

Abstract
We review our baboon models of Escherichia coli sepsis that mimic, respectively, the shock/disseminated intravascular coagulation (DIC) and organ failure variants of severe sepsis, and analyse the pathophysiologic processes that are unique to each. The multi-stage, multi-factorial characteristics of severe sepsis develop as a result of the initial insult, which – depending on its intensity – activates components of the intravascular compartment leading to overwhelming shock/DIC; or initiates a sequence of events involving both the intra- and extravascular (tissues) compartments that lead to organ failure. In the latter case, the disorder passes through two stages: an initial inflammatory/coagulopathic intravascular first stage triggered by E. coli, followed by an extravascular second stage, involving components unique to each organ and triggered by ischemia/reperfusion (oxidative stress and histone release). Although a myriad of overlapping cellular and molecular components are involved, it is the context in which these components are brought into play that determine whether shock/DIC or organ failure predominate. For example, inflammatory and thrombotic responses amplified by thrombin in the first case whereas similar responses are amplified by complement activation products in the second. Rather than blocking specific mediators, we found that attenuation of the thrombin and complement amplification pathways can effectively reverse the shock/DIC and organ failure exhibited by the LD100 and LD50 E. coli models of severe sepsis, respectively. Translation of these concepts to successful intervention in the respective baboon models of E. coli sepsis and the application to their clinical counterparts is described.
Keywords: sepsis, E. coli, staging, animal model, multiple organ failure, complement
Introduction
Severe sepsis is a multi-stage, multi-factorial and life-threatening condition that arises when the innate response of the body to infection injures its own tissues and organs [1]. Sepsis is a major cause of death worldwide and a huge burden for the health care system. Despite important strides made in understanding its pathophysiology, sepsis-related mortality and morbidity remain unacceptably high. Sepsis afflicts over 750,000 patients, kills over 210,000 patients and it costs over 17 billion dollars per year in the United States alone [2].
The editorial foreword of this review series [3] briefly outlined our current understanding of severe sepsis. In this review we focus on the pathophysiology of severe sepsis including its clinical variants, as revealed by the experimental models of E. coli sepsis in baboons that were developed by our group during the last three decades [4-6]. This subject is an especially appropriate topic for this journal as it is devoted to the translation of experimental observations to clinical practice.
Modeling the shock/disseminated intravascular coagulation (DIC) variant of severe sepsis using the LD100 baboon model of E. coli sepsis
Patients with severe sepsis can present with transient organ dysfunction that rapidly improves with antibiotics and supportive therapy, a more profound and persistent organ failure that may progress despite treatment or, in its most virulent form, patients die within hours with massive DIC and cardiovascular collapse that is refractory to therapy [6]. Our initial primate model of E. coli sepsis closely mimics the third presentation [20].
Intravenous challenge with E. coli [1010 colony forming units (cfu)/kg, LD100] induces an immediate innate immune response. Pattern recognition receptors, such as Toll-like receptor (TLR) [7] and soluble recognition molecules, such as complement components [8] rapidly initiate host defense responses [9] after detection of microbial infection or tissue damage. TLR4 in complex with MD-2 and the recognition molecule CD14 binds bacterial lipopolysaccharide (LPS) on immune and other cell types and is the main trigger of the inflammatory response in Gram-negative sepsis [10]. Consequently, activated cells release excessive amounts of pro-inflammatory mediators, such as TNFα, IL-6 and IL-8 [10]. In direct response to the invading bacteria, neutrophils and other inflammatory cells release extracellular traps (ETs) made of DNA and core histones, as well as anti-microbial components (defensins, myeloperoxidase, elastase) that bind, disarm an kill microbes extracellularly, independent of phagocytic uptake [11]. However, ETs and their components have harmful effects for the host, too [12]. Thus, the ET-clustered neutrophils and bacteria end up obstructing vessels thus contributing to ischemia. Released histones and DNA triggers inflammation via TLRs [13]. We have recently shown that histones kill not only the bacteria but also the cells of the host, which then release additional histones, beginning a positive feedback cascade that may cause and amplify organ failure [12]. The inflammatory response up-regulates tissue factor (TF)-dependent coagulation [14, 15] and decreases the anticoagulant [16] and fibrinolytic [17] functions. The loss of this balance results in depletion of platelets, DIC and widespread bleeding [18].
When we developed the LD100 primate model 30 years ago, we assumed that the initial classical inflammatory/coagulopathic response to LD100E. coli would account for both the shock/DIC and the organ failure variants exhibited by the severe sepsis patients. Therefore, we reasoned that these animals might be rescued from both variants by blocking the haemostatic arm of this inflammatory/haemostatic response. However, anticoagulation with agents such as heparin [19], hirudin or active site blocked factor Xa [19, 20], failed to rescue these animals. At about that time we observed that dogs that had undergone extracorporeal perfusion before being exposed to lethal endotoxin were protected [21]. This observation suggested that the extracorporeal perfusion had induced the production of a factor in the blood that protected these animals. By good fortune this observation coincided with the identification of the protein C (PC) activation pathway, by which thrombin in complex with thrombomodulin (TM) turns off its own production [22]. This complex located on the endothelium, in turn activates PC in the circulation, which shuts off further production of thrombin by inactivating factors Va, VIIIa in a negative feedback [23]. We reasoned that when run at the correct speed, the extracorporeal pump induced the production of a sufficient amount of thrombin to trigger the production of activated protein C (aPC) and that this accounted both for not having to add heparin to the perfusion pump, and for the dogs’ survival. We then infused aPC together with LD100E. coli into baboons.
In contrast to similar studies carried out with other anticoagulants, aPC attenuated the responses of the first stage biomarkers, including TNF and fibrinogen and had organ-protective effects, leading to 80% survival benefit [21]. The cytoprotective effects of aPC were later attributed to its signaling via endothelial cell protein C receptor (EPCR)-protease activated receptor 1(PAR-1)-sphingosine 1 phosphate 1 receptor axis. The observations made in our baboon model led to the development of recombinant human aPC (Xigris®) as an effective treatment for severe sepsis patients [24, 25]. The PROWESS trial showed that aPC treatment was associated with an absolute reduction of mortality of 6.1% but the benefit was primarily in the most severely ill patients (APACHE II scores of 25 or higher) [24, 25]. Subsequent studies on low risk and pediatric populations have failed to find a benefit in these groups. As aPC is a potent anticoagulant, the major adverse effects are associated with bleeding. Signaling only aPC mutants with reduced anticoagulant properties are currently under development [26]. These mutants were not tested yet in the baboon model. Since the LD100 is a highly thrombogenic, shock/DIC model, the anticoagulant properties of the aPC may be essential for survival. However, signaling only aPC mutants might be beneficial for preserving organ integrity and function in the LD50 model described below.
Principles arising from the studies carried out in the LD100E. coli model
In addition to identifying a new therapy for severe sepsis patients [25], these early studies led to the confirmation of two important pathophysiologic phenomena. Firstly, a link was found between the initial inflammatory response to bacteria and the subsequent consumptive coagulopathy. This, so-called ‘inflammatory/coagulopathic axis’ was driven by the extrinsic clotting pathway, due to the expression of TF by monocytes/macrophages and the exposure of TF with subendothelial location [18]. Moreover, microbial products such as polyphosphates and omptins can affect the host’s clotting system by triggering clotting via the contact pathways or inactivating natural inhibitors, such as TF pathway inhibitor (TFPI) [27]. The critical insight here was that this coagulopathy involved not only the blood or the vessel wall, but it involved a repertoire of aberrant responses involving components of both compartments.
This set the stage for the second important finding. With the discovery of the regulatory role of the vascular endothelium via TM-PC network [28], it became obvious that the blood and the microvasculature normally functioned as a unit, and that E. coli sepsis was a disorder involving both components of this unit [29]. Thus, sepsis progression targeting the blood-vasculature unit, not only directly elicits the aberrant TF response [30] but also disrupts two critical anticoagulant and anti-inflammatory pathways dependent on TM-PC [24] and TFPI [16], which operate on the surface of the vascular endothelium. This understanding provided the rationale for attempting to treat E. coli sepsis by replacing the component controlling the homeostatic balance disrupted between these two compartments (e.g. aPC [24] and TFPI [31]).
Studies of severe sepsis-induced organ failure using the LD50 baboon model of E. coli sepsis
In the later stages of sepsis, anti-inflammatory mediators, such as IL-10, transforming growth factor β (TGF-β) and IL-13 are produced to compensate for the pro-inflammatory status but this may lead to a hypo-reactive host defense system and immune paralysis with apoptosis of immune cells (for review, see [32]). However, this affects the host’s ability to eradicate other invading pathogens and predisposition to secondary infections. The anti-inflammatory stage is a feedback of the initial overwhelming activation.
Although cardiovascular collapse is the most dramatic manifestation of severe sepsis, the most common complication in septic patients is the development of multi-organ failure (MOF) secondary to hypoperfusion, histone release and intravascular thrombosis. MOF can have a complex and protracted clinical course that is fatal in 30–40% of patients. Mortality increases almost linearly as the number of organ failures accumulates. Usually, single organ failure is associated with a low mortality rate, whereas the majority of patients with 3–4 organ failures will succumb. Survivors of this variant of sepsis can suffer long-term residual disabilities resulting from the sepsis-related tissue and organ damage.
Despite a clear understanding of the inflammatory and coagulation mechanisms triggered during the first stage of severe sepsis, not much is known about the cellular aspects underlying the mechanisms that ultimately lead to organ dysfunction and death. To examine this problem, we developed baboon models of E. coli sepsis, which, depending on the bacterial dose, mimic the different pathophysiological syndromes, including MOF, observed in clinical practice [4, 5]. The administration of 109 cfu/kg E. coli (LD50) produces transient hypotension followed by MOF, which may progress and prove fatal in approximately 50% of the animals. Administration of lower concentrations, 107–108 cfu/kg E. coli (LD10), produces two distinct stages, a brief period of mild hypotension followed by transient organ dysfunction of less severity, which typically resolves within 24 to 48 hrs.
The pathophysiology of the LD50 model also demonstrates a two-stage or two-compartment response, each driven by distinct mechanisms. The first stage is an exacerbated intravascular host defense response to bacterial infection whereas the second stage is an uncontrolled extravascular host recovery response leading to MOF, which is likely driven, at least in part by ischemia-reperfusion (IR) injury. Figure 1 shows a diagrammatic summary of the triggers, responses and potential treatments of the three baboon models and their clinical counterparts. It also reflects how first the intravascular (circulation), then the extravascular (tissue) compartments come into play with increases in the concentration of E. coli, until at the highest concentration, the organ failure responses of the extravascular compartment are completely overridden by the shock/DIC response of the intravascular compartment. These mirror the non-progressive, progressive organ failure and the shock/DIC variants of severe sepsis. This figure also reflects the differences in mechanisms driving these variants in the baboon, indicating that one responds to aPC, while the other responds to C3 convertase inhibitors (see below).
Fig 1.
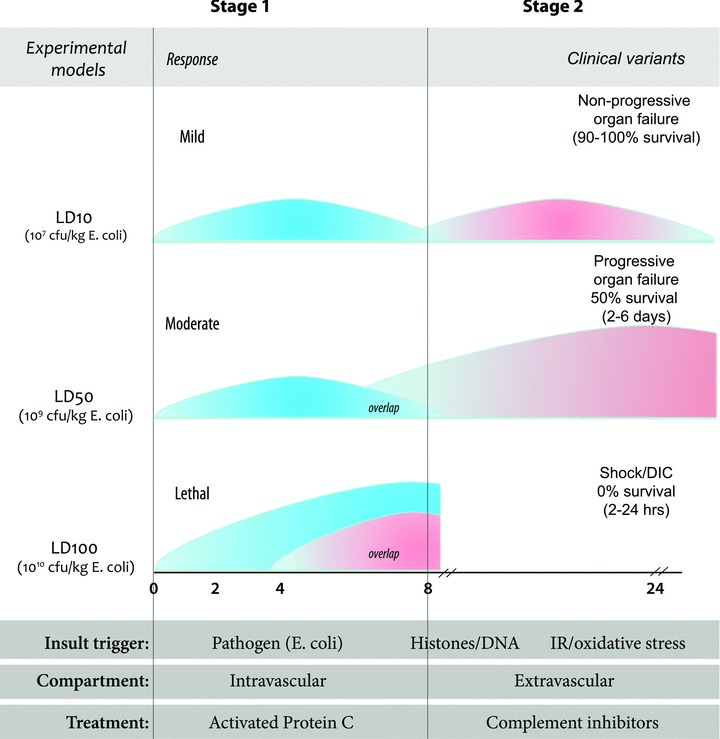
Summary diagram of the three models of E. coli sepsis and their clinical counterparts.
It is well established that while early restoration of blood flow is essential to halt cellular injury, the late reperfusion triggers IR injury [33]. This enhances inflammation and induces oxidative damage in distal organs that were not affected by the initial ischemic insult, thus amplifying the harm and leading to MOF [33]. IR in organs, such as the gastrointestinal tract, triggers the production of acute phase proteins (e.g. CRP), and a second round of inflammatory mediators [34]. IR activates defense mechanisms, including the complement system, leading to widespread deposition of complement in the microcirculation [33] and release of biologically active complement split products (C3a, C5a). In turn, these further enhance cytokine production and elicit excessive, intravascular innate immune responses to bacteria that induce cell/tissue injury, further leading to organ dysfunction, MOF and death.
It is believed that both stages occur also in the more severe (LD100) model of sepsis. However, in the LD100 model, the two stages overlap greatly, making it difficult to distinguish the two as having separate etiologies.
Ischemia-reperfusion and oxidative stress as drivers of the second stage
Clinical experience has shown that some patients who survived shock/DIC after aPC treatment still die later because of organ failure. We also observed that baboons infused with LD50 rather than LD100E. coli could survive the initial inflammatory/coagulopathic response but die later from MOF [4]. Based on our baboon models, we hypothesized that a different pathophysiological mechanism could come into play in these patients. We utilized the LD50E. coli sepsis model to determine the mechanism driving the organ failure that had not responded to aPC.
It should be noted that these concepts evolved gradually over several years. It was only after observing the responses of humans to endotoxin and of baboons to sublethal E. coli that we realized that the responses to lower concentrations exhibited two stages [4]. We established that first stage was dominated by cytokines and inflammatory cells, and the second was dominated by complement activation products and thrombocytopenia in the absence of a consumptive coagulopathy, long after all E. coli organisms had been cleared. We also established that when the concentration of E. coli was increased above LD50 towards LD100, these two stages overlapped resulting in an override and concealment of the second stage events by the residual of the first stage.
Next, we aimed to identify and characterize the dominant processes of the two stages and their impact on survival. We already knew that not all organs are equally affected by sepsis and that animals exposed to comparable doses of E. coli may have opposite clinical outcome: some cope with the challenge, while others die. We used transcriptomics combined with histological and biochemical analysis to investigate if there was a temporal association between events that happens in the organs/tissues and molecular plasma biomarkers over the first 24 hrs in animals infused with LD50E. coli [35]. We observed strong induction of genes involved in tissue remodeling, regeneration and functional recovery at T+24 hrs [35]. Histological analysis also revealed fibroblast proliferation and collagen deposition in the lung, as well as renal microvascular thrombosis. These events occurred after the initial classical inflammatory/coagulant responses had run their course and after the clearance of the bacteria from the circulation had been completed. Intriguing, these events coincided with the appearance of complement activation products and the development of thrombocytopenia [36].
It is likely that these initial changes occurring in organs were masked in the LD100E. coli sepsis models by the severity of the initial inflammatory/coagulopathic events and because we examined the tissues only towards the end of the study, at the time the animals had become moribund.
Retrospective analysis of a large number of plasma samples from LD50 survival experiments established that animals that have a T+5 hrs fibrinogen level lower than 30% of the initial value and FDP higher than 80 μg/dl will have a high probability of death. These findings allowed us to stratify LD50 animals at 24 hrs as probable survivors (LD50-S) and non-survivors (LD50-NS) and to compare the specific pathologies of each group. In contrast to the LD100 animals, which died from events occurring in circulation, LD50-NS died from different pathophysiological processes arising from events within the tissues. In Figure 2, blood analysis showed that LD50-NS reached certain biomarker values – including FDP, elastase, sTM and C5b9 terminal complement complex – during the second stage (8–24 hrs) that resembled those observed during the first stage (2–6 hrs) in the LD100 challenged baboons. In contrast, the LD50-S animals showed significantly lower levels of elastase, sTM and C5b9, and less WBC and platelet consumption than LD50-NS. Moreover, the profound second stage thrombocytopenia observed in LD50-NS was accompanied by a marked rise in prothrombin time (PT) while the LD50-S showed little change in PT values.
Fig 2.
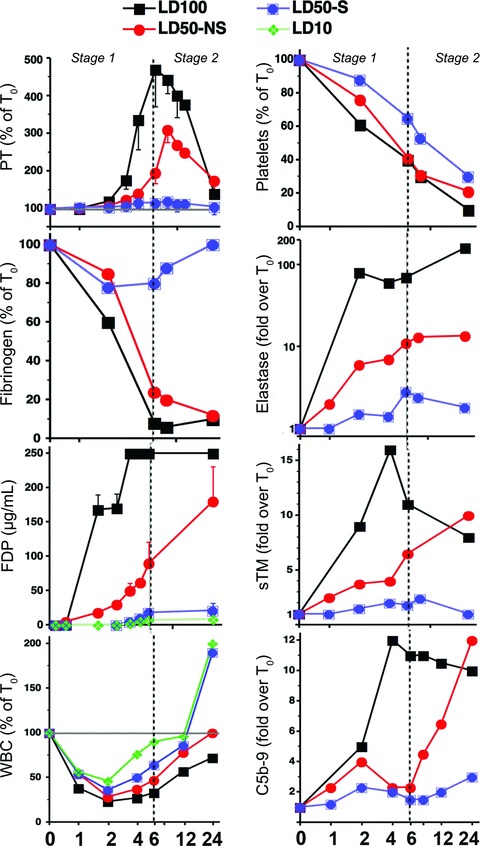
Time-course changes of plasma biomarkers during E. coli sepsis in baboons challenged with LD50 and stratified into potential survivors (LD50-S) and non-survivors (LD50-NS), as compared to the LD100 and LD10 groups.
Postmortem examination at T+24 hrs established that the pathology of the lung in LD50-NS baboons was entirely different from the LD100 baboons (Fig. 3). The former (Fig. 3A–C) displayed extensive fibrocytes, macrophages and Sirius red stained collagen infiltration, as well as edema in the absence of the massive hemorrhage and neutrophil infiltration found in the LD100 model (Fig. 3F), while the LD50-S group (Fig. 3B and D) showed an almost normal structure. This indicates that the lungs of the animals that advanced into the second stage pass the aberrant neutrophil-dominated host defense that occur in the LD100 challenged animals and display a cell repertoire typical for tissue recovery, a different pathology which also is lethal. Furthermore, lung samples collected from LD50-NS versus LD50-S were analysed using genomic approaches combined with structural and biochemical techniques. We have observed that animals with non-survival phenotype had a significantly stronger induction of NFkB signaling than the potential survivors, suggesting a stronger pro-inflammatory response. Furthermore, TGF-β and fibrosis signaling pathways as well as the genes involved in hypoxia, oxidative stress and mitochondrial dysfunction were significantly increased in LD50-NS versus LD50-S (Fig. 4A). Similarly, LD50-NS showed more robust changes of genes involved in G1/S cell cycle transition as well as strong down-regulation of death receptor signaling via anti-apoptotic BID-BCL2 and inhibitor of apoptosis (IAPs) pathways (Fig. 4B; down-regulated genes are shown in green). Correlated with the pathology data, these results implicate the second stage events, such as IR, oxidative stress, mitochondrial dysfunction, apoptosis and fibrosis as key factors in the development of organ dysfunction and therefore as potential therapeutic targets.
Fig 3.
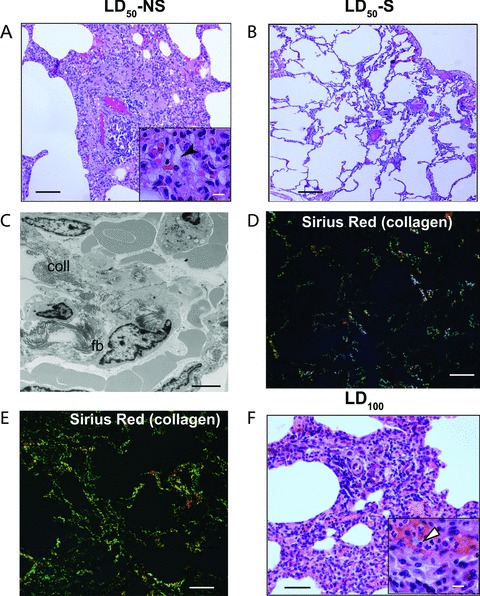
Histopathological features of the lung samples from potential survivors (LD50-S) and non-survivors (LD50-NS), as compared to LD100E. coli challenged baboons. Hematoxylin-eosin staining of the lung samples from the LD50-NS (A), LD50-S (B) and LD100 (F) experimental groups; sirius red staining and epipolarized light imaging of collagen deposition in the samples from LD50-S (D) versus LD50-NS (E); (C) electron micrograph demonstrating the presence of fibroblasts (fb) and collagen (coll) in the interalveolar space, in the lung of animals from the LD50-NS group. Scale bars: (A): 150 μm; (A and F insets): 25 μm; (B, D and E): 250 μm; (C): 5 μm.
Fig 4.
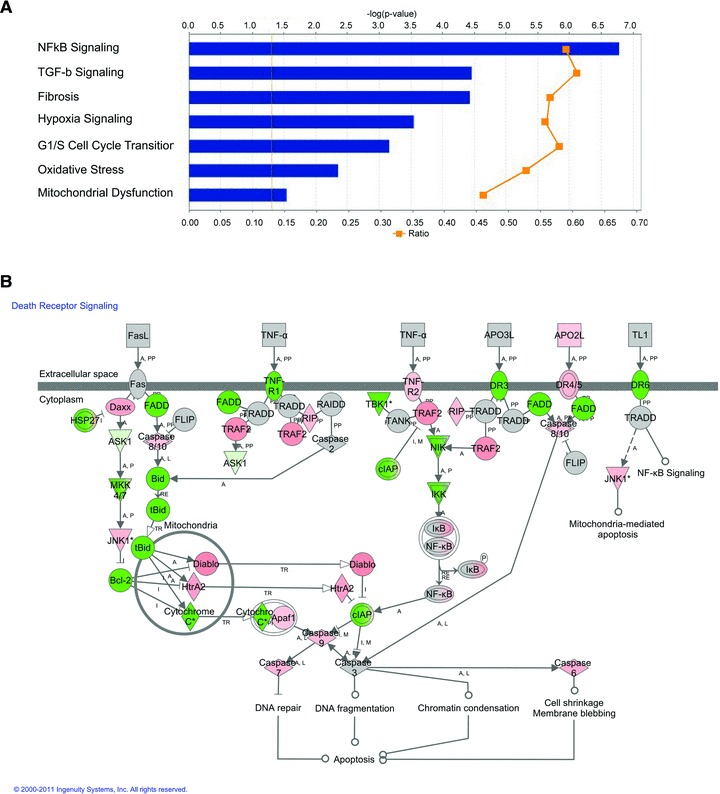
Gene expression analysis of the lung from LD50-S versus LD50-NS baboons using Ingenuity Pathway analysis. (A) Canonical pathways that are differentially induced in non-survivors based on the – log (p) and ratio values (LD50-S:LD50-NS); (B) canonical death receptor signaling pathway demonstrates the inhibition of anti-apoptotic and cell survival genes (BID, BCL2 and cIAP) in non-survivors (LD50-NS), as compared to survivors (LD50-S) challenged with LD50E. coli. Gene expression changes over 2-fold threshold are indicated by colour code: green: downregulation; red: up-regulation.
The fact that complement activation had been observed by Stahl et al. [33] in other models of ischemia reperfusion, and that the rapid production of collagen within 24 hrs also had been observed by Coalson et al. [37] in neonates exposed to oxidative stress suggested that the second ‘organ failure’ stage (ARDS, pulmonary fibrosis etc.) in this LD50E. coli sepsis model might be driven, at least in part, by IR/oxidative stress and that complement activation might also play a role.
Therapeutic targets during the second stage of sepsis
Complement activation
On the basis of the observations showing that organ failure coincided with the generation of complement activation products, thrombocytopenia and the production of lactate, we asked whether the events involving the tissues/organs (extravascular compartment) were functionally linked to events occurring simultaneously in the circulation (intravascular compartment). We hypothesized that complement activation and histone release could represent amplifying loops of the initial injury produced by IR/oxidative stress. If true, blockade of complement activation could suppress both the generation of activation products and the tissue damage induced by IR/oxidative stress.
Three excellent reviews on complement activation and its involvement in the pathophysiology of sepsis were published as part of this series [8, 38, 39], therefore a detailed survey of this topic is out of our purpose. Complement is critical for innate immunity against pathogens but uncontrolled complement activation has been associated with many immuno-inflammatory conditions [8]. Once microbes enter the bloodstream, activated complement system components rapidly opsonize the pathogens [40], thus facilitating their phagocytosis by leukocytes [41]. Complement activation products, in particular the anaphylatoxin C5a are critically involved in the up-regulation of the phagocytosis and induction of the oxidative burst [42, 43]. C3b participates in the formation of C5 convertase, which cleaves C5 to C5a and C5b, the latter becoming part of the terminal C5b-9 complex (TCC) [39]. C3a anaphylatoxin activates platelets, induces their aggregation and recruits leukocytes. Elevated levels of C5a signal through its receptors C5aR (CD88) and C5L2 (GPR77) are contributing to immune paralysis, multi-organ dysfunction, apoptosis and deterioration of the coagulation/fibrinolytic system and contractile dysfunction of the cardiomyocytes (reviewed in [44]). Data from our group show a biphasic activation of the complement cascade in response to sublethal E. coli in baboons, with a maximum peak of TCC occurring during the 2nd stage [45, 46].
Although early increase of complement activation is principally beneficial to the host defense response, complement activation triggered by IR/oxidative stress during the second stage of sepsis contributes to tissue damage. Moreover, complement activation further amplifies the IR and oxidative stress, thus acting as a positive feedback that leads to a subsequent round of inflammatory activity, this time localized in the tissues rather than in the vasculature. Collectively, the interplay between complement activation and oxidative stress leads to pathological responses unique to each tissue or organ, and finally to death in many cases. Clinical studies have shown a consistent association between the extent of complement activation during sepsis and poor outcome [47, 48].
Using the LD50 baboon model of E. coli sepsis, we demonstrated that inhibition of complement activation effectively attenuates inflammatory and haemostatic processes, restores systemic blood pressure and improves organ function during severe sepsis with either early or late treatment regimens [36]. Blocking complement activation at the early phase virtually completely abolished the fall in systemic blood pressure, indicating that complement activation is responsible for one of the most important physiological disturbances during early sepsis. Although it is possible that complement inhibitors may be used during the first stage of sepsis, if administered together with antibiotics or blockers of CD14-TLR signaling [49, 50], we are particularly excited by the protective effect of the complement inhibitor when administered during the second stage, as this indicates that complement activation during this time-frame contributes to disease progression towards organ failure and death. The fact that late intervention of complement activation (5 hrs post-challenge) still provides organ protection is important, as most septic patients receive medical attention after the debut of the disease. These findings are particularly notable as aPC, the only approved agent for the treatment of sepsis, has therapeutic efficiency in baboons only when administered 1–2 hrs post-challenge.
Circulating histones
Recently we have shown that histones, a group of basic proteins located mainly in the nucleus, are released in the extracellular environment in response to inflammatory challenges and contribute to endothelial dysfunction, organ failure and death during sepsis [12, 51].
Traditionally, histones are known as major components of nucleosomal structures in eukaryotic cells. There is growing evidence that histones are present not only inside, but also outside the nucleus, and these extracellular histones appear to have other functions, such as playing a role in the innate immune defense systems. Support for this idea comes from the observations that histones are among the most potent bactericidal proteins in eukaryotic cells [52] and that peptides derived from histones are produced and secreted in epithelia, where they act as barriers to microbial invasion in frogs, fish and mammals [53]. Arg-rich histones (H3 and H4) exhibit stronger anti-microbial activities than Lys-rich histones (H1, H2A and H2B) towards E. coli and other species by binding to the bacterial cell surface, thus causing damage to the osmotic barrier and cell respiration [54]. Like other anti-microbial peptides, histones are hydrophobic and cationic and thus electrostatically attracted to negatively charged phospholipid groups of the external leaflet of the microbial cytoplasmic membrane, where they form amphiphilic helical conformations [54].
We have shown that extracellular histones are released into the circulation from damaged cells during the transition from the first to the second stage in non-survivor LD50E. coli baboons [12]. Besides their anti-bacterial roles, the extracellular histones have major cytotoxic effects in eukaryotic cells, thus inducing extensive ‘collateral damage’ to the host tissue. To examine this possibility we infused histones and histone/DNA complexes into mice and later into baboons. The infusion of low concentrations of histones induced thrombocytopenia while higher concentrations were lethal. Among histones, the arginine-rich H3 and H4 are the most cytotoxic [12]. Intravenously administered histones have lethal effects that mimic the pathophysiological features of sepsis in mice. Administration of neutralizing anti-histone monoclonal antibodies provided significant protection, rescuing mice from lethal sepsis. Moreover, aPC cleaves histones and reduces their toxicity, which may explain why it is effective in septic patients while other anticoagulants are not [12].
Our current hypothesis is that histones released under certain conditions can combine with IR/oxidative stress to exacerbate organ failure through generation of additional complement activation products as well as through inflammatory events induced through the Toll/NFKB pathway [55].
Lessons learned from the baboon models of sepsis
Various small animal models involving exogenous administration of toxins, viable pathogens or alteration of the animal’s protective barriers have contributed to our current understanding of host responses to infection (for review see [56]). However, data from small animal rodent models of shock and sepsis are difficult to transpose to the disease in humans [57]. Baboons and humans have a high degree of genetic similarity and their haemostatic and cardiovascular responses to sepsis are similar [57]. Moreover, our baboon models mimic the human septic response with respect to the immuno-inflammatory parameters (e.g. late-phase sepsis immunosupression) and apoptosis of specific immunocompetent cells.
Our intravenous infusion models involve challenges with live E. coli, one of the most common pathogens casing sepsis in humans, and allow precise control of the infecting bacterial strain, infusion timing and bacterial load. The baboon model allows bridging and validation of data obtained in small animals to the settings of human sepsis. The original studies using aPC for sepsis therapy in baboons [24] were particularly important for the subsequent clinical development of aPC for use in humans. Additionally, due to the close homology between baboon and human proteins, most human-specific antibodies and biological reagents can be successfully used in this model.
By varying the amount of E. coli administered from 107 to 1010 cfu/kg we have created primate models, which mimic the different clinical presentations of patients with severe sepsis. Our initial studies utilized a high dose (1010 cfu/kg) of E. coli to create a model of refractory hypotension and consumptive coagulopathy that was rapidly fatal. These studies helped establish the link between inflammation and coagulation and ultimately led to the development of aPC for the treatment of sepsis.
Subsequent studies infusing a lower dose of E. coli permitted us to distinguish the initial hypotensive and coagulopathic response from the ensuing organ injury that developed after the hypotension and coagulation abnormalities had resolved. This led to the concept that sepsis is a two-stage disease in which processes that are initiated within the intravascular compartment during stage 1 extend into the extravascular compartment during stage 2 (Fig. 5).
Fig 5.
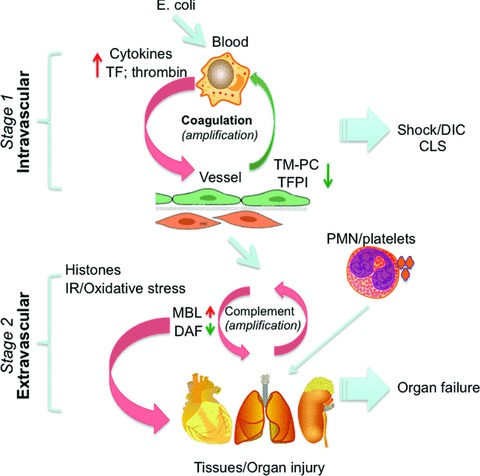
Diagram illustrating the mechanisms controlling the stage-specific pathophysiologic responses in the baboon models of severe sepsis. The first stage (top) inflammatory and coagulopathic responses are induced by E. coli and involves cytokine and TF expression by leukocytes. These responses are further amplified by thrombin that overrides the PC and TFPI regulatory networks. During the second stage (bottom), organ-specific injury responses to oxidative stress and histones are further amplified by complement activation products that act on cellular components (platelets, PMNs) recruited from the intravascular compartment.
While 107 cfu/kg E. coli challenge produced mild organ dysfunction that rapidly resolved, infusing a higher dose (109 cfu/kg) produced a model of persistent multi-organ failure that allowed us to identify factors that contribute to MOF. During stage 2, the cell organelles actively participate in a second round of aberrant responses, instead of passively disintegrating due to lack of oxygen. In this case, the second stage tissue organ failure response to oxidative stress and circulating histones is a consequence of the first stage circulatory/shock/DIC response to LD50E. coli.
As shown in Figure 5, it is likely that the tissue damage involving this extravascular compartment following oxidative stress/histones promotes acute respiratory distress and fibrosis in the lungs, thrombotic angiopathy in the kidney and apoptosis in lymphoid organs. Our studies show that the initial aberrant tissue-specific responses are further amplified by complement activation products and histones and in turn by other components (platelets, PMNs) recruited from the intravascular compartment [36].
The fact that different pathological processes are driving stage 1 and stage 2 of sepsis suggests that different inhibitors of inflammatory amplification loops would be effective in stage 1 and stage 2. Activated protein C is most effective when administered within 24 hrs of the onset of severe sepsis in patients, which corresponds to our observation that it must be given early to prevent death in our LD100 model. Patients with DIC, a marker of stage 1 disease, appear to benefit most from aPC treatment. Once sepsis has progressed to stage 2 in which oxidative stress and histone release contribute to complement activation and organ failure, direct blockade of C3 convertase with an inhibitor such as compstatin will be more effective.
Acknowledgments
The authors would like to thank Miss Catalina Lupu for editorial assistance.
Conflict of interest
FBT is an inventor on a patent on the use of activated protein C in sepsis and received royalties from Eli Lilly. FBT, GK and FL are co-inventors on a patent application on the use of complement inhibitors to prevent organ failure in sepsis.
Funding source: This work was supported by the grants GM037704, 1RC1GM09739 and 1R01GM097747 from the National Institutes of Health.
References
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Lupu F. “Crossroads in Sepsis Research” review series overview of the pathophysiology of sepsis. J Cell Mol Med. 2008;12:1072–3. doi: 10.1111/j.1582-4934.2008.00366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor FB., Jr Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care Med. 2001;29:S78–89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 5.Taylor FB, Jr, Wada H, Kinasewitz G. Description of compensated and uncompensated disseminated intravascular coagulation (DIC) responses (non-overt and overt DIC) in baboon models of intravenous and intraperitoneal Escherichia coli sepsis and in the human model of endotoxemia: toward a better definition of DIC. Crit Care Med. 2000;28:S12–9. doi: 10.1097/00003246-200009001-00004. [DOI] [PubMed] [Google Scholar]
- 6.Taylor FB, Jr, Kosanke S, Randolph M, et al. Retrospective description and experimental reconstitution of three different responses of the baboon to lethal. E. coli. Circ Shock. 1994;42:92–103. [PubMed] [Google Scholar]
- 7.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–63. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 8.Markiewski MM, DeAngelis RA, Lambris JD. Complexity of complement activation in sepsis. J Cell Mol Med. 2008;12:2245–54. doi: 10.1111/j.1582-4934.2008.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–20. doi: 10.1111/j.1582-4934.2009.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 11.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–21. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Evankovich J, Yan W, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9. Hepatology. 2011;54:999–1008. doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor FB, Chang AC, Peer G, et al. Active site inhibited factor VIIa (DEGR VIIa) attenuates the coagulant and interleukin-6 and -8, but not tumour necrosis factor, responses of the baboon to LD100 Escherichia coli. Blood. 1998;91:1609–15. [PubMed] [Google Scholar]
- 15.Lupu C, Westmuckett AD, Peer G, et al. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of Escherichia coli sepsis. Am J Pathol. 2005;167:1161–72. doi: 10.1016/S0002-9440(10)61204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang H, Ivanciu L, Popescu N, et al. Sepsis-induced coagulation in the baboon lung is associated with decreased tissue factor pathway inhibitor. Am J Pathol. 2007;171:1066–77. doi: 10.2353/ajpath.2007.070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Boer JP, Creasy AA, Chang A, et al. Activation patterns of coagulation and fibrinolysis in baboons following infusion with lethal or sublethal dose of Escherichia coli. Circ Shock. 1993;39:59–67. [PubMed] [Google Scholar]
- 18.Taylor FB., Jr The inflammatory-coagulant axis in the host response to gram-negative sepsis: regulatory roles of proteins and inhibitors of tissue factor. New Horiz. 1994;2:555–65. [PubMed] [Google Scholar]
- 19.Coalson JJ, Benjamin B, Archer LT, et al. Prolonged shock in the baboon subjected to infusion of E. coli endotoxin. Circ Shock. 1978;5:423–37. [PubMed] [Google Scholar]
- 20.Taylor FB, Jr, Chang AC, Peer GT, et al. DEGR-factor Xa blocks disseminated intravascular coagulation initiated by Escherichia coli without preventing shock or organ damage. Blood. 1991;78:364–8. [PubMed] [Google Scholar]
- 21.Hinshaw LB, Chang AC, Beller-Todd BK, et al. Extracorporeal perfusion without exogenous anticoagulation: its protective role in endotoxin shock. Circ Shock. 1982;9:281–95. [PubMed] [Google Scholar]
- 22.Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1982;257:859–64. [PubMed] [Google Scholar]
- 23.Esmon CT, Taylor FB, Jr, Snow TR. Inflammation and coagulation: linked processes potentially regulated through a common pathway mediated by protein C. Thromb Haemost. 1991;66:160–5. [PubMed] [Google Scholar]
- 24.Taylor FB, Jr, Chang A, Esmon CT, et al. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79:918–25. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor FB, Kinasewitz G. Activated protein C in sepsis. J Thromb Haemost. 2004;2:708–17. doi: 10.1111/j.1538-7836.2004.00751.x. [DOI] [PubMed] [Google Scholar]
- 26.Weiler H, Ruf W. Activated protein C in sepsis: the promise of nonanticoagulant activated protein C. Curr Opin Hematol. 2008;15:487–93. doi: 10.1097/MOH.0b013e32830abdf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yun TH, Morrissey JH. Polyphosphate and omptins: novel bacterial procoagulant agents. J Cell Mol Med. 2009;13:4146–53. doi: 10.1111/j.1582-4934.2009.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J Biol Chem. 1982;257:7944–7. [PubMed] [Google Scholar]
- 29.Taylor FB., Jr Response of anticoagulant pathways in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27:619–31. doi: 10.1055/s-2001-18872. [DOI] [PubMed] [Google Scholar]
- 30.Taylor FB., Jr Role of tissue factor and factor VIIa in the coagulant and inflammatory response to LD100 Escherichia coli in the baboon. Haemostasis. 1996;26:83–91. doi: 10.1159/000217246. [DOI] [PubMed] [Google Scholar]
- 31.Randolph MM, White GL, Kosanke SD, et al. Attenuation of tissue thrombosis and hemorrhage by ala-TFPI does not account for its protection against E. coli-a comparative study of treated and untreated non-surviving baboons challenged with LD100 E. coli. Thromb Haemost. 1998;79:1048–53. [PubMed] [Google Scholar]
- 32.Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435–44. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stahl GL, Xu Y, Hao L, et al. Role for the alternative complement pathway in ischemia/reperfusion injury. Am J Pathol. 2003;162:449–55. doi: 10.1016/S0002-9440(10)63839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Boer JP, Creasey AA, Chang A, et al. Activation of the complement system in baboons challenged with live Escherichia coli: correlation with mortality and evidence for a biphasic activation pattern. Infect Immun. 1993;61:4293–301. doi: 10.1128/iai.61.10.4293-4301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu H, Tang Y, Ivanciu L, et al. Temporal dynamics of gene expression in the lung in a baboon model of E. coli sepsis. BMC Genomics. 2007;8:58. doi: 10.1186/1471-2164-8-58. . doi: 10.1186/1471-2164-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silasi-Mansat R, Zhu H, Popescu NI, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002–10. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coalson JJ, Kuehl TJ, Prihoda TJ, et al. Diffuse alveolar damage in the evolution of bronchopulmonary dysplasia in the baboon. Pediatr Res. 1988;24:357–66. doi: 10.1203/00006450-198809000-00017. [DOI] [PubMed] [Google Scholar]
- 38.Ward PA, Gao H. Sepsis, complement and the dysregulated inflammatory response. J Cell Mol Med. 2009;13:4154–60. doi: 10.1111/j.1582-4934.2009.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. 2008;12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newman SL, Mikus LK. Deposition of C3b and iC3b onto particulate activators of the human complement system. Quantitation with monoclonal antibodies to human C3. J Exp Med. 1985;161:1414–31. doi: 10.1084/jem.161.6.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol. 2004;41:1089–98. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 42.Mollnes TE, Brekke OL, Fung M, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100:1869–77. [PubMed] [Google Scholar]
- 43.Brekke OL, Christiansen D, Fure H, et al. The role of complement C3 opsonization, C5a receptor, and CD14 in E. coli-induced up-regulation of granulocyte and monocyte CD11b/CD18 (CR3), phagocytosis, and oxidative burst in human whole blood. J Leukoc Biol. 2007;81:1404–13. doi: 10.1189/jlb.0806538. [DOI] [PubMed] [Google Scholar]
- 44.Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004;4:133–42. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 45.Taylor FB, Jr, Hack E, Lupu F. Observations on complement activity in the two-stage inflammatory/hemostatic response in the baboon and human models of E. coli sepsis and endotoxemia. Adv Exp Med Biol. 2006;586:203–16. doi: 10.1007/0-387-34134-X_14. [DOI] [PubMed] [Google Scholar]
- 46.Riedemann NC, Ward PA. Complement in ischemia reperfusion injury. Am J Pathol. 2003;162:363–7. doi: 10.1016/S0002-9440(10)63830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakae H, Endo S, Inada K, et al. Serum complement levels and severity of sepsis. Res Commun Chem Pathol Pharmacol. 1994;84:189–95. [PubMed] [Google Scholar]
- 48.Hack CE, Nuijens JH, Felt-Bersma RJ, et al. Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am J Med. 1989;86:20–6. doi: 10.1016/0002-9343(89)90224-6. [DOI] [PubMed] [Google Scholar]
- 49.Thorgersen EB, Hellerud BC, Nielsen EW, et al. CD14 inhibition efficiently attenuates early inflammatory and hemostatic responses in Escherichia coli sepsis in pigs. FASEB J. 2010;24:712–22. doi: 10.1096/fj.09-140798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mollnes TE, Christiansen D, Brekke OL, et al. Hypothesis: combined inhibition of complement and CD14 as treatment regimen to attenuate the inflammatory response. Adv Exp Med Biol. 2008;632:253–63. [PubMed] [Google Scholar]
- 51.Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost. 2011;9:182–8. doi: 10.1111/j.1538-7836.2011.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirsch JG. Bactericidal action of histone. J Exp Med. 1958;108:925–44. doi: 10.1084/jem.108.6.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim HS, Yoon H, Minn I, et al. Pepsin-mediated processing of the cytoplasmic histone H2A to strong antimicrobial peptide buforin I. J Immunol. 2000;165:3268–74. doi: 10.4049/jimmunol.165.6.3268. [DOI] [PubMed] [Google Scholar]
- 54.Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets. 2008;8:195–205. doi: 10.2174/1871526510808030195. [DOI] [PubMed] [Google Scholar]
- 55.Xu J, Zhang X, Monestier M, et al. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–65. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 57.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81:137–43. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]