

Abstract
Interferon-γ (IFNγ) plays a major role during host defense against Mycobacterium tuberculosis (Mtb). T cells produce IFNγ in response to IL-12 and IL-18 secreted from Mtb infected macrophages. IFNγ in turn, induces nitric oxide secretion in macrophages that kills Mtb. IFNγ knock-out mice are thus hyper-susceptible to tuberculosis. We reported earlier that Complement C5 deficient (C5-/-) congenic mice are more susceptible to tuberculosis and showed reduced IL-12 synthesis in their macrophages. Using C5-/- congenic mice that carry a deletion in the C5 gene and the wild type C5+/+ mice, we demonstrate here that, the C5-/-derived CD3+ T cells, have an additional defect in the synthesis of IFNγ. C5-/- T cells produced lower levels of IFNγ upon stimulation by antigen presenting cells (APCs) infected with Mtb or when stimulated directly with a combination of IL-12 and IL-18. The latter was in part due to a reduced phosphorylation of STAT-4 following IL-12/IL-18 stimulation. Addition of C5a peptide to IL-12/IL-18 partially restored STAT4 phosphorylation and IFNγ synthesis in C5-/- T cells indicating that IL-12/IL-18 mediated signaling within CD3+ T cells involves C5a peptide. Finally, C5-/- T cells derived from M.bovis BCG or Mtb infected mice showed a reduced expression of T-bet (T-box expressed in T cells) transcription factor, which correlated well with a reduced T cell secretion of IFNγ. Since T-bet mediated IFNγ synthesis facilitates Th1 expansion, C5-/- mouse derived T cells appear to have an intrinsic defect in the production of IFNγ, which is related to C5 deficiency and this may explain their increased susceptibility to infection with Mtb and BCG.
Keywords: Mycobacterium tuberculosis, Complement, Interferon-γ, anaphylatoxin C5a, Mouse, IL-12 and IL-18
1. Introduction
Tuberculosis is the leading cause of death in the world due to a single infectious agent and accounts for about 2 million deaths per year. One third of the world's population is latently infected with Mycobacterium tuberculosis (Mtb), and five to ten percent of these individuals reactivate to clinical disease. Immunity to tuberculosis is primarily T cell mediated and the use of various mouse models has enabled the characterization of multiple cytokines and cell populations in host defense[1,2]. A key mediator of ant-tuberculosis immunity is IFNγ. Macrophages infected with Mtb release IL-12 and IL-18, that in turn, activate T cells to produce IFNγ [3]. While Mtb can replicate within naïve macrophages, IFNγ activates the inducible nitric oxide synthase in macrophages to produce nitric oxide and kill intracellular Mtb[4]. Thus, IFNγ and IL-12 knockout mice are hyper susceptible to tuberculosis [2,5]. Furthermore, mice treated with inhibitors of NO synthesis are also more susceptible to tuberculosis [6]. IL-12 and IFNγ are therefore major regulators of immunity to tuberculosis. In addition, interaction between Mtb infected macrophages and T cells results in the release of a variety of mediators such as IL-1β, IL-6, IL-10 and TNF-α [7]. These cytokines play various regulatory roles, culminating in either control or enhancement of Mtb growth within macrophages.
We previously reported that, C5 deficient A/J and congenic C5 deficient B/10 derived mice (C5-/-) are more susceptible to tuberculosis, compared to C5 intact, C57Bl/6 mice or B.10 derived C5 sufficient congenic mice (C5+/+)[8,9]. Lungs of infected C5-/- mice contain larger bacterial loads after aerosol or intravenous infections. Interestingly, other C5 deficient strains such as DBA/2 and SWR strains have also been reported to be hyper-susceptible to tuberculosis [10] [11]. C5 deficient A/J and C5-/- mice show poor granuloma formation needed for containment of the infection and instead show a pneumonitis. Macrophages secrete a C5 peptidase, which cleaves C5 into C5a and C5b [12]. The most potent anaphylatoxin of the complement system is C5a, the 14 kDa cleavage product of C5. We found that Mtb infected C5 sufficient (C5+/+ and C57Bl/6) macrophages secrete and cleave C5 to C5a peptide [8]. Macrophages also express C5a receptor (C5aR) and we found that C5a-C5aR signaling along with stimulation of the TNF-α receptor regulates the production of IL-12 by macrophages through a feedback mechanism [8]. IL-12 is known to induce CD4 and CD8 T cells to secrete IFNγ via a signal transducer and activator of transcription 4 (STAT4) dependent pathway [13]. We and Karp et al. reported that C5 deficient macrophages secrete reduced levels of IL-12 and that such macrophages were also deficient in the production of TNF-α and IL-1β [8,14].
Since lL-12 induced IFNγ synthesis in T cells is well established, in our initial studies, we were surprised to note that reduced IL-12 mRNA of lungs of Mtb infected C5 deficient mice was not consistent with pulmonary expression of mRNA for IFNγ. Indeed, mRNA messages for IFNγ were comparable between the lungs of congenic C5 mice at certain time points after aerosol induced tuberculosis [9]. However, IFNγ can be produced by a variety of immune cells including MHC-II restricted CD4 T cells, MHC-I restricted CD8 T cells, CD1d restricted NKT cells and even certain types of dendritic cells or macrophages [15]. Furthermore, IFNγ producing T cells can have different effects depending upon the kinetics of expansion, and homing during the various phases of tuberculosis. CD4 T cells are thought to be effective against acute tuberculosis while CD8 T cells have been proposed to prevent reactivation of latent tuberculosis [16,17]. NKT cells have been reported to play an emerging protective role during latent stages of infection[18]. We therefore sought to characterize the ability of purified CD3+ T cells to secrete IFNγ during C5 deficiency using in vitro and in vivo models. We now demonstrate that C5-/- T cells show a defect in the production of IFNγ, when activated with mycobacteria infected macrophages, recombinant proteins of IL-12 or IL-18 and during in vivo mycobacterial infection.
2. Results
Differential IFNγ response of T cells from C5 congenic mice to activation from Mtb infected macrophages
Mice were immunized with Mtb and purified CD3+ T cells from spleens allowed to react in vitro with macrophages infected with Mtb. IFNγ levels were determined and T cells analyzed by flow cytometry to correlate with the CD4 and CD8 phenotype. Fig.1A shows that, C5-/- T cells produce lower levels of IFNγ compared to C5+/+ T cells when primed from APCs, even when they were activated at comparable numbers. T cells overlaid on naïve macrophages secreted insignificant IFNγ. Fig.1B illustrates that the reduced IFNγ is likely due to reduced expansion of CD4+IFNγ cells of C5-/- mice during coculture. Data on of three similar experiments are summarized in Fig.1C. CD8+IFNγ+ T cells showed less significant differences between C5-/- and C5+/+ mice.
Figure 1. Complement C5 sufficient B10.D2-H2d H2-T18cHc1/C5+/+J (C5+/+) and deficient B10.D2-H2d H2-T18cHc0/C5-/-J (C5-/-) mice derived T cells show a differential Interferon-γ response to activation from macrophages.


A: M.tuberculosis immunized spleens of C5+/+ and C5-/- mice were dissected and depleted of non-T-cells. Equal numbers of CD3+ bead purified T-cells were layered on Mtb infected or naïve syngenic macrophages for 72 h. Supernatants were collected and analyzed for IFNγ levels using sandwich ELISA. T-cells from C5+/+ mice produce significantly more IFNγ than their C5-/- counterparts (p values; t test, 3 experiments). B: T cells from the above experiment obtained on day 3, were stained for intracellular IFNγ using antibody conjugates in combination with CD4 or CD8 surface phenotype and analyzed using BD Facscan and Cellquest software. Histograms illustrate that C5-/- T cells show a reduced expansion into CD4+IFNγ+ T cells. C: Data of T cells stained for cytokines from one of three similar experiments are summarized.
C5+/+ macrophages are able to compensate for the defect in IFNγ secretion of C5-/- T cells
To determine if the reduced levels of IFNγ in C5-/- T cells could be restored, C5+/+ and C5-/-macrophages were infected separately with Mtb and allowed to cross-present antigens against both C5+/+ and C5-/- T-cells. Interestingly, overlay of the C5-/- T cells on the C5+/+ macrophages partially rescued the IFNγ response (Fig.2A). Since IL-12 primes IFNγ response in T cells, the levels of IL-12p70 in the supernatants were measured. ELISA confirmed that C5-/- macrophage-T cell cocultures contained reduced IL-12p70 compared those of C5+/+ cocultures (Fig.2B).
Figure 2. C5+/+ macrophages are able to compensate for the defect in C5-/- macrophages.

A: C5+/+ and C5-/- macrophages were infected separately with Mtb and allowed to cross-activate C5-/- and C5+/+ T-cells from Mtb immunized mice. IFNγ response measured over time shows that C5+/+ macrophages are able to induce IFNγ in C5-/- T-cells that were reduced for this cytokine, when activated with C5-/- macrophages. Conversely, C5+/+ T-cells primed with C5-/-macrophages show far reduced response when compared to their response primed with C5+/+ macrophages (p values, t test; 2 experiments). B: Supernatants of macrophage-T cell cocultures contained reduced levels of IL-12p70 (pg/mL ± SEM from duplicate experiments) when C5-/-APCs, which secrete less IL-12p70. Culture supernatants of experiment shown inn panel A were titrated for IL-12p70 using a sandwich ELISA kit from R and D systems (p values; t test).
C5-/- T cells secrete less IFNγ upon direct activation with IL-12 and IL-18
Since IL-12 was reduced in macrophage-T cell cocultures as shown above, the goal here was to determine if C5-/- T cells could be directly activated with IL-12 to produce sufficient IFNγ. Initial studies with IL-12 alone were not that effective in inducing copious IFNγ in T cells (not shown). Previous studies have shown that a combination of IL-12 plus IL-18 is most optimal for inducing IFNγ via STAT4 activation [19]. Equal numbers of purified CD3+ T cells were therefore activated in vitro with IL-12 or IL-18 or their combination and T cell supernatants were tested for IFNγ. Fig.3A shows that, even after activation with IL-12 and IL-18, IFNγ levels were significantly reduced in C5-/- T cells. This suggested that the defect in IFNγ synthesis of C5-/- T cells is likely not because of a decreased signaling from macrophage mediated IL-12 signaling but, an intrinsic defect also exists in T cells.
Figure 3. C5-/- T cells have an intrinsic defect and secrete less IFNγ upon in vitro activation with IL-12 and IL-18.


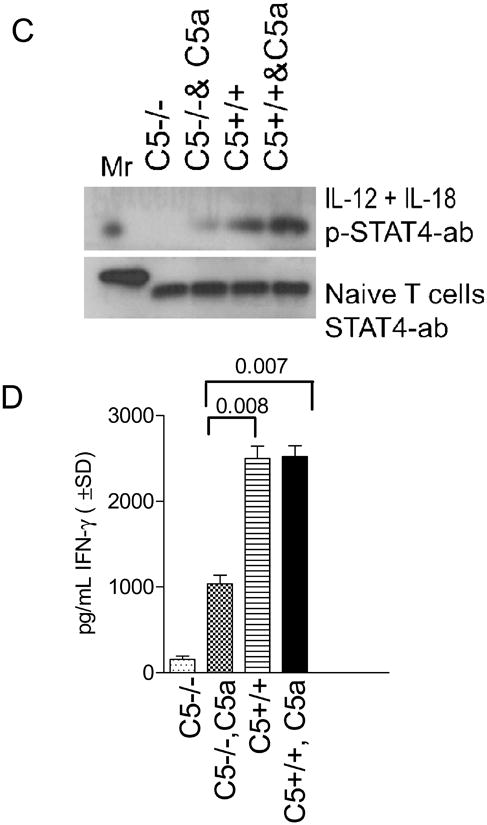
A: CD3 bead purified C5+/+ and C5-/- T cells from naïve mice were activated in vitro with 5 ng/mL of IL-12 and 50 ng/mL of IL-18 and T cell supernatants tested for IFNγ using sandwich ELISA. IFNγ levels are shown at two time points. C5-/- T cells secrete reduced levels of IFNγ compared to C5+/+ T cells (* p< 0.002 vs. all other groups at the same time point; t test). B: Reduced IFNγ response is not due to a reduced expression of IL-12R1β. T cells from the experiment above were stained for IL-12R1β using antibodies conjugated to FITC and analyzed in a BD Facscan using Cellquest software. C5+/+ and C5-/- T cells show a comparable expression of IL-12R1β when activated with cytokines IL-12 and IL-18. Both CD4 and CD8 T cells showed a similar pattern (not shown). C: CD3 bead purified T cells from C5+/+ and C5-/- mice were tested naïve with anti-STAT4 antibody (bottom lane) or stimulated for 15 min with IL-12 and IL-18 in the presence or absence of C5a peptide and assayed for phosphorylated STAT4 (p-STAT4) using western blot. C5-/- T cells show a lack of phosphorylation at 15 min, which is partially restored after the addition of C5a peptide (100 ng/mL; pSTAT-4 migrates at a higher mass). D: Replicate aliquots of T cells activated as in panel C, were incubated for an additional 24 h and supernatants tested for IFNγ using sandwich ELISA. Addition of C5a peptide to IL-12 and IL-18 enhances IFNγ levels in C5-/- T cells but the levels are still lower than that in C5+/+ T cells (p values, t test).
Since IL-12 activates IFNγ synthesis binding to the IL-12R1β and phosphorylating STAT-4, T cells were surface stained for IL-12R1β. Fig.3B indicates that C5-/- and C5+/+ T cells had a comparable distribution of IL-12R1β. Next, T cells were activated with a combination of IL-12 and IL-18 in the presence or absence of 100 ng/mL of C5a peptide. The cell lysates were then examined for native STAT-4 protein and the phosphorylated protein in western blot studies. Fig.3C (bottom lane) shows the constitutive expression of STAT-4 where C5-/- and C5+/+ T cells had comparable levels of STAT4. Addition of cytokines to T cells induced rapid phosphorylation of STAT4 (pSTAT4) in C5+/+ T cells (top lane) and the band was denser when external C5a was added. However, C5-/- T cells did not show significant pSTAT4 bands with IL-12 /IL-18 and a less dense band was evident after addition of C5a peptide. Since T cells to not produce C5a peptide, we conclude from this experiment that, cytokines induce defective phosphorylation of STAT4 in C5-/- T cells, which can be partially restored with C5a. Murine T cells are thought to express C5a-R and we suggest that the externally added C5a can have an activation effect on C5-/- T cells.
Finally, replicate aliquots of treated T cells were also incubated for an additional 24 hr and supernatants tested for IFNγ using ELISA. Addition of C5a to IL-12 and IL-18 enhanced IFNγ levels but these levels were still lower than those produced by C5+/+ T cells (Fig. 3D). Addition of increasing amounts of C5a peptide did not result in a better phosphorylation or better yield of IFNγ more than the levels illustrated in Fig.3C or 3D (not shown). These studies suggest that the defect in IFNγ synthesis by C5-/- T cells is likely related to a defective intracellular signaling by IL-12 and IL-18 that can be partially corrected by addition of C5a peptide.
Defect in IFNγ synthesis of C5-/-T cells is associated with a decreased expansion of T-box expressed in T cell (T-bet) transcription factor
In mouse T cells, T-bet regulates IFNγ synthesis through several pathways [20]. To determine if the defect in IFNγ synthesis was related to T-bet expression and C5 deficiency, the following experiments were carried out.
In vitro recall of T-bet response using T cells from BCG immunized mice
CD3+ splenic T cells were obtained from BCG infected mice and overlaid on BCG infected macrophages in vitro. 4 hours later, T cells were stained for intracellular expression of T-bet in CD4+ T cells using fluorescent antibodies. In this in vitro model of macrophage activation, only immune T cells overlaid on BCG infected macrophages up-regulate T-bet but not those overlaid on naïve macrophages (Fig.4A-E). Data show that C5+/+ T cells markedly up regulate T-bet expression upon activation from BCG infected macrophages, while C5-/- T cells show reduced T-bet. Fig.4F summarizes data from three experiments (p value, t test).
Figure 4. Mycobacterium bovis BCG infection in C5-/-mice is associated with a decreased expression of T-box expressed in T cell (T-bet) transcription factor.
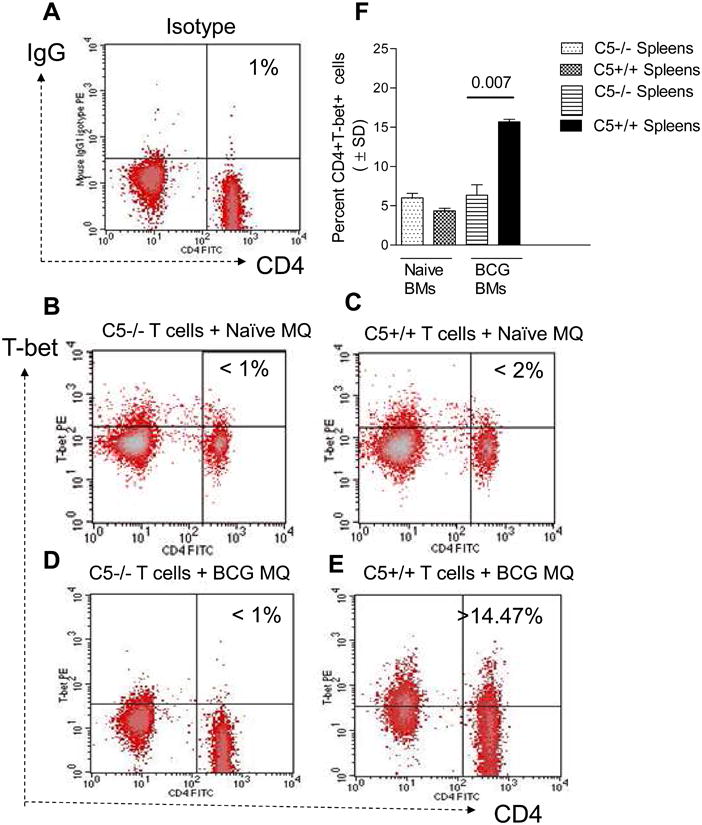
A-E: C5+/+ and C5-/-mice were immunized with 106 CFU of BCG i.p. and spleens harvested 14 days later. CD3 purified T cells were overlaid on naïve macrophages or those infected with BCG and 4 hours later, T cells were stained for intracellular expression of T-bet in CD4 T cells using fluorescent antibodies and flow cytometry. A: T cells do not up regulate T-bet when overlaid on naïve BMs (panels B and C) and C5+/+ but not C5-/- T cells show an up-regulation when cocultured with BCG infected BMs (panels D and E). F: Data generated from triplicate experiments (T cells from 3 mice each) show that C5-/- T cells from BCG immunized mice show a markedly reduced expansion of T-bet in CD4 T cells compared to C5+/+ T cells from BCG immunized (p values, t test)
In vivo expression of T-bet and IFNγ response in M.tuberculosis infected mice
To investigate whether the decreased expression of T-bet and IFNγ occurred in vivo, C5 congenic mice were infected via aerosol with Mtb-Erdman and lungs were analyzed for T cell expression of T-bet and IFNγ using flow cytometry on day 21 post-infection. This time point was chosen to avoid apoptosis of T cells that occurs around 4 weeks of infection. Organs were also harvested at the same time and processed for colony counts of Mtb. Fig.5A shows that C5-/- mice show a better growth of Mtb in both spleens and lungs, consistent with our previously published observations [8,9]. Fig.5B shows data indicating absolute numbers of CD4+T-bet+ IFNγ+ T cells calculated from flow cytometric analysis of cells stained from lungs and spleens. On day 21 post infection, C5-/- mouse lungs as well as spleens contained fewer CD4+T-bet+ IFNγ+ T cells than C5+/+ T cells. Thus, reduced T-bet expression of T cells in C5-/- mice correlated with an increase in the bacterial counts of Mtb in the organs of these mice. Data for T-bet among CD8 T cells were generally comparable between the C5-/- and C5+/+ mice (not shown).
Figure 5. Decreased expansion of IFNγ positive T cells in C5-/- mice during experimental tuberculosis is associated with decreased T-bet and increased susceptibility to tuberculosis.
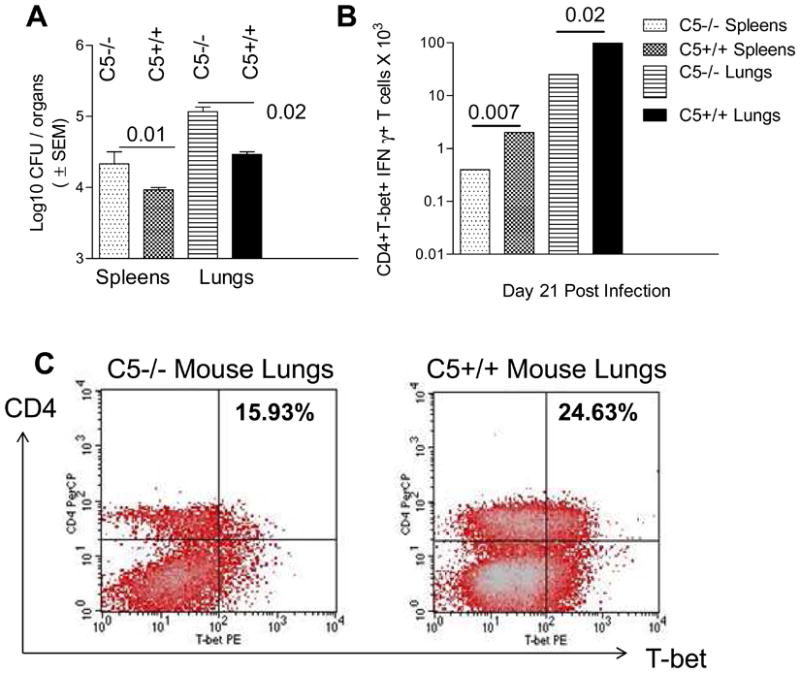
Mice were infected via aerosol with 100 CFU of Mtb-Erdman and on day 21 post-infection, lungs and spleens harvested for colony counts of Mtb on 7H11 agar. A: Spleens and lungs of C5-/- mice contain larger number of organisms compared to C5+/+ mice (n=3 mice per group, p values, ANOVA; 4 mice per group on day 21). B: Splenic and lung derived T cells from mice collected on day 21 post infection were stained for intracellular expression of T-bet and IFNγ using flow cytometry and three colored antibodies. C5-/- T cells show markedly reduced IFNγ and T-bet levels in CD4 T cell population compared to CD4 T cells from C5+/+ mice (p values, t test). No significant differences in T-bet IFNγ were found among CD8 T cells (not shown). C: Histograms illustrate the staining pattern for T-bet for T cells from lungs of Mtb infected C5-/- and C5+/+ mice on day 21 post infection.
3: Discussion
In our earlier studies, C5 deficient macrophages (C5-/-, A/J) were found to secrete lower levels of pro-inflammatory cytokines such as IL-12, IL-1β, IL-6, and TNF-α [8,9]. They were also deficient in chemokine secretion, which correlated with the reduced ability of these mice to form granulomas in the lungs after Mtb infection. Related studies indicated that, C5 deficient macrophages are also defective in killing intracellular Mtb due to reduced reactive oxygen species (ROS)-dependent mechanisms [21]. We anticipated defects in T cell function of C5 deficient mice since, IL-12 induces IFNγ, IL-18 promotes helper T cell activation and IL-2 secretion [22]. In addition, TNF-α can regulate T cells through apoptosis [23]. However, preliminary studies on Mtb infected C5 congenic mice indicated comparable levels of IFNγ mRNA in the lungs [9]. RT-PCR of mRNA messages in whole lungs measures global IFNγ messages and may not identify defects in specific cell populations. In this investigation, we used analyzed purified CD4 and CD8 T cells in vitro and in vivo for their ability to synthesize IFNγ.
Purified C5-/- mouse T cells produced less IFNγ in response to Mtb infected macrophages in vitro suggesting an IL-12 dependent defect. However, even priming with copious amounts of IL-12 and IL-18 showed a persisting defect in IFNγ synthesis. IL-12 and IL-18 activate IFNγ through phosphorylation of STAT4 and this step was found defective in C5-/- T cells [19]. Addition of C5a partially restored pSTAT4 activity and IFNγ synthesis, implicating C5a as a potential regulator of IFNγ synthesis of T cells. We therefore report a novel observation in this study. We noted that C5+/+ macrophages were able to reconstitute the IFNγ response of C5-/- T cells. While macrophages can release C5a to activate T cells, this remains a complex system where T cell activation may occur due to multiple mechanisms, obscuring intrinsic defects in IFNγ secretion by CD3+ T cells of C5-/- mice. One possibility is that C5-/- T cells may not have an initial C5aR expression, but may develop the capacity to express sufficient C5aR only over time. Long term or in vivo studies are perhaps required to answer this question. Finally, T-bet, a major transcription factor that regulates Th1 immunity through IFNγ synthesis was also found reduced in mice with C5 deficiency. Reduced T-bet and IFNγ levels of lungs during active tuberculosis correlated with enhanced growth of Mtb (Fig.5).
These observations suggest an important role of C5 during immune responses to tuberculosis in mice, although it is unclear at the moment, where C5a mechanistically fits into the regulation of IFNγ synthesis. Although mycobacteria infected macrophages secrete IL-12 and IL-18 to prime for IFNγ+ secretion, IFNγ synthesis in T cells involves several mechanisms that seem to cross-regulate each other. For example, IL-12 and IL-18 trigger IFNγ synthesis through STAT4 phosphorylation [19]. The resulting IFNγ promotes additional T-bet expression via activation of STAT-1 [20]. T-bet in turn, induces the expression of IL-12Rβ2 subunit required for T cells to respond to IL-12 [20]. IL-12 itself has been found to induce T-bet synthesis in murine T cells [24]. Thus, a complex feed-back regulatory mechanism appears to be in place between mycobacteria infected macrophages and Th1 type of T cells.
C5a peptide has been previously shown to affect the synthesis of many macrophage derived cytokines like IL-12, IL-6, IL-1β and TNF-α as well as several chemokines [8,9]. Many recent studies show that C5a plays an emerging role in the regulation and maturation of dendritic cells and activation as well as recruitment of T cells in mice [25] [26]. This study demonstrates that C5a can affect the IFNγ secretion in T cells. Thus, C5a appears to affect both the innate arm of immunity involving antigen presenting cells like macrophages as well as the acquired arm of immunity mediated by T cells.
4. Materials and Methods
Mice and Bacteria
Four to eight week old B10.D2-H2d H2-T18cHc1/C5+/+J (C5+/+) and B10.D2-H2d H2-T18cHc0/C5-/-J (C5-/-) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed under specific pathogen free conditions in accordance with IACUC approved procedures. Stock strains of Mtb (H37Rv, ATCC 27294) and BCG (Pasteur, ATCC 35734) were cultured in 7H9 broth to early log phase and organisms were washed and suspended in sterile saline before sonication at 4 watts for 30 sec to disrupt clumps. They were matched to McFarland#1 and used for infections. Aliquots were plated for determination of CFU counts.
Cytokines and reagents
IL-12 and IL-18 were used at concentrations of 5 ng/ml and 50 ng/ml, respectively (Cell Sciences Inc. MA, USA). Human recombinant C5a peptide was used for in vitro complementation assays (Sigma Chemical Co. USA). It was tested by limulus lysate technique and found to contain endotoxin < 10 pg/mL. It was used a dose of 100 ng/mL.
Mouse infections
a) BCG or Mtb infections: For in vitro antigen presentation assays, mice were immunized with 106 CFU of mycobacteria via i.p. injection once. At weekly intervals, CD3 purified T cells from spleens were overlaid on bone-marrow derived macrophages (BMs) infected with appropriate mycobacteria for intracellular expression of T-bet or IFNγ using flow cytometry. b) Mtb infections: Mice were infected via aerosol with 100 CFU of Mtb as published [8]. Mice were sacrificed for CFU counts of organisms in spleens and lungs. T cells were purified from lungs and spleens for cytometric analysis.
Bone marrow-derived macrophages
Macrophages were obtained culturing the marrow derived cells after lysis in ACK Buffer for 5 minutes in Iscoves' modification of DMEM (IDMEM) supplemented with 10% heat inactivated FBS, penicillin and gentamycin, and 10 ng/mL GM-CSF. After 7-10 days, non-adherent cells were removed and macrophages incubated in GM-CSF free medium for 1 day before use in infections and overlay experiments. Macrophages were F4/80+/MOMA1+/CD11c- as evaluated by flow cytometry.
Isolation of T cells from spleens and lungs
T cells from naive or mycobacteria immunized or infected mice were purified from mouse organs using CD3 beads from Miltenyi Inc, CA, USA. Spleens were teased in ACK lysis buffer and washed before T cell purification. PBS lavaged lungs from mice were minced and incubated with calcium and magnesium free HBSS, pH 7.2 containing EDTA (1 mM), dextrose, 100 U per ml collagenase (Type VII) and 50 μg/mL elastase (Sigma Chemical Co, USA) at 37°C and 5% CO2 for 45 minutes on a shaker. The suspension was then passed through a cell strainer with 45 μM pore size and single cells lysed in ACK buffer and washed. Single cell suspensions were then used to purify T cells using CD3 beads.
In vitro activation assays
Macrophages were infected with BCG at a MOI of 1:1 and incubated for 4 hours at 37°C and 5% C02. Macrophage monolayers were then washed 3 times with PBS to remove non-phagocytosed bacteria. T cells were then overlaid for activation using 5 ×106 T cells per 1×106 macrophages of the monolayer. T cells were stained for IFNγ and T-bet at time intervals as below. Supernatants were measured for IFNγ and IL-12p70 using an ELISA kit (R&D Systems).
Flow Cytometry
T cells were suspended in Staining Buffer (PBS containing 0.1% BSA) and stained at 4°C using primary antibodies, isotype controls conjugated to fluorochrome, or primary unconjugated antibodies followed by a fluorochrome conjugated secondary antibody. The intracellular staining was performed as follows. T cells harvested from co-cultures were treated for 4 hours with Golgi Stop to block protein secretion from cells (BD Biosciences). Fc Receptor Block (CD16/32, Caltag Laboratories) was added to cells and incubated on ice for 15 minutes. The appropriate surface antibodies were added to each sample (CD4 FITC, clone RM4-5, Caltag Laboratories; CD8α PerCP, 53-6.7, BD Biosciences) and incubated for 30 minutes in the dark. The cells were washed and fixed with 2% paraformaldehyde for 30 minutes, followed by addition of antibodies for intracellular targets (IFNγ FITC, clone XMG1.2, Caltag Laboratories; T-bet unconjugated, clone 4B10, Santa Cruz Biotechnology) diluted according to manufacturers' recommendations in a perm buffer containing 0.1% Triton-X 100, 0.1% Saponin, 0.01% Digitonin in PBS for 18 hours at 4oC. The samples were washed in Staining Buffer and resuspended in 2% paraformaldehyde or were incubated for 30 minutes with the PE conjugated secondary antibody (anti-mouse IgG1 PE, clone A85-1, BD Biosciences) cells were analyzed using a BD FacSort and Cellquest software (BD Biosciences).
Western blot
CD3 purified T cells were incubated in the presence of a mix of IL-12 and IL-18 with or without 100 ng/mL C5a peptide and after 15 min, pelleted and suspended in SDS lysis buffer. The proteins were estimated and lysates separated in 10% SDS gels and transferred to PVDF membranes. Lysates from naive T cells were first blotted to localize non phosphorylated STAT4 with an antibody that labeled a single band around 87 kDa. A cytokine activated T cell lysate blot was then labeled with anti STAT4, stripped and labeled with a phospho-STAT4 antibody (both antibodies were from Santa Cruz Biotechnology, CA, USA). Blots were developed using a chemiluminescence kit (ECL, Amersham Inc.). Experiments were repeated three times. Phospho-blots required 5 times more protein load than the unactivated T cell lysates. pSTAT4 also migrated at a higher mass.
Acknowledgments
This investigation was supported by NIH NHLBI 68520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cooper AM, Flynn JL. The protective immune response to Mycobacterium tuberculosis. Curr Opin Immunol. 1995;7:512–516. doi: 10.1016/0952-7915(95)80096-4. [DOI] [PubMed] [Google Scholar]
- 2.Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 3.Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams LB, Fukutomi Y, Krahenbuhl JL. Regulation of murine macrophage effector functions by lipoarabinomannan from mycobacterial strains with different degrees of virulence. Infect Immun. 1993;61:4173–4181. doi: 10.1128/iai.61.10.4173-4181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawakami K, Kinjo Y, Uezu K, Miyagi K, Kinjo T, Yara S, Koguchi Y, Miyazato A, Shibuya K, Iwakura Y, Takeda K, Akira S, Saito A. Interferon-gamma production and host protective response against Mycobacterium tuberculosis in mice lacking both IL-12p40 and IL-18. Microbes Infect. 2004;6:339–349. doi: 10.1016/j.micinf.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Shiloh MU, Nathan CF. Reactive nitrogen intermediates and the pathogenesis of Salmonella and mycobacteria. Curr Opin Microbiol. 2000;3:35–42. doi: 10.1016/s1369-5274(99)00048-x. [DOI] [PubMed] [Google Scholar]
- 7.Flynn JL. Immunology of tuberculosis and implications in vaccine development. Tuberculosis (Edinb) 2004;84:93–101. doi: 10.1016/j.tube.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Jagannath C, Hoffmann H, Sepulveda E, Actor JK, Wetsel RA, Hunter RL. Hypersusceptibility of A/J mice to tuberculosis is in part due to a deficiency of the fifth complement component (C5) Scand J Immunol. 2000;52:369–379. doi: 10.1046/j.1365-3083.2000.00770.x. [DOI] [PubMed] [Google Scholar]
- 9.Actor JK, Breij E, Wetsel RA, Hoffmann H, Hunter RL, Jr, Jagannath C. A role for complement C5 in organism containment and granulomatous response during murine tuberculosis. Scand J Immunol. 2001;53:464–474. doi: 10.1046/j.1365-3083.2001.00902.x. [DOI] [PubMed] [Google Scholar]
- 10.Medina E, North RJ. Genetically susceptible mice remain proportionally more susceptible to tuberculosis after vaccination. Immunology. 1999;96:16–21. doi: 10.1046/j.1365-2567.1999.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner OC, Keefe RG, Sugawara I, Yamada H, Orme IM. SWR mice are highly susceptible to pulmonary infection with Mycobacterium tuberculosis. Infect Immun. 2003;71:5266–5272. doi: 10.1128/IAI.71.9.5266-5272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molendijk WJ, van Oudenaren A, van Dijk H, Daha MR, Benner R. Complement split product C5a mediates the lipopolysaccharide-induced mobilization of CFU-s and haemopoietic progenitor cells, but not the mobilization induced by proteolytic enzymes. Cell Tissue Kinet. 1986;19:407–417. doi: 10.1111/j.1365-2184.1986.tb00738.x. [DOI] [PubMed] [Google Scholar]
- 13.Carter LL, Murphy KM. Lineage-specific requirement for signal transducer and activat of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J Exp Med. 1999;189:1355–1360. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karp CL, Grupe A, Schadt E, Ewart SL, Keane-Moore M, Cuomo PJ, Kohl J, Wahl L, Kuperman D, Germer S, Aud D, Peltz G, Wills-Karp M. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol. 2000;1:221–226. doi: 10.1038/79759. [DOI] [PubMed] [Google Scholar]
- 15.Bogdan C, Schleicher U. Production of interferon-gamma by myeloid cells - fact or fancy? Trends Immunol. 2006;27:282–290. doi: 10.1016/j.it.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Flynn JL, Chan J. Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr Opin Immunol. 2003;15:450–455. doi: 10.1016/s0952-7915(03)00075-x. [DOI] [PubMed] [Google Scholar]
- 17.Flynn JL, Chan J. What's good for the host is good for the bug. Trends Microbiol. 2005;13:98–102. doi: 10.1016/j.tim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Chackerian A, Alt J, Perera V, Behar SM. Activation of NKT cells protects mice from tuberculosis. Infect Immun. 2002;70:6302–6309. doi: 10.1128/IAI.70.11.6302-6309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto N, Nakahira M, Ahn HJ, Micallef M, Hamaoka T, Kurimoto M, Fujiwara H. Differential requirements for JAK2 and TYK2 in T cell proliferation and IFN-gamma production induced by IL-12 alone or together with IL-18. Eur J Immunol. 2003;33:243–251. doi: 10.1002/immu.200390027. [DOI] [PubMed] [Google Scholar]
- 20.O'Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 21.Daniel S, Singh CR, Lindsey DR, Dhandayuthapani S, Hunter RL, Jagannath C. The decreased bactericidal function of Complement C5 deficient macrophages is in part due to a defective assembly of phagocyte oxidase and delivery of reactive oxygen species to mycobacterial phagosomes (in press) Journal of Immunology. 2006 doi: 10.4049/jimmunol.177.7.4688. [DOI] [PubMed] [Google Scholar]
- 22.Chang JF, Thomas CA, 3rd, Kung JT. Mitogen-induced IL-2 production and proliferation at defined stages of T helper cell development. J Immunol. 1991;147:860–866. [PubMed] [Google Scholar]
- 23.Roberts AI, Devadas S, Zhang X, Zhang L, Keegan A, Greeneltch K, Solomon J, Wei L, Das J, Sun E, Liu C, Yuan Z, Zhou JN, Shi Y. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol Res. 2003;28:285–293. doi: 10.1385/IR:28:3:285. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Galan MC, Bream JH, Farr A, Young HA. Synergistic effect of IL-2, IL-12, and IL-18 on thymocyte apoptosis and Th1/Th2 cytokine expression. J Immunol. 2005;174:2796–2804. doi: 10.4049/jimmunol.174.5.2796. [DOI] [PubMed] [Google Scholar]
- 25.Soruri A, Riggert J, Schlott T, Kiafard Z, Dettmer C, Zwirner J. Anaphylatoxin C5a induces monocyte recruitment and differentiation into dendritic cells by TNF-alpha and prostaglandin E2-dependent mechanisms. J Immunol. 2003;171:2631–2636. doi: 10.4049/jimmunol.171.5.2631. [DOI] [PubMed] [Google Scholar]
- 26.Tsuji RF, Kawikova I, Ramabhadran R, Akahira-Azuma M, Taub D, Hugli TE, Gerard C, Askenase PW. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J Immunol. 2000;165:1588–1598. doi: 10.4049/jimmunol.165.3.1588. [DOI] [PubMed] [Google Scholar]