

Abstract
The human intestine harbors a large number of bacteria that are constantly interacting with the intestinal immune system, eliciting non-pathological basal level immune responses. Increasing evidence points to dysbiosis of microbiota in the intestine as an underlying factor in inflammatory bowel disease susceptibility. Loss of function mutations in NOD2 are among the stronger genetic factors linked to ileal Crohn’s disease. Indeed, Nod2 is a key regulator of microbiota in the intestine, as microflora in the terminal ileum is dysregulated in Nod2-deficient mice. Nod2 is highly expressed in Paneth cells, which are responsible for the regulation of ileal microflora by anti-microbial compounds, and Nod2-deficient ileal intestinal epithelia are unable to kill bacteria efficiently. It is therefore likely that NOD2 mutations in Crohn’s disease may increase disease susceptibility by altering interactions between ileal microbiota and mucosal immunity.
Keywords: NLR proteins, Nod2, commensal microflora, Crohn’s disease, Paneth cells
Introduction
The mammalian gastrointestinal tract is colonized by a diverse microbial population. Having a symbiotic relationship with the host, the microbiota play a pivotal role in both physiological and pathophysiological conditions. In the human gut, a constant homeostasis is maintained by the stringent regulation of microbial load and the immune response generated against it. In a healthy state, these colonized bacteria play an important role in host metabolism. When the epithelial barrier is disrupted, however, they induce an uncontrolled inflammatory condition. Breakdown of this balance by dysbiosis or dysregulation of immune responses may increase susceptibility to chronic inflammatory conditions like Crohn’s disease (CD) [1–3].
Invasion of microbes into the host tissue is detected by a family of highly conserved receptors called pattern recognition receptors (PRRs) [4]. PRRs sense signature patterns associated with microbes, frequently termed PAMPs (pathogen associated molecular patterns) [4], and prepare the host to combat the invasion. Two major groups of PRRs are cell surface and endosomal Toll-like receptors (TLRs) and cytoplasmic NLR (Nucleotide binding domain, Leucine-rich repeats) proteins. Among the NLRs, Nod2 was found to be strongly associated with ileal CD in humans [5, 6]. Although the mechanism by which Nod2 contributes to CD pathogenesis is not fully known, loss-of-function Nod2 mutations have been suggested to alter host-microbial interactions by various mechanisms [7, 8].
Nod2, a member of the NLR family
NLR proteins in the cytoplasm, consisting of 22 members in humans and about 34 in mice, are prevalent on a wide variety of cells, including immune and epithelial cells [9]. NLR proteins play an important role in the recognition of and host defense against pathogens or extracellular danger signals [10]. These proteins are characterized by a tripartite domain structure [11]. They have a centrally located NACHT domain (present in neuronal apoptosis inhibitor protein (NAIP), the major histocompatibility complex (MHC) class II transactivator (CIITA), HET-E and TP1), involved in oligomerization and activation [12, 13]. At the C-terminus they contain leucine-rich repeats (LRRs) acting as sensors of PAMPs, and at the N-terminus they have a variable effector domain. The latter can consist of either caspase recruitment domain (CARD), pyrin domain (also known as PAAD, PYD or DAPIN) [14–20], acidic domain or baculovirus inhibitor repeat (BIR) domains [21, 22].
Nod1 and Nod2 were the first NLRs reported to function as intracellular microbial recognition molecules. Both belong to the CARD subfamily of NLRs (NLRC or NLR family CARD domain containing), with Nod2 having two CARDs at the N terminus [23–25]. Unlike Nod1, the expression of Nod2 is restricted mainly to dendritic cells [26], macrophages [25], Paneth cells [27] and intestinal, lung and oral epithelial cells [28–30]. Nod2 is also expressed at low levels in T cells [31]. Nod2 confers resistance to a wide variety of bacteria as it recognizes MDP (muramyl dipeptide), which consists of N-acetyl muramic acid-L-Ala-D-Glu, a moiety shared by both Gram-positive and Gram-negative bacteria cell wall [32, 33]. Interestingly, despite the presence of peptidoglycans, not all bacterial species can elicit a strong Nod2 response [34], suggesting a specificity of Nod2 towards bacterial stimuli. Despite the fact that the exact mechanism behind Nod2 activation is still unknown, a current model hints at a similarity to that of APAF-1, a CARD and NBD containing protein required for programmed cell death through a mitochondria-dependent pathway (Figure 1)[35]. Under steady-state, Nod2 is restrained in an inactive conformation through auto-inhibition of the NACHT domain by the LRRs. Experimentally, it has been shown that the C-terminal LRR fragment of Nod2 interacts with the N-terminal CARD-NACHT fragment when they are co-expressed in cells. This interaction is disrupted in the presence of MDP [36, 37]. After ligand recognition, Nod2 undergoes a conformational change and oligomerization of the NACHT domain. This leads to the exposure of the CARD domain and recruitment of the serine-threonine kinase RIP2 (also known as RICK/CARDIAK/RIP2K) through CARD/CARD interactions [24, 25]. Upon activation, RIP2 switches on the downstream NF-κB and MAPK signaling cascades. RIP2 activates IKK, either directly or by recruitment of the transformation growth factor β-activated kinase (TAK1), leading to the phosphorylation and subsequent degradation of IκB and resulting in the release and nuclear translocation of NF-κB [38, 39]. These pathways culminate in the induction of pro-inflammatory cytokines and chemokines. The MAPK pathway is also activated by RIP2 and TAK1, although the details of this pathway remain to be elucidated [40–42]. Several proteins such as the nuclear protein GRIM 19; a PDI domain and LRR containing protein Erbin; TGF-β-activated kinase 1 (TAKI); and the GTPase-activating protein Centaurin β1 were proposed to have role in the regulation of Nod2 signaling, but their involvement is not yet understood. Erbin has also been proposed to have a negative regulatory role on Nod2 activation. It has been shown to interact with the CARD domain of Nod2 and inhibit MDP-mediated NF-κB activation [43, 44].
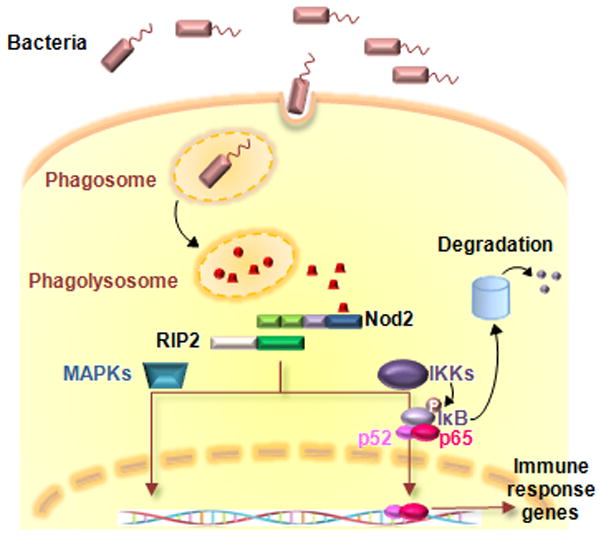
Figure 1.
MDP-induced Nod2 signaling pathway.
Enzymatic breakdown of bacterial cell wall peptidoglycan in the phagolysosome may generate active moieties including muramyl dipeptide (MDP), which is recognized by NOD2 through the leucine-rich repeat (LRR). NOD2 recruits RIP2 kinase through CARD-CARD interactions and RIP2 subsequently activates the IKK complex resulting in phosphorylation, ubiquitination and degradation of IκB, and nuclear translocation of NF-κB transcription factors. NOD2 activation also results in the activation of MAP kinase pathways through RIP2. These signaling cascades result in the induction of various immune response gene such as proinflammatory cytokines and defensins.
Crosstalk between Nod2 and commensal bacterial flora
The mammalian intestinal tract can be presented as an ideal model of mutualism, where homeostasis is maintained by the peaceful coexistence between the host immune system and the intestinal microbial flora. The system has evolved and has been perfected over the ages to allow both host and microbiota to benefit from this balanced relationship [45]. The bacterial flora is assured of a stable habitat, and in exchange it aids host metabolic pathways with an array of enzymes and other bacterial products. Moreover, the commensal microbiota competes with pathogenic invaders and thus wards off the latter from colonizing the host intestinal tract [45, 46]. Intestinal commensals also help in the development and maturation of the host immune system, as evident from the poorly developed systemic and mucosal immune systems in germ-free animals [47–49]. This not only protects the host from pathogenic intruders but also plays an essential role in keeping the commensal load under check. Nod2 has recently gained much attention as a component of the host immune system that plays a crucial role in regulating host-microbial interaction [50].
Compared to the large intestine, the small intestine harbors a significantly lower number of bacteria, and the anti-bacterial function of crypts in the small intestine may be a major contributing factor. In fact, Paneth cells, specialized secretory epithelial cells of the small intestinal crypts, synthesize and secrete several anti-microbial peptides [51, 52] and express Nod2 at high levels [53]. We have shown that Nod2 is an important regulator of crypt antimicrobial function [50]. The Nod2 ligand, MDP, was found to effectively induce the bactericidal activity of crypts. Isolated crypts, when incubated in the presence of active forms of MDP, and not its inactive chiral isomer MDPDD or MDPLL, induced effective killing of both gram-positive and gram-negative bacteria in a bacterial killing assay. Although MyD88, a downstream adaptor of TLR, is important for the Paneth cell function and transcription of MyD88-dependent antimicrobial compounds [54], MDP-induced bactericidal activity is independent of the TLR-MyD88 pathway, as MyD88-deficient crypts efficiently kill bacteria upon MDP stimulation. The role of Nod2 in anti-microbial activity of crypts was further confirmed by the obliteration of bacterial killing in Nod2- and Rip2-deficient mice. Nod2-deficient crypts stimulated with MDP or a nonspecific inducer of secretion, carbamylcholine (CCH), could not induce effective bacterial-killing activity. Deletion of Nod2 affects the killing of both Gram-negative and Gram-positive bacteria, as confirmed by performing the killing assay on 3 different bacterial strains: E. coli, Salmonella typhimurium and Listeria monocytogenes. The effect of Nod2 is not specific to MDP-induced signaling and may rather be a general effect on Paneth cell secretion or composition, since Nod2-deficient crypts were unable to kill bacteria efficiently even upon CCH stimulation, a nonspecific inducer of secretion. Deletion of Rip2, a downstream kinase of Nod2, also showed a reduced ability to kill bacteria, indicating that the Nod2-Rip2 signaling pathway regulates the function of Paneth cells.
Bacteroides, Firmicutes, Proteobacteria and Actinobacteria comprise 99% of the commensal microbial flora [2], and their load is regulated by multitude of factors such as genetic background, diet and interaction between commensal and pathogenic bacteria [55]. Looking at animals sharing a congenic C57BL/6 genetic background, the same parents, the same cages and the same diet, there was a significant rise in the amount of Bacteroides, Firmicutes and Bacillus in the terminal ileum of Nod2-deficient mice compared to wild-type littermate controls. Such differences were, however, less evident in the feces, probably because the expression of Nod2 is localized to Paneth cells, which are mainly localized in the terminal ileum [53, 56], and Nod2-mediated control has little effect on the regulation of bacterial load in the lower colon. Another study did observe differences in the Bacteroides content of feces in addition to the terminal ileum [57], suggesting that perhaps the effects in the terminal ileum may extend to the colon, although those differences are significantly less pronounced in feces than in the ileum [57]. These observations unambiguously delineate the role of Nod2 in the management of bacterial flora in the terminal ileum. Nod2 not only regulates the colonization of commensal bacteria but also suppresses the load of pathogenic bacteria in the terminal ileum. Inoculation with the opportunistic pathogen Helicobacter hepaticus resulted in a much higher bacterial load in both the terminal ileum and feces of Nod2-deficient mice, which showed poor bacterial clearing capacity compared to wild-type controls. However, this regulation is not merely a one-sided affair. While Nod2 plays a vital role in regulating intestinal microbiota, commensal bacteria in the gut also play a part in controlling the expression of Nod2 and Rip2. This is evident from the poor expression profiles of these genes in germ-free mice in contrast to conventional specific pathogen-free (SPF) mice of congenic background. Moreover, complementation of the germ-free mice with a single non-pathogenic bacteria, either Lactobacillus plantarum or E. coli strain Nissle 1917, increased the expression of Nod2 and Rip2. These results highlight the significance of commensal microbiota in regulating Nod2 expression and elucidate how a balanced interaction between bacterial flora and the host immune system contributes to maintenance of intestinal homeostasis.
These findings, taken together, show the importance of the Nod2-Rip2 pathway in maintaining a much-needed homeostasis between the host immune system and the bacterial flora[50]. The commensals positively regulate the expression of Nod2, which in turn negatively controls the gut microbiota, keeping the bacterial load under check. A disruption in this balanced relationship leads to dysbiosis, which is known to underlie the pathogenesis of CD (Figure 2) [2]. Apart from Nod2-Rip2, another pathway that plays a vital role in protecting the gut from invaders is the TLR-MyD88 pathway [54]. Vaishnava et. al. has described that intestinal Paneth cells can directly sense enteric bacteria through MyD88-dependent TLR activation and trigger the expression of anti-bacterial compounds. Although both pathways lead to protection of the gut, their mode of action is different. While the Nod2-Rip2 pathway mainly acts by maintaining the commensal flora at a steady-state, MyD88 plays a more direct role, where Paneth cell-specific MyD88 signaling inhibits bacterial penetration of the host tissue [54]. Several studies have reported dysbiosis of microbial composition in CD patients, and metagenomic study showed a general decrease in the biodiversity of fecal microbiota in these patients [58–60]. Thus, the role of Nod2 in the context of commensal microbiota regulation is extremely important, as the dysregulation of microbial flora seems to be one the most significant contributing factors in the development of CD.
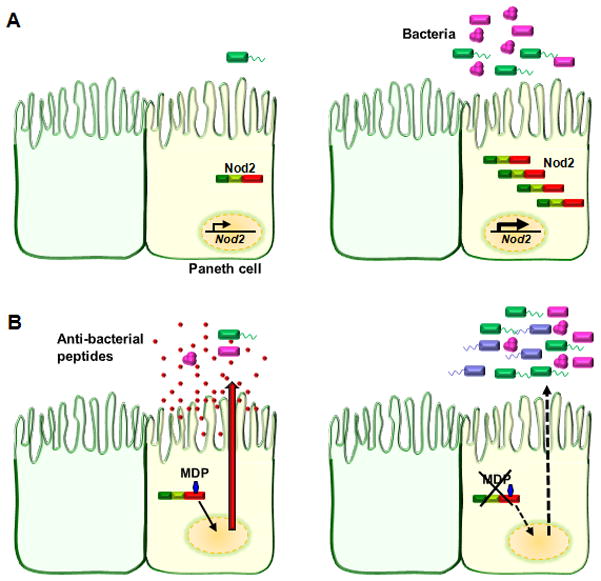
Figure 2.
Inter-regulation of Nod2 and commensal bacteria in the gut.
A) Expression of Nod2 in ileal Paneth cells with reduced bacterial flora or in the absence of commensal bacteria (as in germ free mice) is low (left panel), but its expression is induced by bacterial colonization of the gut mucosa (right panel).
B) In the presence of functional Nod2, Paneth cells sense the presence of bacteria or bacterial antigen and release anti-microbial peptides, which keep the intestinal flora in check and aid in maintaining homeostasis (left panel). Deletion of Nod2 or the presence of a Nod2 “loss of function” mutation renders Paneth cells nonfunctional. Lack of Paneth cell-derived anti-bacterial compounds leads to unchecked bacterial colonization of the intestinal mucosa and breakdown of homeostasis (right panel).
Crohn’s disease pathogenesis: the role of NOD2
CD, a chronic inflammatory bowel disease (IBD), is a multifactorial disorder of the gastrointestinal tract. It is histologically characterized by massive transmural infiltration of lymphoctes and macrophages with granuloma. Although CD can affect any part of the gastrointestinal tract, inflammation is mainly isolated to the small intestine and colon. Uncontrolled mucosal inflammatory responses against gut microbial flora due to the disruption of the intestinal epithelial barrier and mucosal immunity play important roles in the pathogenesis of CD. Under normal physiological conditions, gut microbial flora colonize a nutrient rich area and, in turn, contribute to host metabolism and the proper development and maturation of the mucosal immune system [61]. Dysregulation or disruption of this physiological homeostasis due to genetic or environmental factors can initiate uncontrolled, chronic inflammation of the gut, leading to CD [62]. The initial detection of microbial components in the gut by PRRs like TLRs and NLRs triggers the host innate immune system.
Through familial clustering, studies of monozygotic twins and linkage analysis, it has been revealed that several genetic loci, termed IBD1–30 [63], have a significant association with CD. Furthermore, Franke et al. reported 71 confirmed CD susceptibility loci through meta-analysis of CD genome-wide association studies [64]. One significant breakthrough in the study of CD is the identification of NOD2 as a CD susceptible gene through mapping of the IBD1 locus. NOD2 was identified as the first gene to be strongly associated with ileal CD susceptibility in North American and European populations [5, 6]. NOD2, encoded by the NOD2/Card15 gene, is located on human chromosome 16q12. In genetic studies of CD patients, three main variants, or polymorphisms, in the NOD2 gene were identified as risk factors for CD: i) a frame shift mutation at position 1007 (1007fs) that results in partial translation of the LRRs and early termination; ii) a glycine to arginine conversion at amino acid residue 908 (G908R); iii) an arginine to tryptophan conversion at amino acid residue 702 (R702W) [5, 6, 65]. All three variants alter the C-terminal portion of NOD2, which is within or close to the LRR domain.
Mechanistic models: Nod2 mutations causing Crohn’s disease
Depending on the animal model used, three hypotheses have been proposed for the mechanism by which NOD2 mutations contribute to CD pathogenesis (Table). The first proposed a “gain-of-function” observation with Nod2 mutations. A mouse model was developed with a mutation at the residue corresponding to the CD L1007fs mutation, leading to the murine frameshift mutation NOD23020insC. Macrophages isolated from Nod22939iC mutant mice and stimulated with MDP resulted in increased NF-κB and IL-1β secretions when compared to those of wild-type mice. Moreover, upon oral treatment with dextran sodium sulfate (DSS), Nod22939iC mutant mice showed increased IL-1β and inflammation in the colon compared to wild-type mice [66]. Interestingly, this data contradicts the “loss-of-function” observed in CD patients with Nod2 mutations, which results in decreased NF-kB activation [32, 33, 67–69]. Also, dendritic cells from CD patients homozygous for the L1007fs NOD2 mutation fail to produce cytokines and up-regulate the co-stimulatory molecules CD80 and CD86 upon MDP stimulation, showing a deficiency in the activation of the adaptive immune system [70]. These indicate that the mechanism behind inflammation in the Nod22939iC mice differs from that involved in human CD. The second animal model for the development of CD proposes that Nod2 acts as a negative regulator of TLR2 signaling. Splenic macrophages from these Nod2 deficient mice produced increased levels of IL-12 upon TLR2 stimulation through activation of the NF-κB subunit c-Rel [71, 72]. Therefore, it was proposed that elevated amounts of IL-12 promote IFN-γ production by T and NK cells and promotes enhanced growth and differentiation towards T helper type 1 (Th1) effector cells, thus promoting pathogenesis of CD [71, 73]. However, a major concern regarding this proposed model for CD development arises from contradicting observations by other groups. Uehara et al. showed a synergistic effect of MDP with synthetic ligands for TLR2 and TLR4 in the production of IL-8 by epithelial cells [30]. Furthermore, Nod2-deficient macrophages had normal responses when stimulated with the TLR2 ligand [74]. Stimulation of blood mononuclear cells from CD patients, harboring either wild-type or heterozygous Nod2 mutations, with the TLR2 ligand resulted in a synergistic production of TNF-α[68]. Recently, another group reported that NOD2 and TLR2 function independently in a murine model of arthritis, where they showed that a deficiency in NOD2 did not alter TLR2-dependent joint inflammation elicited by the synthetic TLR2 agonist, Pam3CSK4 [75]. Thus, negative regulation of TLR2 by NOD2 is not a universal phenomenon and could be dependent on factors such as genetic background,, cell type and type and/or length of infection. In continuation, Watanabe et al. reported that administration of MDP protects mice from the development of experimental colitis by downregulating multiple TLR responses. Their study, and others, showed that pre-stimulation of cells with MDP reduces cytokine responses induced by multiple TLR ligands or whole bacteria [76, 77]. The above observation could be due to cross-tolarization of TLRs by chronic Nod2 stimulation rather than negative regulation as reported by Hedl et al[78]. They found that pretreatment with MDP significantly decreased production of the proinflammatory cytokines TNF-α, IL-8, and L-1β upon Nod2, TLR4, and TLR2 restimulation in primary human monocyte-derived macrophages [78].
Table.
Possible mechanistic hypotheses of Nod2-associated CD pathogenesis
| Hypothesis | Phenotypes | Reference |
|---|---|---|
| “Gain-of-function” Nod2 mutation | MDP stimulation of macrophages from Nod22939iC mutant mice resulted in increased NF-κB and IL-1β secretions when compared to those of wild-type mice. Oral treatment of Nod22939iC mutant mice with dextran sodium sulfate (DSS), showed increased IL-1β and inflammation in the colon compared to wild-type mice. | [66] |
| Nod2, negative regulator of TLR2 signaling | Splenic macrophages from Nod2 deficient mice produced increased levels of IL-12 upon TLR2 stimulation through activation of the NF-κB subunit c-Rel. | [71, 73] |
| Nod2 mutation leads to Impaired Paneth cell function | NOD2 mutations affect the expression of small anti- bacterial peptides from Paneth cells, increasing susceptibility to abnormal infection and inflammation in the gut. | [74, 81, 82] |
| Nod2 mutations lead to the suppression of IL-10 expression | 3020insC Nod2 protein actively inhibits IL-10 transcription by blocking the phosphorylation of ribonucleoprotein hnRNP-A1 via the mitogen activated protein kinase p38. | [87] |
| Nod2 mediated autophagy | Nod2 directly interact with ATG16L1 for its recruitment at the site of bacterial entry, induce autophagy and regulates bacterial clearance. | [89–91] |
The third model proposes that mutations in NOD2 may result in altered mucosal host-microbial interactions [7]. Paneth cells, specialized epithelial cells of the intestinal crypts of Lieberkühn, express high levels of Nod2 [53, 56]. Paneth cells release anti-microbial peptides in the intestinal lumen following stimulation with bacterial products such as the Nod2 ligand MDP [79] and play an important role in the innate regulation of gut microbiota. NOD2 mutations were found to affect the expression of small anti-bacterial peptides, which may increase the susceptibility to abnormal infection and inflammation of the gut. Crohn’s NOD2 mutations are associated with the development of small intestinal (ileal) lesions, which correspond to the location of Paneth cells [80]. The link between Nod2 mutations and Paneth cell-derived defensins was discovered through a number of human and rodent studies [74, 81, 82]. Reduced mRNA expression of Paneth cell-derived α-defensins in Nod2 knockout mice was reported by our group. Moreover these mice were more susceptible to oral infection with Listeria monocytogenes. We also showed that intestinal crypts lacking Nod2 are unable to kill bacteria efficiently, and increased loads of commensal and pathogenic bacteria are detected in the terminal ileum of Nod2-deficient mice [50]. In humans, CD patients with ileal involvement showed reduced levels of the Paneth cell-derived human α-defensins HD-5 and 6 [82–84]. This decrease in the amount of α-defensins occurs irrespective of Nod2 mutations, but levels are much more drastically reduce in CD patients harboring NOD2 mutations [82]. Among CD patients with mutations in NOD2, all had a decrease in the expression of α-defensins, and those with the common frame-shift mutation NOD23020insC had an approximately 3-fold more pronounced decrease than others [82]. Recently Simms et al. reported that the decreased expression of α-defensins in CD patients is a consequence of inflammation and is independent of Nod2 mutations, proposing that loss of antimicrobial function might be secondary to active CD rather than being responsible for the inflammation [83]. In response to the above report, Bevins et al. proposed that the decreased expression of α-defensins in ileal CD, whether or not there was an identified mutation in NOD2, was independent of tissue inflammation [85]. These diverse research endeavors reveal the complex manifestation of CD. The conceptual belief that CD may result from abnormal host-microbe interactions is very appealing, but a good animal model to study that is yet lacking. We have recently reported a mouse model of ileal CD in Nod2-deficient mice that has many similarities with the human CD manifestation [86]. We showed that deficiency of Nod2 in mice leads to the development of granulomatous inflammation in the ileum upon Helicobacter hepaticus inoculation and that proper Nod2 function in non-hematopoietic cells of the gut is important. We were also successful in demonstrating that restoration of Paneth cell bacterial killing activity in Nod2-deficient mice can exhibit protection against ileal inflammation induced by H. hepaticus. Therefore, bacterial killing controlled by Nod2 expression in Paneth cells plays an important role in ileal mucosal homeostasis.
Newly proposed models
Recently, different research groups have proposed different working models for the association of Nod2 with CD. One model proposes that Nod2 mutations lead to the suppression of IL-10 expression [87]. Previously, Netea et. al. reported that peripheral blood mononuclear cells (PBMC) from CD patients with the 3020insC frameshift mutation showed a defect in the production of IL-10 upon stimulation with the TLR2 ligand Pam3CSK4 [88]. Noguchi et al. showed that the 3020insC Nod2 protein actively inhibits IL-10 transcription by blocking the phosphorylation of ribonucleoprotein hnRNP-A1 via the mitogen activated protein kinase p38 [87]. However, the same group also showed that the reduction in IL-10 production by cells carrying the NOD2 mutation is specific to MDP stimulation and that the same cells produced IL-10 normally upon TLR simulation. Together, these results suggest that the reduction of IL-10 production might be simply due to the absence of MDP sensing by the mutation in Nod2 [87]. Three interesting reports have also surfaced linking Nod2 with autophagy [89–91]}. Autophagy is a survival mechanism induced by starvation where cell components are broken down to provide nutrients. The ubiquitin-like autophagy-related proteins (ATG)5, ATG7 and ATG16L1 are components of a large protein complex essential for autophagy and, interestingly, polymorphisms of ATG16L were found to be associated with CD. Cooney et. al. showed that Nod2, in the presence of MDP, induces autophagy in dendritic cells dependent on Rip2, ATG5, ATG7 and ATG16L1. They further showed that dendritic cells from patients harboring CD-associated NOD2 or ATG16L1 mutations are defective in bacterial handling and autophagy induction [89]. Travassos et al. showed that both Nod1 and Nod2, independent of RIP2 and NF-κB, are important for bacterial handling through autophagy, where they directly interact with ATG16L1 for its recruitment at the site of bacterial entry [90]. These studies draw similar inferences but propose contradictory mechanisms, one being RIP2 dependent, the other being RIP2 independent. A third report by Homer et al. showed that anti-bacterial activity was disrupted in epithelial cells harboring the CD-associated ATG16L1 variant upon Nod2 mediated MDP stimulation [91]. Interestingly, this study showed that bacterial handling by monocytic cells in response to MDP is not altered in the CD-associated ATG16L1 variant. These independent studies tried to show a functional link between Nod2 and ATG16L1, which have a close association with CD susceptibility and thereby combine the two strongest risk factors of CD.
Perspectives
The studies using Nod2 deficient mice showed that Nod2 is critical for the function of bactericidal activity of ileal crypts and the regulation of microbiota of the ileum. Therefore, it is tempting to speculate that ileal CD caused by Nod2 mutations is due to the dysfunction of Paneth cells (Figure 1). In this scenario, microbiota in the terminal ileum is tightly regulated by bacterial killing activity at the terminal ileum in healthy individuals. This restricts both pathogenic and non-pathogenic bacteria as well as bacterial antigens that constitutively stimulate the intestinal immune system. In the intestines of Nod2-deficient mice or CD patients with NOD2 mutations, Paneth cells are unable to control microbiota due to impaired bactericidal activity. This may lead to bacterial overload in the terminal ileum. An increase in the load of microbiota alone is likely not sufficient to induce chronic inflammation and may simply enhance “physiological inflammation,” which is tightly regulated by various immune mechanisms. However, in conjunction with other genetic, environmental and immunological factors, dysbiosis of bacteria may significantly increase susceptibility to ileal inflammation by enhancing stimulation of the intestinal immune system beyond the controlled physiological threshold, leading to pathological changes and finally to chronic inflammation in CD. Although the studies of Nod2 deficient mice and CD patients for α-defensin gene expression support this scenario, an actual confirmation would require further research into the association among microbial flora communities, NOD2 genotype and inflammation.
A finding of interest is that Nod2-deficient crypts have greatly reduced bacterial killing activity. In mouse and humans, Nod2 apparently regulates only a limited subgroup of defensins in Paneth cells. Paneth cells also express other compounds such as lysozyme, RegIIIγ, Phospholipase A or cathelicidins, whose expression is Nod2-independent. However, Nod2-deficient crypts are unable to kill several strains of both Gram-positive and negative-bacteria. It would be interesting to know if the function of Nod2 in Paneth cells is limited to the regulation of subsets of defensins or may have a more general role in the Paneth cell function.
Although more studies are needed to fully explain the association of Nod2 function and intestinal microbiota, recent studies have clearly shown the importance of Nod2 in mediating interactions between microbiota and the intestinal immune system. Since dysbiosis of commensal flora and reduced defensins are associated with CD, it is tempting to postulate that the regulation of microbiota using defensins or other anti-bacterial compounds may be useful as therapeutic tools for prophylactic purposes or the treatment of actual symptoms in individuals with a susceptible genetic background. Defensins and other intestinal anti-bacterial compounds are natural “antibiotics,” and it is unlikely that intestinal bacteria flora will acquire further resistance, as defensins have been an effective bacterial killing tool throughout the co-evolution of host and microbiota. Further studies on CD-associated microbiota and the generation of appropriate models will be needed.
Acknowledgments
The authors thank Yuen-Joyce Liu for critical reading. This work was supported by grants from the NIH (K.S.K. R01DK074738) and the Crohn’s and Colitis Foundation of America (K.S.K.). K.S.K. is a recipient of the Investigator Award from the Cancer Research Institute and the Claudia Adams Barr Award.
Abbreviations used
- NLR proteins
nucleotide binding domain (NBD) leucine rich repeat (LRR) proteins
- CD
Crohn’s disease
- MDP
muramyl dipeptide
Footnotes
Conflict of interest
The authors have no conflicting financial interests.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 3.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 5.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 6.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 7.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nature reviews. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 8.Abraham C, Cho JH. Functional consequences of NOD2 (CARD15) mutations. Inflammatory bowel diseases. 2006;12:641–650. doi: 10.1097/01.MIB.0000225332.83861.5f. [DOI] [PubMed] [Google Scholar]
- 9.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 11.Inohara N, Nunez G. Cell death and immunity: NODs: intracellular proteins involved in inflammation and apoptosis. Nature reviews. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 12.Koonin EV, Aravind L. The NACHT family - a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci. 2000;25:223–224. doi: 10.1016/s0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- 13.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 14.Bertin J, DiStefano PS. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 2000;7:1273–1274. doi: 10.1038/sj.cdd.4400774. [DOI] [PubMed] [Google Scholar]
- 15.Inohara N, Nunez G. The NOD: a signaling module that regulates apoptosis and host defense against pathogens. Oncogene. 2001;20:6473–6481. doi: 10.1038/sj.onc.1204787. [DOI] [PubMed] [Google Scholar]
- 16.Martinon F, Hofmann K, Tschopp J. The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr Biol. 2001;11:R118–120. doi: 10.1016/s0960-9822(01)00056-2. [DOI] [PubMed] [Google Scholar]
- 17.Fairbrother WJ, Gordon NC, Humke EW, O’Rourke KM, Starovasnik MA, Yin JP, Dixit VM. The PYRIN domain: a member of the death domain-fold superfamily. Protein Sci. 2001;10:1911–1918. doi: 10.1110/ps.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlowski K, Pio F, Chu Z, Reed JC, Godzik A. PAAD - a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem Sci. 2001;26:85–87. doi: 10.1016/s0968-0004(00)01729-1. [DOI] [PubMed] [Google Scholar]
- 19.Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci. 2001;26:475–481. doi: 10.1016/s0968-0004(01)01905-3. [DOI] [PubMed] [Google Scholar]
- 20.Staub E, Dahl E, Rosenthal A. The DAPIN family: a novel domain links apoptotic and interferon response proteins. Trends Biochem Sci. 2001;26:83–85. doi: 10.1016/s0968-0004(00)01717-5. [DOI] [PubMed] [Google Scholar]
- 21.Birnbaum MJ, Clem RJ, Miller LK. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol. 1994;68:2521–2528. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, Keilty JJ, Gosselin ML, Robison KE, Wong GH, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 24.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Nunez G. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 25.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 26.Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun. 2005;73:7967–7976. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voss E, Wehkamp J, Wehkamp K, Stange EF, Schroder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem. 2006;281:2005–2011. doi: 10.1074/jbc.M511044200. [DOI] [PubMed] [Google Scholar]
- 28.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 29.Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100–3111. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Uehara A, Sugawara Y, Kurata S, Fujimoto Y, Fukase K, Kusumoto S, Satta Y, Sasano T, Sugawara S, Takada H. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol. 2005;7:675–686. doi: 10.1111/j.1462-5822.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, Fernandez-Luna JL. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 32.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 33.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 34.Hasegawa M, Yang K, Hashimoto M, Park JH, Kim YG, Fujimoto Y, Nunez G, Fukase K, Inohara N. Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem. 2006;281:29054–29063. doi: 10.1074/jbc.M602638200. [DOI] [PubMed] [Google Scholar]
- 35.Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol. 2008;83:13–30. doi: 10.1189/jlb.0607402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, Ghosh P, Moran A, Predergast MM, Tromp G, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. Embo J. 2004;23:1587–1597. doi: 10.1038/sj.emboj.7600175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 39.Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 40.Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 41.Windheim M, Lang C, Peggie M, Plater LA, Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem J. 2007;404:179–190. doi: 10.1042/BJ20061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.da Silva Correia J, Miranda Y, Leonard N, Hsu J, Ulevitch RJ. Regulation of Nod1-mediated signaling pathways. Cell Death Differ. 2007;14:830–839. doi: 10.1038/sj.cdd.4402070. [DOI] [PubMed] [Google Scholar]
- 43.Kufer TA, Kremmer E, Banks DJ, Philpott DJ. Role for erbin in bacterial activation of Nod2. Infect Immun. 2006;74:3115–3124. doi: 10.1128/IAI.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P, Borg JP, Nunez G. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem. 2005;280:40301–40309. doi: 10.1074/jbc.M508538200. [DOI] [PubMed] [Google Scholar]
- 45.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nature reviews. 10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 46.Macpherson AJ, Uhr T. Compartmentalization of the mucosal immune responses to commensal intestinal bacteria. Annals of the New York Academy of Sciences. 2004;1029:36–43. doi: 10.1196/annals.1309.005. [DOI] [PubMed] [Google Scholar]
- 47.Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol. 1963;42:471–483. [PMC free article] [PubMed] [Google Scholar]
- 48.Macpherson AJ, Hunziker L, McCoy K, Lamarre A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 2001;3:1021–1035. doi: 10.1016/s1286-4579(01)01460-5. [DOI] [PubMed] [Google Scholar]
- 49.Macpherson AJ, Martinic MM, Harris N. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol Life Sci. 2002;59:2088–2096. doi: 10.1007/s000180200009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS. Nod2 is required for the regulation of commensal microbiota in the intestine. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ouellette AJ, Bevins CL. Paneth cell defensins and innate immunity of the small bowel. Inflammatory bowel diseases. 2001;7:43–50. doi: 10.1097/00054725-200102000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Wehkamp J, Fellermann K, Herrlinger KR, Bevins CL, Stange EF. Mechanisms of disease: defensins in gastrointestinal diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:406–415. doi: 10.1038/ncpgasthep0265. [DOI] [PubMed] [Google Scholar]
- 53.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nunez G, Keshav S. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 54.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Begue B, Dumant C, Bambou JC, Beaulieu JF, Chamaillard M, Hugot JP, Goulet O, Schmitz J, Philpott DJ, Cerf-Bensussan N, et al. Microbial induction of CARD15 expression in intestinal epithelial cells via toll-like receptor 5 triggers an antibacterial response loop. J Cell Physiol. 2006;209:241–252. doi: 10.1002/jcp.20739. [DOI] [PubMed] [Google Scholar]
- 56.Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut. 2003;52:1591–1597. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, Schreiber S, Rosenstiel P. Nod2 is essential for temporal development of intestinal microbial communities. Gut. Mar 18; doi: 10.1136/gut.2010.216259. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Dore J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflammatory bowel diseases. 2006;12:106–111. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- 59.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Umesaki Y, Setoyama H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes Infect. 2000;2:1343–1351. doi: 10.1016/s1286-4579(00)01288-0. [DOI] [PubMed] [Google Scholar]
- 62.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 63.Shih DQ, Targan SR, McGovern D. Recent advances in IBD pathogenesis: genetics and immunobiology. Current gastroenterology reports. 2008;10:568–575. doi: 10.1007/s11894-008-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature genetics. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O’Morain C, Gassull M, Binder V, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 67.Netea MG, Ferwerda G, de Jong DJ, Girardin SE, Kullberg BJ, van der Meer JW. NOD2 3020insC mutation and the pathogenesis of Crohn’s disease: impaired IL-1beta production points to a loss-of-function phenotype. Neth J Med. 2005;63:305–308. [PubMed] [Google Scholar]
- 68.Netea MG, Ferwerda G, de Jong DJ, Werts C, Boneca IG, Jehanno M, Van Der Meer JW, Mengin-Lecreulx D, Sansonetti PJ, Philpott DJ, Dharancy S, Girardin SE. The frameshift mutation in Nod2 results in unresponsiveness not only to Nod2- but also Nod1-activating peptidoglycan agonists. J Biol Chem. 2005;280:35859–35867. doi: 10.1074/jbc.M504924200. [DOI] [PubMed] [Google Scholar]
- 69.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 70.Kramer M, Netea MG, de Jong DJ, Kullberg BJ, Adema GJ. Impaired dendritic cell function in Crohn’s disease patients with NOD2 3020insC mutation. J Leukoc Biol. 2006;79:860–866. doi: 10.1189/jlb.0805484. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 negative regulator of Toll-like receptor 2 is a-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 72.Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 73.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nature reviews. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 74.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 75.Rosenzweig HL, Jann MJ, Vance EE, Planck SR, Rosenbaum JT, Davey MP. Nucleotide-binding oligomerization domain 2 and Toll-like receptor 2 function independently in a murine model of arthritis triggered by intraarticular peptidoglycan. Arthritis Rheum. 62:1051–1059. doi: 10.1002/art.27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. The Journal of clinical investigation. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petnicki-Ocwieja T, DeFrancesco AS, Chung E, Darcy CT, Bronson RT, Kobayashi KS, Hu LT. Nod2 suppresses Borrelia burgdorferi mediated murine Lyme arthritis and carditis through the induction of tolerance. PloS one. 2011;6:e17414. doi: 10.1371/journal.pone.0017414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 80.Gasche C, Grundtner P. Genotypes and phenotypes in Crohn’s disease: do they help in clinical management? Gut. 2005;54:162–167. doi: 10.1136/gut.2003.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut. 2008;57:903–910. doi: 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- 84.Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, Vatn MH, Wehkamp J. Defective Paneth Cell-Mediated Host Defense in Pediatric Ileal Crohn’s Disease. Am J Gastroenterol. 2009;105:452–459. doi: 10.1038/ajg.2009.643. [DOI] [PubMed] [Google Scholar]
- 85.Bevins CL, Stange EF, Wehkamp J. Decreased Paneth cell defensin expression in ileal Crohn’s disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut. 2009;58:882–883. discussion 883–884. [PubMed] [Google Scholar]
- 86.Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, Kobayashi KS. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proceedings of the National Academy of Sciences of the United States of America; pp. 14739–14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Noguchi E, Homma Y, Kang X, Netea MG, Ma X. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol. 2009;10:471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, Drenth JP, Van der Meer JW. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol. 2004;34:2052–2059. doi: 10.1002/eji.200425229. [DOI] [PubMed] [Google Scholar]
- 89.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature medicine. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 90.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 91.Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139:1630–1641. 1641 e1631–1632. doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]