

Abstract
Glioblastoma is the most common primary brain tumor with the most dismal prognosis. It is characterized by extensive invasion, migration, and angiogenesis. Median survival is only 15 months due to this behavior, rendering focal surgical resection ineffective and adequate radiotherapy impossible. At this moment, several ion channels have been implicated in glioblastoma proliferation, migration, and invasion. This paper summarizes studies on potassium, sodium, chloride, and calcium channels of glioblastoma. It provides an up-to-date overview of the literature that could ultimately lead to new therapeutic targets.
1. Introduction
Glioblastoma (astrocytomas, WHO grade IV) is the most aggressive primary brain tumor. With an incidence of 3.5 per 100,000 people per year, it may affect children, adults, and elderly. However, it preferentially affects adults between 45 and 75 years of age [1].
Glioblastomas can either present themselves as primary glioblastomas (95%), which manifest de novo and lack precursor tumors, or secondary glioblastomas. These tumors have progressed from less malignant glioma [2].
Surgery is the initial intervention when a patient has been diagnosed with a brain tumor. This is needed to obtain a histological diagnosis and reduces the space-occupying effect of the tumor. However, in glioblastoma, surgery is of limited therapeutic value, as complete resection is impossible due to the extensive invasive, and migratory behavior of glioblastoma cells. This renders radiotherapy ineffective as well. The current treatment is concomitant administration of temozolomide and radiotherapy. However, median survival is only 15 months [3].
The understanding of molecular alterations in signaling pathways and the consequent pathology in glioblastoma has greatly increased in the last years due to the availability of new techniques, such as genome-wide sequencing. One of the pathways that are frequently affected in glioblastoma includes channels involved in transport of sodium, potassium, and calcium ions [4]. The present paper provides an overview of the current evidence of the involvement of these ion channels in glioblastoma in terms of gliomagenesis, glioma progression, and their effect on prognosis. Because of the progression of lower-grade glioma to glioblastoma, the involvement of ion channels in high-grade glioma is discussed as well. Finally, the application of these insights is discussed in the light of future prospects for experimental and clinical practice.
2. Ion Channels and Glioblastoma
Glial cells express a variety of ion channels [5]. Recently, genome-wide analyses of glioblastoma became available. A survey of the coding sequence of 20,661 genes in glioblastoma genomes has implicated many new gene alterations. One cluster of mutated genes reported was that of ion channel genes. Of the 555 genes involved in potassium, sodium, chloride, calcium and other ion transport, 55 mutations were detected to affect 90% of the glioblastoma samples studied [4].
Ion channels are thought to facilitate progression through cell cycle checkpoints and thereby are required for cell proliferation. This process most probably occurs via modulation of the resting membrane potential. For example, progression through the G1/S checkpoint is correlated with increased potassium K+ channel activity and momentary hyperpolarization [6]. To illustrate this, iberiotoxin, a pharmacological inhibitor of big conductance K+ channels, arrests glioma cells in S phase of the cell cycle [7]. On the other hand, transient depolarization facilitated by Cl− channels is observed at the G2/M checkpoint [6]. It is for these reasons that uncontrolled ion channel activity can contribute to oncogenesis.
As stated before, the prognosis of glioblastoma is abysmal due to its invasive migration, which renders surgical resection ineffective. Ion channels may contribute to this invasion and migration. They influence shape and volume of cancer cells by affecting ion and water transport over the plasma membrane. Ion channels thereby facilitate invasive migration through the sinuous extracellular space of brain tissue [8–13] (Figure 1). In addition, ion channels may be functionally involved in proliferation [7, 14, 15]. It is for these reasons that ion channels may contribute to the malignant behavior of glioblastoma cells. Therefore, ion channels may be novel therapeutic targets in the treatment of glioblastoma.
Figure 1.
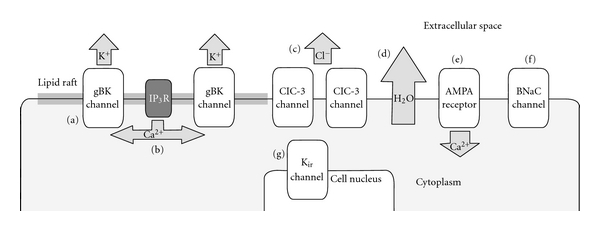
gBK channels (a) facilitate an increased outwardly K+ current. Ca2+ input for gBK channels is provided by IP3R (b). ClC-3 channels facilitate an increased Cl− outwardly current. (a) and (c) facilitate increased H2O movement through osmosis (d) over the plasma membrane. Glioblastoma AMPA receptors (e) lack the GluR2 subunit and therefore have increased Ca2+ permeability. Amiloride-sensitive BNaC channels (f) are expressed in glioblastoma. Kir channels are mislocalized to the cell nucleus (g), diminishing inwardly rectifying K+ currents.
2.1. Potassium Channels
2.1.1. Ca2+-Activated K+ Channels
Ca2+-activated K+ channels facilitate outwardly rectifying potassium currents and respond to Ca2+ concentrations. Increases in the intracellular [Ca2+] shift the voltage dependence of Ca2+-activated K+ channels to more negative potentials.
One of these channels, big conductance K+ channels or BK channels, is widely expressed in excitable and non-excitable cells. BK channels respond to both membrane voltage potentials and intracellular [Ca2+] [16].
Specific overexpression of BK channels has been observed in biopsies of patients with malignant gliomas, compared with nonmalignant human cortical tissues. In addition, expression levels correlate positively with the malignancy grade of the tumor [16]. Lastly, BK currents in glioma cells are more sensitive to intracellular [Ca2+] compared to BK channels in healthy glial cells [17, 18].
BK channels can express a variety of electrophysiological properties, which is due to alternative splicing of their α-subunits. The increased sensitivity to intracellular [Ca2+] is found in a novel splice isoform of hSlo, the gene that encodes the α-subunits. This BK channel isoform has exclusively been observed to be expressed in glioma. In addition, glioma most likely only expresses this new isoform, as the classical BK channel has not been found in gliomas yet. These findings led to the term glioma BK (gBK) channel (Figure 1).
gBK channels have been suggested as a candidate channel for providing the electrochemical driving force for ion movement needed for the release of cytoplasmic water and cell shrinkage which in turn facilitates the extensive migrating behavior of glioblastoma cells. First of all, the effect of menthol was studied, an agonist of transient receptor potential melastatin 8 (TPRM8) ion channels, which increases intracellular [Ca2+], which in turn activates gBK channels. Menthol stimulated glioma cell migration [11, 12]. In addition, administration of paxilline and tetraethylammonium, both gBK channel inhibitors, inhibited migration [11, 12]. These findings make a role of gBK channels in the migration of glioblastoma cells probable. However, the effect of gBK channel knockdown has yet to be investigated, leaving room for doubt.
In contrast to the better studied role that gBK channels have in glioblastoma cell invasive migration, there is discussion whether or not gBK channels contribute to proliferation. Some studies have implicated gBK channels in the proliferation of glioblastoma cells. Glioblastoma cells were exposed to pharmacological inhibitors of gBK channels, such as iberiotoxin, paxilline, tetraethylammonium, and penitrem A. After 3 to 5 days, growth inhibition and even tumor shrinkage were observed in vitro [7, 14, 15]. In contrast, more recent literature contradicts these findings and suggests that gBK channels are not required for proliferation or even have antitumorigenic properties. It has been found that pharmacological inhibitors suppress glioblastoma cell growth at concentrations far higher than concentrations that were sufficient to inhibit gBK channel activity. Low concentrations that were sufficient to inhibit these channels did not affect glioblastoma cell growth in vitro. In addition, downregulation of gBK channels using gene-specific siRNAs reduced K+ current densities, but caused no changes in proliferation [19]. This argues against a critical role for gBK in glioblastoma proliferation.
Inositol 1,4,5-triphosphate receptors (IP3Rs) may provide Ca2+ for gBK channels (Figure 1). These receptors are localized close to and are linked with gBK channels in dedicated plasma membrane domains called lipid rafts. Disruption of these lipid rafts with methyl-β-cyclodextrin disturbs the connection between gBK channels and Ca2+. This disturbance was restored by inclusion of Ca2+ in the pipette solution during the whole cell patch-clamp experiments. This suggests that the disturbance was not caused by destruction or calcium desensitization of the gBK channels [20].
2.1.2. Inwardly Rectifying K+ Channels
Glioma cells both in vitro and in situ are characterized by depolarized resting membrane potentials of about −20 to −40 mV [21]. In addition, they express increased outwardly rectifying K+ currents [16]. This contrasts the very negative resting membrane (−80 to −90 mV) and large inwardly rectifying K+ currents that characterize normal glial cells [22]. These findings have led to the assumption that glioma cells express a decreased density of inwardly rectifying K+ channels (Kir) compared to normal brain tissue.
However, western blot analysis of D54 glioblastoma cell lines showed only slightly lowered expression of Kir2.1, normal expression of Kir4.1, and increased expression of Kir2.3 and Kir3.1 as compared to normal astrocytes, while electrophysiological recording found no Kir current. Immunocytochemistry placed suspicion on mislocalization of Kir channels in glioblastoma cells. Immunostaining of Kir2.3 and Kir4.1 predominantly labeled the nucleus of glioblastoma cells (Figure 1), while expression in normal astrocytes was diffuse over the plasma membrane [23]. The intracellular localization of Kir channels may explain why glioblastoma cells express a depolarized resting membrane potential and decreased inwardly rectifying K+ current.
Kir channels in glial cells are specifically known for two functions: buffering of extracellular K+ and establishment of a very negative resting potential [24]. Glial cells perform K+ uptake from the extracellular space and redistribute K+ ions toward areas where the extracellular [K+] is lower. This process includes diffusion from cell to cell through gap junctions. Eventually, K+ is released into blood vessels. This K+ buffering by glial cells is essential for neuronal homeostasis, as elevated extracellular [K+] would depolarize neurons, preventing them from firing action potentials. Kir channels in healthy glial cells seem to be fitted very well for this task, as they have a large open probability at resting potential. This means that at resting potential, a relatively high number of Kir channels are open. In addition, with increasing extracellular [K+] their conductance increases as well, which makes them perfectly suited for the task of correcting an excess of K+ in the extracellular space [22]. Mislocalization of Kir channels in glioblastoma cells (Figure 1) renders these cells insufficient for these tasks, resulting in accumulation of K+ in the extracellular space. This accumulation occurs in concurrence with epileptic seizures [25], although the underlying mechanism is not entirely clear. Peritumoral epileptic seizures are often seen in glioblastoma patients, perhaps facilitated by mislocalization of Kir channels. On the other hand, it is likely that very few to no neurons survive around glioblastoma cells due to their invasive behavior. Moreover, increased concentrations of glutamate in gliomas have been reported [26]. These arguments, supported by the excitotoxic properties of high glutamate concentrations to neurons, render high glutamate release a more logical suspect of causing epileptical seizures than high [K+] in the extracellular space.
On the other hand, mislocalized Kir channels can contribute indirectly to epileptic seizures as they fail in their function of establishing a very negative resting potential. At the depolarized resting potentials of −20 to −40 mV that characterize glioma cells [21], the Na+ gradient across the plasma membrane is diminished. As a consequence, Na+-dependent glutamate transporters become inactive, increasing the extracellular glutamate concentration and thereby possibly causing epileptic seizures [23].
2.1.3. Ether À Go-Go K+ Channels
Other candidate channels possibly responsible for the depolarized resting membrane potential in glioblastoma cells are the ether à go-go 1 (EAG1) and ether à go-go related 1 (ERG1) channels. Expression of these channels is upregulated in glioblastoma. Depolarized resting membrane potential allows large hyperpolarizations, which provide a driving force for Ca2+ entry. Ca2+ is necessary for cell-cycle progression. In this way, EAG1 and ERG1 channels can contribute to gliomagenesis.
Several studies have described expression levels of hEAG1 and hERG1 in glioblastoma, which encodes EAG1 and ERG1 channels, respectively. However, the results are contradictory. Patt et al. reported low expression of hEAG1 and hERG1 in 5 glioblastoma samples compared to healthy brain tissue. Expression was elevated in low-grade gliomas [27]. This contradicts with the hypothesis above. In contrast, Masi et al. found elevated expression of hERG1 in 26 glioblastoma samples, supporting the hypothesis above [28]. The observation by Masi et al. is supported by other literature, reporting increased ERG1 mRNA expression, elevated protein levels and high densities of ERG1 channels in other tumors, such as colorectal cancer [29], endometrial adenocarcinoma [30], and myeloid leukemia [31].
ERG1 activity has also been reported to be correlated with induction of vascular endothelial growth factor (VEGF) secretion, thereby contributing to angiogenesis [28]. Primary glioblastomas are characterized by extensive neoangiogenesis [32].
2.2. Chloride Channels
2.2.1. ClC Family Channels
Besides the gBK channel discussed earlier, the ClC-3 chloride channel is another important candidate channel to facilitate migrating behavior of glioblastoma cells [10]. ClC-3 channel expression was, together with that of ClC-2 and ClC-5, indeed high in glioblastoma cell lines and biopsy tissue compared to healthy glial cells. Whole cell patch-clamp recordings in combination with channel-specific antisense oligonucleotides showed that ClC-3 channels facilitated outwardly rectifying current. On the other hand, ClC-2 channels facilitated inwardly rectifying currents [33]. As a result of the relative overexpression of ClC-3 as compared to ClC-2 [33], the outwardly rectifying current may prevail over the inwardly rectifying current. As a consequence the glioblastoma cell depolarizes, which is needed to pass the G2/M checkpoint in the cell cycle [6].
In addition, the overexpression of both channels equips glioblastoma cells with an improved ability to transport Cl−. This may facilitate rapid changes in cell size and shape as glioblastoma cells invade through sinuous extracellular brain spaces [33].
Inhibition of ClC-3 channels using chlorotoxin indeed markedly but not completely inhibited glioblastoma cell invasion in vitro. The same effect was accomplished using ClC-3 siRNA knockdown. In addition, the nonspecific ClC-blocker 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) completely inhibited glioblastoma cell invasion [9]. Limitations of this study include the possibility that the glioblastoma cell was overdosed with NPPB and invasion was stopped by cytotoxic levels of NPPB. It is unclear whether concentrations of NPPB were just sufficient to block ClC channels. This doubt was also raised in studies where gBK channel inhibition correlated with proliferation [19]. Secondly, NPPB has also been reported to be a Ca2+-activated K+-channel inhibitor [34]. This fact may compromise the implication of ClC-3 in glioblastoma cell invasion.
ClC-3 is regulated through phosphorylation via Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII was infused intracellularly to D54 glioblastoma cells via a patch-clamp pipette, increasing Cl− currents 3-fold. In addition, administration of autocamtide-2, a CaMKII-specific inhibitor, inhibited this current. To confirm the relation between ClC-3 and CaMKII, ClC-3 was shown to be knocked down after CaMKII modulation of Cl− currents was lost. Furthermore, immunohistochemistry colocalized ClC-3 with CaMKII. Interestingly, inhibition of CaMKII in ClC-3-expressing cells reduced glioblastoma cell invasion to the same extent as direct inhibition of ClC-3 [35]. These findings suggest that CaMKII is a molecular link between intracellular [Ca2+] changes and ClC-3 conductance required for cell movement during invasive migration in glioblastoma.
2.3. Calcium Channels
Ca2+ is required by glioblastoma cells as a second messenger to support cell migration. Oscillatory changes in intracellular [Ca2+] that correlate with cell invasion and migration have been observed. It has been hypothesized that these changes in intracellular [Ca2+] activate ClC-3 channels through CaMKII. This in turn may initiate glioblastoma invasion [35, 36].
Glioblastoma cells express Ca2+ permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) glutamate receptors (Figure 1). These glutamate receptors have become Ca2+ permeable due to the lack of the GluR2 subunit as they have been assembled entirely of GluR1 and/or GluR4 subunits. AMPA receptors containing GluR2 subunits show little Ca2+ permeability, while those lacking GluR2 subunits exhibit high Ca2+ permeability due to a deformed pore with an aberrant size and polarity. Adenovirus-mediated transfer of the GluR2 cDNA decreased intracellular [Ca2+], inhibited cell migration and induced apoptosis. However, overexpression of Ca2+ permeable AMPA receptors increased migration and proliferation of glioblastoma cells in vitro. GluR2 was indeed not expressed in most glioblastoma surgical samples. These findings implicate Ca2+-permeable AMPA receptors in proliferation, migration and invasion of glioblastoma [8]. It can be hypothesized that this is due to increased intracellular [Ca2+], which in turn facilitates increased activity of gBK and ClC-3 channels.
The same theory applies to a study in which inhibition of IP3R subtype 3 (IP3R3) was achieved using caffeine. Inhibition of IP3R3 led to decreased intracellular [Ca2+]. This was associated with inhibited migration of glioblastoma cells in vitro. In addition, an increase of mean survival was observed after caffeine was administered to a mouse xenograft model of glioblastoma. These mice had a 6 μg/mL serum caffeine concentration. This is approximately the same concentration in people that drink two to five cups of coffee a day. These findings suggest that IP3R3 can serve as a possible therapeutic target [13]. It would be interesting to investigate whether large cohort studies can associate coffee consumption with a lower incidence of glioblastoma or prolonged survival. However, such information is not available yet. With a yearly glioblastoma incidence of 3.5 per 100,000 people, study sizes are probably not large enough to show such results. The effect of caffeine on the survival of glioblastoma mouse models is interesting as caffeine is regarded as “not classifiable as to its carcinogenicity to humans” by the WHO, meaning that there is contradictory evidence about its carcinogenic hazard [37].
The Ca2+-permeable transient receptor potential canonical channel protein 6 (TRPC6) has been implicated in glioma proliferation. TRPC6 is overexpressed in human glioma cells, and inhibition suppressed intracellular [Ca2+] and cell growth and induced cell cycle arrest at the G2/M phase. In mouse models with xenografted human tumors, inhibition of TRPC6 reduced tumor volume and increased mean survival [38].
2.4. Sodium Channels
In contrast to mutations found in potassium and calcium ion channels, sodium channel mutations correlated with shorter survival in a univariate analysis [39]. The authors reported that all samples with IDH1 mutations did not have any sodium channel mutations. However, this association was not significant. This may be due to the small sample size of the study (21 patients).
IDH1 encodes for an enzyme that functions at a crossroads of cellular metabolism. Mutations in IDH1 have been identified to be associated with a specific subgroup of glioblastoma patients who are younger and have a prolonged survival [4, 40]. After correction for IDH1, the difference in survival between patients with mutated and unmutated sodium channels dropped to nonsignificant levels. Further research is needed to investigate the association between mutations in IDH1 and sodium channels and whether the effect of sodium channel mutations on survival is independent of IDH1 mutation status.
Furthermore, among the patients with mutations in sodium channels, the mutations were scattered over the different genes. All 14 sodium channel genes were mutated only once among the 21 patients except for SCN9A, which was mutated twice. In addition, of the 12 patients with sodium channel mutations, only 2 had mutations in more than one gene [39]. This could suggest a similar function, and therefore mutual exclusivity among these mutations. This is supported by the fact that of the 14 studied genes, 12 genes were from the SCN or SLC subset classes with 5 and 7 genes, respectively. A similar scattering among the studied genes and clustering in gene families was observed in potassium and calcium channel mutation analyses [39].
Moreover, the effect of sodium channel inhibitors on glioblastoma cell growth was studied. Digoxin and ouabain were administered to 2 glioblastoma cell lines in vitro. Both drugs showed antiproliferative effects and toxicity against the cell lines. Furthermore, cells treated showed an apoptotic phenotype under the light microscope [39]. These findings are supported by the fact that the antiproliferative effect in cancer of cardiac glycosides is well known [41, 42]. Furthermore, they may be neuroprotective [43]. The underlying mechanism of these side effects has not been clarified yet, although inhibition of sodium channels in brain tissue could be the cause of this.
Another study observed an inward, amiloride-sensitive Na+ current in glioblastoma cell lines and tumor samples. These currents were not observed in normal astrocytes or low-grade astrocytomas. Currently, brain Na+ channels (BNaCs) are the only amiloride-sensitive Na+ channels identified in the brain. PCR analyses indeed demonstrated the presence of BNaC mRNA in these tumors [44].
Finally, the effect of Psalmotoxin 1, an inhibitor of cation currents mediated by acid-sensing ion channels, was studied using the whole-cell patch-clamp technique. This toxin inhibited Na+ currents in glioblastoma cell lines and human glioblastoma samples, but not in normal human astrocytes [45]. Since this effect can only be measured using electrophysiological experiments, the diagnostic value seems low. However, if the Na+ current facilitated by acid-sensing ion channels proves essential for glioblastoma cells in vivo, inhibition of Psalmotoxin may serve as a possible future therapy.
3. Future Therapeutic Targets
3.1. Ion Channels
Given the important role that gBK and ClC-3 channels are thought to have in glioma invasion and migration, these channels may render a promising therapeutic target to render glioblastoma less aggressive. However, even if in vivo inhibition of gBK and ClC-3 channels can inhibit glioblastoma invasion and migration, these future therapies probably have to be administered at an early stage in order to make a difference in the treatability by resection and radiotherapy.
Taken the central role of intracellular [Ca2+] in ClC-3 and gBK channel activity into account, influencing glioblastoma Ca2+ homeostasis may be a target of future therapies. This can be accomplished by inhibition of Ca2+-permeable AMPA receptors, for example by adenovirus-mediated transfer of GluR2 cDNA [8]. Another possible therapy is inhibition of IP3R3 using caffeine [13]. However, the carcinogenic hazard of caffeine is not clear yet [37]. On the other hand, Ca2+ coupling to ClC-3 and gBK channels can be disturbed by inhibiting CaMKII [35] or lipid rafts, [20] respectively. In addition, inhibition of TRPC6 has shown promising results both in vitro and in vivo in mouse models.
The finding that glioma cells' depolarized resting membrane potential and their inability to maintain K+, Na+ and glutamate homeostasis are caused by mislocalization of Kir channels suggests that these channels function at a crossroads of cellular homeostasis and basic electrophysiological functions in glioma cells [23]. Correction of this mislocalization could therefore serve as a possible therapeutic target. However, the underlying mechanism of this mislocalization is currently unknown, making therapy uncertain in the near future.
Recently, the antiproliferative effect of cardiac glycosides on glioblastoma cell growth in vitro was studied. Digoxin and ouabain proved useful inhibitors of cell proliferation [39]. However, concentrations that provide an anticancer effect are high and induce severe cardiovascular side effects. Therefore, their development as anticancer agents has been limited thus far. Chemical modification is needed to increase affinity for tumor sodium channels and decrease affinity for cardiac sodium channels [42].
3.2. Chemotherapeutic Agents
Tetrandrine is an inhibitor of gBK channels. It therefore may inhibit the extensive invasion of glioblastoma cells. Moreover, tetrandrine has cytotoxic effects. Furthermore, it exacerbates radiation-induced cell-cycle perturbation, thereby inducing apoptosis and radiosensitization in glioblastoma cells. In addition, it has antiangiogenic effects. These capabilities render it a possible useful therapy to treat glioblastomas, especially combined with radiotherapy or other chemotherapeutic agents [46]. The combination of classical cytotoxic, apoptotic, radiosensitization and antiangiogenic effects with inhibition of gBK channels is promising. However, the effect of tetrandrine has thus far only been studied in rats [47]. Furthermore, tetrandrines' large arsenal of possible therapeutic targets in gliomas may impede to find an optimal dosage.
The blood-brain tumor barrier is an important hurdle to overcome in glioblastoma treatment. Temozolomide, a chemotherapeutic agent that was discussed earlier, is currently, together with radiotherapy, the golden standard in glioblastoma treatment. However, temozolomide crosses the blood-brain tumor barrier insufficiently to have a significant impact on patient survival. The same accounts for trastuzumab, which may be especially effective in a distinct glioblastoma subgroup (neural, as discussed by Verhaak et al. [48]) when combined with temozolomide. Therefore, both drugs were coinfused with minoxidil sulfate, an ATP-sensitive potassium channel (KATP) activator. In mice, this indeed resulted in improved selective drug delivery to glioblastoma. The underlying mechanism is not completely understood, but it involves formation of brain vascular endothelial transcytotic vesicles to facilitate absorption of the drug [49].
3.3. Limitations of Cell Lines
Currently, most research in the field of ion channels in glioma is conducted on glioma cell lines. However, several experiments have shown that established glioblastoma cell lines resemble glioblastomas in patients very poorly when compared at the level of DNA alterations or gene expression profiles [50]. With this in mind, results from experiments conducted with cell lines should always be put into the right context. In vitro studies in human glioblastoma samples or in vivo studies in animal xenograft models remain needed.
4. Conclusion
At this moment, we have increased our understanding of the molecular mechanisms involving ion channels underlying the invasive migration of glioblastoma. However, in contrast to many other forms of cancer and considering the genetic research in glioblastoma, consequences for treatment are lagging behind. Under the shadow of the large and extensive research on genetic alterations and its effects on therapy responses in glioblastoma [51], it is doubtful whether any therapies involving ion channels will ever see the light. Our current understanding of ion channels in glioblastoma will most probably lead to drugs that can be given concomitantly with chemotherapeutic agents to increase their effectivity, such as the discussed coinfusion with minoxidil sulfate. In addition, the current knowledge of the involvement of BK channels, ClC-3 channels and intracellular [Ca2+] homeostasis in glioblastoma invasion and migration would justify the first steps in drug development targeting these aspects.
Conflict of Interests
The author declared that there is no conflict of interrests.
Acknowledgment
The author likes to thank Dr. Ronald Wilders for his helpful suggestions and critical reading of the paper.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathologica. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. American Journal of Pathology. 2007;170(5):1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verkhratsky A, Steinhäuser C. Ion channels in glial cells. Brain Research Reviews. 2000;32(2-3):380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- 6.Blackiston DJ, McLaughlin KA, Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle. 2009;8(21):3519–3528. doi: 10.4161/cc.8.21.9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weaver AK, Liu X, Sontheimer H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. Journal of Neuroscience Research. 2004;78(2):224–234. doi: 10.1002/jnr.20240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nature Medicine. 2002;8(9):971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 9.Lui VCH, Lung SSS, Pu JKS, Hung KN, Leung GKK. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Research. 2010;30(11):4515–4524. [PubMed] [Google Scholar]
- 10.Soroceanu L, Manning TJ, Jr., Sontheimer H. Modulation of glioma cell migration and invasion using Cl− and K+ ion channel blockers. Journal of Neuroscience. 1999;19(14):5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wondergem R, Bartley JW. Menthol increases human glioblastoma intracellular Ca2+, BK channel activity and cell migration. Journal of Biomedical Science. 2009;16(1, article no. 90) doi: 10.1186/1423-0127-16-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wondergem R, Ecay TW, Mahieu F, Owsianik G, Nilius B. HGF/SF and menthol increase human glioblastoma cell calcium and migration. Biochemical and Biophysical Research Communications. 2008;372(1):210–215. doi: 10.1016/j.bbrc.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 13.Kang SS, Han KS, Ku BM, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Research. 2010;70(3):1173–1183. doi: 10.1158/0008-5472.CAN-09-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basrai D, Kraft R, Bollensdorff C, Liebmann L, Benndorf K, Patt S. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. NeuroReport. 2002;13(4):403–407. doi: 10.1097/00001756-200203250-00008. [DOI] [PubMed] [Google Scholar]
- 15.Weaver AK, Bomben VC, Sontheimer H. Expression and function of calcium-activated potassium channels in human glioma cells. GLIA. 2006;54(3):223–233. doi: 10.1002/glia.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ransom CB, Sontheimer H. BK channels in human glioma cells. Journal of Neurophysiology. 2001;85(2):790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- 17.Ransom CB, Liu X, Sontheimer H. BK channels in human glioma cells have enhanced calcium sensitivity. GLIA. 2002;38(4):281–291. doi: 10.1002/glia.10064. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Chang Y, Reinhart PH, Sontheimer H. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. Journal of Neuroscience. 2002;22(5):1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdullaev IF, Rudkouskaya A, Mongin AA, Kuo YH. Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulate cell proliferation. PLoS One. 2010;5(8) doi: 10.1371/journal.pone.0012304. Article ID e12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weaver AK, Olsen ML, McFerrin MB, Sontheimer H. BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: a novel mechanism for coupling [Ca2+]i to ion channel activation. Journal of Biological Chemistry. 2007;282(43):31558–31568. doi: 10.1074/jbc.M702866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bordey A, Sontheimer H. Electrophysiological properties of human astrocytic tumor cells in situ: enigma of spiking glial cells. Journal of Neurophysiology. 1998;79(5):2782–2793. doi: 10.1152/jn.1998.79.5.2782. [DOI] [PubMed] [Google Scholar]
- 22.Newman EA. Inward-rectifying potassium channels in retinal glial (Muller) cells. Journal of Neuroscience. 1993;13(8):3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen ML, Sontheimer H. Mislocalization of Kir channels in malignant glia. GLIA. 2004;46(1):63–73. doi: 10.1002/glia.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. Journal of Neurophysiology. 1966;29(4):788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 25.Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. Journal of Neurophysiology. 1988;59(1):259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- 26.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Research. 1999;59(17):4383–4391. [PubMed] [Google Scholar]
- 27.Patt S, Preußat K, Beetz C, et al. Expression of ether à go-go potassium channels in human gliomas. Neuroscience Letters. 2004;368(3):249–253. doi: 10.1016/j.neulet.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Masi A, Becchetti A, Restano-Cassulini R, et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. British Journal of Cancer. 2005;93(7):781–792. doi: 10.1038/sj.bjc.6602775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lastraioli E, Guasti L, Crociani O, et al. herg1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Research. 2004;64(2):606–611. doi: 10.1158/0008-5472.can-03-2360. [DOI] [PubMed] [Google Scholar]
- 30.Cherubini A, Taddei GL, Crociani O, et al. HERG potassium channels are more frequently expressed in human endometrial cancer as compared to non-cancerous endometrium. British Journal of Cancer. 2000;83(12):1722–1729. doi: 10.1054/bjoc.2000.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pillozzi S, Brizzi MF, Balzi M, et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia. 2002;16(9):1791–1798. doi: 10.1038/sj.leu.2402572. [DOI] [PubMed] [Google Scholar]
- 32.Godard S, Getz G, Delorenzi M, et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Research. 2003;63(20):6613–6625. [PubMed] [Google Scholar]
- 33.Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expression of voltage-gated chloride channels in human glioma cells. Journal of Neuroscience. 2003;23(13):5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fioretti B, Castigli E, Calzuola I, Harper AA, Franciolini F, Catacuzzeno L. NPPB block of the intermediate-conductance Ca2+-activated K+ channel. European Journal of Pharmacology. 2004;497(1):1–6. doi: 10.1016/j.ejphar.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 35.Cuddapah VA, Sontheimer H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. Journal of Biological Chemistry. 2010;285(15):11188–11196. doi: 10.1074/jbc.M109.097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. Journal of Membrane Biology. 2000;176(1):31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- 37.IARC. Agents Classified by the IARC Monographs. 1–102. IARC; 2011. [Google Scholar]
- 38.Ding X, He Z, Zhou K, et al. Essential role of trpc6 channels in G2/M phase transition and development of human glioma. Journal of the National Cancer Institute. 2010;102(14):1052–1068. doi: 10.1093/jnci/djq217. [DOI] [PubMed] [Google Scholar]
- 39.Joshi AD, Parsons DW, Velculescu VE, Riggins GJ. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Molecular Cancer. 2011;10, article 17 doi: 10.1186/1476-4598-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldin GG, Safa AR. Digitalis and cancer. Lancet. 1984;1(8386):p. 1134. doi: 10.1016/s0140-6736(84)92556-x. [DOI] [PubMed] [Google Scholar]
- 42.Mijatovic T, Van Quaquebeke E, Delest B, Debeir O, Darro F, Kiss R. Cardiotonic steroids on the road to anti-cancer therapy. Biochimica et Biophysica Acta. 2007;1776(1):32–57. doi: 10.1016/j.bbcan.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Wang JKT, Portbury S, Thomas MB, et al. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(27):10461–10466. doi: 10.1073/pnas.0600930103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bubien JK, Keeton DA, Fuller CM, et al. Malignant human gliomas express an amiloride-sensitive Na+ conductance. American Journal of Physiology. 1999;276(6):C1405–C1410. doi: 10.1152/ajpcell.1999.276.6.C1405. [DOI] [PubMed] [Google Scholar]
- 45.Bubien JK, Ji HL, Gillespie GY, et al. Cation selectivity and inhibition of malignant glioma Na+ channels by Psalmotoxin 1. American Journal of Physiology. 2004;287(5):C1282–C1291. doi: 10.1152/ajpcell.00077.2004. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Tseng SH. The potential of tetrandrine against gliomas. Anti-Cancer Agents in Medicinal Chemistry. 2010;10(7):534–542. doi: 10.2174/187152010793498609. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y, Chen JC, Tseng SH. Tetrandrine suppresses tumor growth and angiogenesis of gliomas in rats. International Journal of Cancer. 2009;124(10):2260–2269. doi: 10.1002/ijc.24208. [DOI] [PubMed] [Google Scholar]
- 48.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ningaraj NS, Sankpal UT, Khaitan D, Meister EA, Vats T. Activation of KATP channels increases anticancer drug delivery to brain tumors and survival. European Journal of Pharmacology. 2009;602(2-3):188–193. doi: 10.1016/j.ejphar.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 50.Li A, Walling J, Kotliarov Y, et al. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Molecular Cancer Research. 2008;6(1):21–30. doi: 10.1158/1541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
- 51.Clarke J, Butowski N, Chang S. Recent advances in therapy for glioblastoma. Archives of Neurology. 2010;67(3):279–283. doi: 10.1001/archneurol.2010.5. [DOI] [PubMed] [Google Scholar]