCR product was fractionated on 8% Polyacrylamid gel and visualized after staining by AgNo3.
Statistical analysis
Statistical analysis was performed using the Sigma Stat 2.0 software. Allelic and genotypic frequencies were obtained by direct counting. Hardy–Weinberg equilibrium was tested using a χ2 goodness-of-fit test. Statistical significance was set at P< 0.05. The odds ratio (OR) was calculated at 95% CI.
Results
A total of 323 DNA samples prepared from peripheral blood of case and control groups were studied using the PCR and Restriction Fragment Length Polymorphism (RFLP) methods. The samples consisted of 160 AD patients (94 women, 66 men, mean age: 76.6 ± 7.75yr) and 163 healthy controls (95 women, 68 men, mean average: 75.29 ± 6.75 yr). The difference between mean age, gender and education of case and control groups were non-significant according to the statistical analysis (Table 1).
Table 1.
Comparison of mean age, sex and education levels between AD cases and control subjects using t-test and χ2 test analysis.
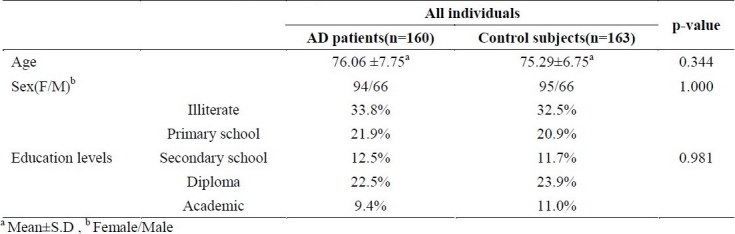
The PCR amplification of TNF-α, -308G/A polymorphic site, produced a 144 base-pair oligonucleotide which in normal form was cleaved by NCO1 restriction enzyme at restriction site “C’CATGG” giving rise to two fragments of 126 and 18 bp(). Homozygous samples for wild type (TNF-α I) and mutated alleles showed a single band of 126 bp and 144 bp on polyachrilamid gel respectively, while heterozygous subjects demonstrated two bands of 144 and 126 bp (figure 1).
Figure 1.
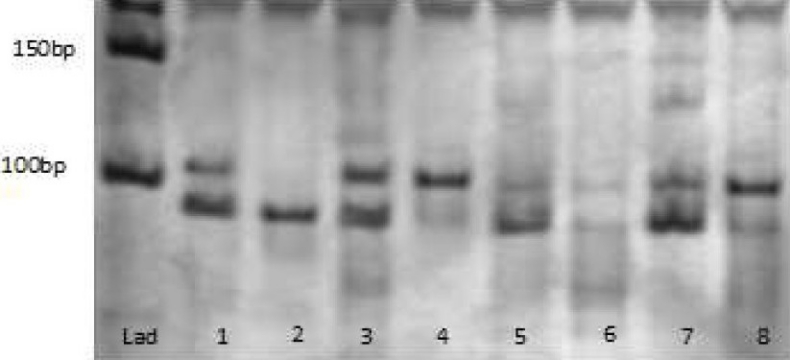
The PCR yields a 107-base-pair (bp) product; the NcoI restriction enzyme cleaves the wild-type (TNF1) allele (restriction sequence: C’CATGG), giving rise to two different fragments of 87 and 20 bp. Thus, in subjects homozygous for the wild-type (TNF1) allele, a single band of 87 bp is observed (lane 2,3); in heterozygous subjects (TNF1/TNF2), two bands of 107 and 87 bp (lane 1,3,6,7); and in subjects homozygous for the mutated (TNF2) allele, a band of 107 bp (lane 4,8).
Amplification of TNF-α, -863C/A polymorphic sequence by PCR, produced a DNA sequence of 125 base pair length. The tail restriction enzyme cleaved the mutated allele (-863 A) in restriction site (ACGT’). Homozygous for the wild-types allele (-863C) demonstrated a single band of 125 bp on polyachrilamid gel, heterozygous subjects (-863 C/A) formed two bands of 104 and 125 bp and homozygous for the mutated allele (-863 A) showed a single band of 104 bp (figure 2).
Figure 2.
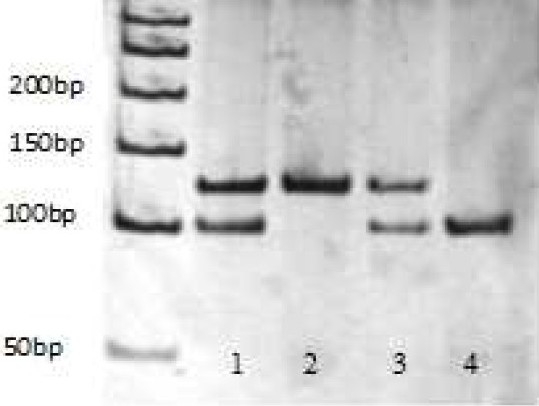
The PCR yields a 125-base-pair product; the TaiI restriction enzyme cleaves the mutated (-863A) allele (restriction sequence: ACGT), giving rise to two different fragments of 104 and 21 bp. Thus, in subjects homozygous for the wild-type (-863C) allele, a single band of 125 bp is observed (lane 2); in heterozygous subjects (-863C/A), two bands of 104 and 125 bp (lane 1,3); and in subjects homozygous for the mutated (-863A) allele, a band of 104 bp (lane 4).
The statistic analysis of patients, genotypes and allele frequencies (Table 2) indicated a significant difference in TNF-α, -308 G/A geno-type and allele frequencies between the AD patients and healthy subjects (allele A frequency in AD patient was 44.69% compared to 6.25% in control subjects).
Table 2.
TNF-α -308G/A allele and genotype frequencies (%) in AD patients and healthy controls
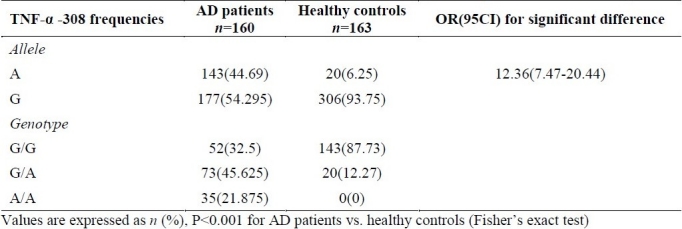
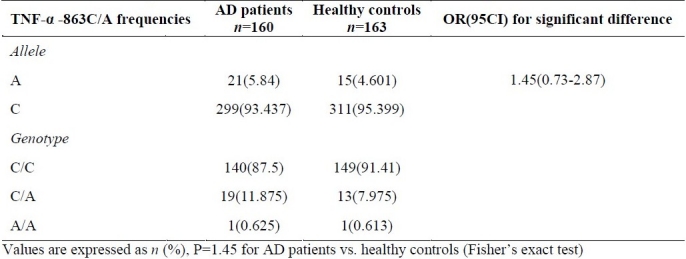
Table 3 indicates a non-significant difference between TNF-α -863 C/A genotype and allele frequencies of the AD patients and healthy subject (allele A frequency was 5.84% in AD patient and 4.601% in control subjects).
Table 3.
TNF-α -863C/A allele and genotype frequencies (%) in AD patients and healthy controls
Discussion
Previous studies indicate that individuals who use anti-inflammatory drugs are at a decreased risk of developing AD.26 An increasing amount of basic and clinical evidence have also shown that inflammatory processes and resulting glial activation play a role in the pathogenesis of AD.27,28 A number of studies suggest that inhibition of the inflammatory cytokines such as TNF-α may hold promise as a potential approach to treatment of AD.29
TNF-α is one of the main pro-inflammatory cytokines that plays a fundamental role in inflammatory responses.30 This cytokine has surprisingly controversial actions in the nervous system, since it has been claimed to be trophic or killer for neurons.30 This dictotomy could be also explained by the fact that TNF-α elicits its biological effects through the activation of two distinct receptors: the P55 TNF-α receptor (Type I TNFR) and the P75 TNF-α receptor (Type II TNFR). Type I TNFR contains an intracellular death domain and contributes to cell death when activated; in contrast, type II TNFR is likely to play a trophic or protective role in neuronal survival.30 Elevated levels of TNF-α have been implicated in pathogenesis of Alzheimer disease31 Polymorphisms in the promoter of the TNF-α gene have been reported to affect the transcription rate and the release of this cytokine. Among the best studied, the –308 polymorphism have been associated with an increased transcriptional activity and TNF-α release, whereas the –863 polymorphism have been associated with a reduced transcriptional activity. A growing body of evidence indicates that these polymorphisms may affect susceptibility to different diseases.23
In the present study the -308G/A polymorphism of the promoter region of TNF-α gene was shown to be a genetic marker for susceptibility to AD in Azeri Turk population. Our results are in agreement with the results reported for the other ethnic groups. A report about the population of Southern China suggests that the -308G/A polymorphism of TNF-α gene might be a risk factor for AD in this ethnicity. Similar results have been reported for the Chilean population32.
To determine whether particular combination of genes would contribute to the risk of AD, the polymorphisms of 9 genes including TNF-α was studied in a research in USA. Analysis of patient's genotypes and phenotypes revealed that the younger patients are heterozygous for TNF -308A/G allele, suggesting that this polymorphism could affect the age of onset in patients.33 A similar study in Spain showed the effect of -308G/A polymorphism of TNF-α gene on the age of onset of LOAD.34
The association of -863 polymorphism with AD was not proved in the present study. This finding had been previously reported by others.32
In the above mentioned study by USA research group both of the youngest and oldest subjects, were heterozygous for -863 (AC) TNF promoter polymorphisms.33 Similarly, studies in Spain and Italy have also reported that there is no association between TNF-α -863C/A and AD.
Regarding to the different roles of -308 G/A and -863 C/A polymorphisms on the levels of TNF-α transcription, a negative association is expected between Alzheimer's disease and -863 C/A polymorphism. However our results did not reveal such an association.
Authors’ Contributions
All the authors have carried out the study, participated in the design of the study and acquisition of data performed the statistical analysis and wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
Conflict of Interests Authors have no conflict of interests.
References
- 1.Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s. Neurobiol Aging. 1998;19(3):173–89. doi: 10.1016/s0197-4580(98)00052-9. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88(9):1337–42. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3(3):186–91. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 4.Culpan D, MacGowan SH, Ford JM, Nicoll JA, Griffin WS, Dewar D, et al. Tumour necrosis factor-alpha gene polymorphisms and Alzheimer's disease. Neurosci Lett. 2003;350(1):61–5. doi: 10.1016/s0304-3940(03)00854-1. [DOI] [PubMed] [Google Scholar]
- 5.Lio D, Annoni G, Licastro F, Crivello A, Forte GI, Scola L, et al. Tumor necrosis factor-alpha -308A/G polymorphism is associated with age at onset of Alzheimer's disease. Mech Ageing Dev. 2006;127(6):567–71. doi: 10.1016/j.mad.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62(11):1984–9. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 8.Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26(27):7212–21. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70(1):1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 10.Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. 1989;86(19):7606–10. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monning U, Sandbrink R, Banati RB, Masters CL, Beyreuther K. Transforming growth factor beta mediates increase of mature transmembrane amyloid precursor protein in microglial cells. FEBS Lett. 1994;342(3):267–72. doi: 10.1016/0014-5793(94)80514-8. [DOI] [PubMed] [Google Scholar]
- 12.McGeer PL, Rogers J, McGeer EG. Neuroimmune mechanisms in Alzheimer disease pathogenesis. Alzheimer Dis Assoc Disord. 1994;8(3):149–58. doi: 10.1097/00002093-199408030-00001. [DOI] [PubMed] [Google Scholar]
- 13.Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68(22):1902–8. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 14.Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10(2):119–30. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 15.Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflammation and Alzheimer's disease: critical roles for cytokine/A beta-induced glial activation, NF-kappa B, and apolipoprotein E. Neurobiol Aging. 2000;21(3):427–32. doi: 10.1016/s0197-4580(00)00143-3. [DOI] [PubMed] [Google Scholar]
- 16.Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333(19):1242–7. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 17.Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61(3):255–60. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 18.Kroeger KM, Carville KS, Abraham LJ. The -308 tumor necrosis factor-alpha promoter polymorphism effects transcription. Mol Immunol. 1997;34(5):391–9. doi: 10.1016/s0161-5890(97)00052-7. [DOI] [PubMed] [Google Scholar]
- 19.Straub RE, MacLean CJ, Kendler KS. The putative schizophrenia locus on chromosome 6p: a brief overview of the linkage studies. Mol Psychiatry. 1996;1(2):89–92. [PubMed] [Google Scholar]
- 20.Jongeneel CV, Briant L, Udalova IA, Sevin A, Nedospasov SA, Cambon-Thomsen A. Extensive genetic polymorphism in the human tumor necrosis factor region and relation to extended HLA haplotypes. Proc Natl Acad Sci U S A. 1991;88(21):9717–21. doi: 10.1073/pnas.88.21.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hajeer AH, Hutchinson IV. Influence of TNFalpha gene polymorphisms on TNFalpha production and disease. Hum Immunol. 2001;62(11):1191–9. doi: 10.1016/s0198-8859(01)00322-6. [DOI] [PubMed] [Google Scholar]
- 22.Feldmann M, Maini RN. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9(10):1245–50. doi: 10.1038/nm939. [DOI] [PubMed] [Google Scholar]
- 23.Fargion S, Valenti L, Dongiovanni P, Fracanzani AL. TNFalpha promoter polymorphisms. Methods Mol Med. 2004;98:47–58. doi: 10.1385/1-59259-771-8:047. [DOI] [PubMed] [Google Scholar]
- 24.Wilson AG, di Giovine FS, Duff GW. Genetics of tumour necrosis factor-alpha in autoimmune, infectious, and neoplastic diseases. J Inflamm. 1995;45(1):1–12. [PubMed] [Google Scholar]
- 25.Doody RS, Massman P, Dunn JK. A method for estimating progression rates in Alzheimer disease. Arch Neurol. 2001;58(3):449–54. doi: 10.1001/archneur.58.3.449. [DOI] [PubMed] [Google Scholar]
- 26.Aisen PS. The potential of anti-inflammatory drugs for the treatment of Alzheimer's disease. Lancet Neurol. 2002;1(5):279–84. doi: 10.1016/s1474-4422(02)00133-3. [DOI] [PubMed] [Google Scholar]
- 27.McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28(5):639–47. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Streit WJ, Kincaid-Colton CA. The brain's immune system. Sci Am. 1995;273(5):54–61. doi: 10.1038/scientificamerican1195-54. [DOI] [PubMed] [Google Scholar]
- 29.Reale M, Iarlori C, Gambi F, Feliciani C, Salone A, Toma L, et al. Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. J Neuroimmunol. 2004;148(1-2):162–71. doi: 10.1016/j.jneuroim.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, et al. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24(7):1760–71. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, et al. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129(2):318–20. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- 32.Di BD, Candore G, Franceschi C, Licastro F, Colonna-Romano G, Camma C, et al. Systematic review by metaanalyses on the possible role of TNF-alpha polymorphisms in association with Alzheimer's disease. Brain Res Rev. 2009;61(2):60–8. doi: 10.1016/j.brainresrev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Randall CN, Strasburger D, Prozonic J, Morris SN, Winkie AD, Parker GR, et al. Cluster analysis of risk factor genetic polymorphisms in Alzheimer's disease. Neurochem Res. 2009;34(1):23–8. doi: 10.1007/s11064-008-9626-8. [DOI] [PubMed] [Google Scholar]
- 34.Alvarez V, Mata IF, Gonzalez P, Lahoz CH, Martinez C, Pena J, et al. Association between the TNFalpha-308 A/G polymorphism and the onset-age of Alzheimer disease. Am J Med Genet. 2002;114(5):574–7. doi: 10.1002/ajmg.10515. [DOI] [PubMed] [Google Scholar]