

Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating and neurodegenerative disease initiated by autoreactive T cells. Mgat5, a gene in the Asn (N-) linked protein glycosylation pathway, associates with MS severity and negatively regulates experimental autoimmune encephalomyelitis (EAE) and spontaneous inflammatory demyelination in mice. N-glycan branching by Mgat5 regulates interaction of surface glycoproteins with galectins, forming a molecular lattice that differentially controls the concentration of surface glycoproteins. T-cell receptor signaling, T-cell proliferation, TH1 differentiation, and CTLA-4 endocytosis are inhibited by Mgat5 branching. Non-T cells also contribute to MS pathogenesis and express abundant Mgat5 branched N-glycans. Here we explore whether Mgat5 deficiency in myelin-reactive T cells is sufficient to promote demyelinating disease. Adoptive transfer of myelin-reactive Mgat5−/− T cells into Mgat5+/+ versus Mgat5−/− recipients revealed more severe EAE in the latter, suggesting that Mgat5 branching deficiency in recipient naive T cells and/or non-T cells contribute to disease pathogenesis.
1. Introduction
Multiple sclerosis (MS) is a complex trait disease where multiple genetic and environmental factors combine to influence susceptibility to disease [1]. Concordance rates in monozygotic twins is ~30%, an ~300-fold higher risk than the general population risk of ~0.1% [2]. However, Baranzini et al. recently reported that they failed to observe sequence differences in the genome, epigenome, or transcriptome of monozygotic twins discordant for MS [3], implicating direct environmental impact on genetic risk. The disparate prevalence of MS along north-south gradients implicates various environmental factors as well, including sunshine exposure, diet, and Vitamin D3 status [4]. Genomewide association studies (GWAS) and candidate gene investigations have identified a number of genes associated with MS susceptibility [5–9]. A recent GWAS for variants regulating MS severity identified Mgat5, a gene encoding an enzyme in the Asn (N-) linked protein glycosylation pathway [10]. Mgat5 catalyzes the addition of β1,6-GlcNAc to N-glycan intermediates on glycoproteins in the Golgi apparatus (Figure 1) [11, 12]. Indeed, we have recently shown that multiple genetic and environmental risk factors converge to dysregulate N-glycosylation in MS [13]. N-glycan branching by Mgat5 and related enzymes determines binding avidity of surface glycoproteins for galectins, interactions that form a molecular lattice at the cell surface [14]. The galectin-glycoprotein lattice regulates cell growth and differentiation by altering the concentration of surface glycoproteins [15]. Mice deficient in Mgat5 display enhanced delayed-type hypersensitivity, spontaneous kidney autoimmunity, and increased susceptibility to experimental autoimmune encephalomyelitis (EAE) [16]. Furthermore, mouse strains susceptible to EAE (PL/J, SJL, and NOD) display reduced N-glycan branching in T cells compared with strains resistant to EAE (129/Sv, BALB/c, and B10.S) [17]. The PL/J strain displays the lowest levels of N-glycan branching with mass spectroscopy and enzyme assays demonstrating deficiencies in multiple N-glycosylation pathway enzymes. A small minority of aged PL/J mice develop a spontaneous late-onset clinical disease manifested by inflammatory demyelination and neurodegeneration, phenotypes markedly enhanced by Mgat5+/− and Mgat5−/− genotypes in a gene dose-dependent manner. Mgat5−/− PL/J mice with spontaneous disease display features of chronic MS, including slow progressive paralysis, tremor, focal dystonic posturing, paroxysmal dystonia, neuronophagia, and axonal damage in demyelinated lesions and normal white matter [17–19]. Increasing N-glycan branching in T cells by metabolically increasing availability of substrate (i.e., UDP-GlcNAc) to Mgat5 in the Golgi suppresses autoimmune pathogenesis. In vitro supplementation of encephalitogenic T cells with the simple sugar N-acetylglucosamine (GlcNAc), which enhances metabolic supply of UDP-GlcNAc to Mgat5, reduced the incidence and severity of EAE following adoptive transfer of the cells into naïve recipient mice [20]. Oral GlcNAc also reduced the development of spontaneous diabetes in nonobese diabetic mice [20].
Figure 1.
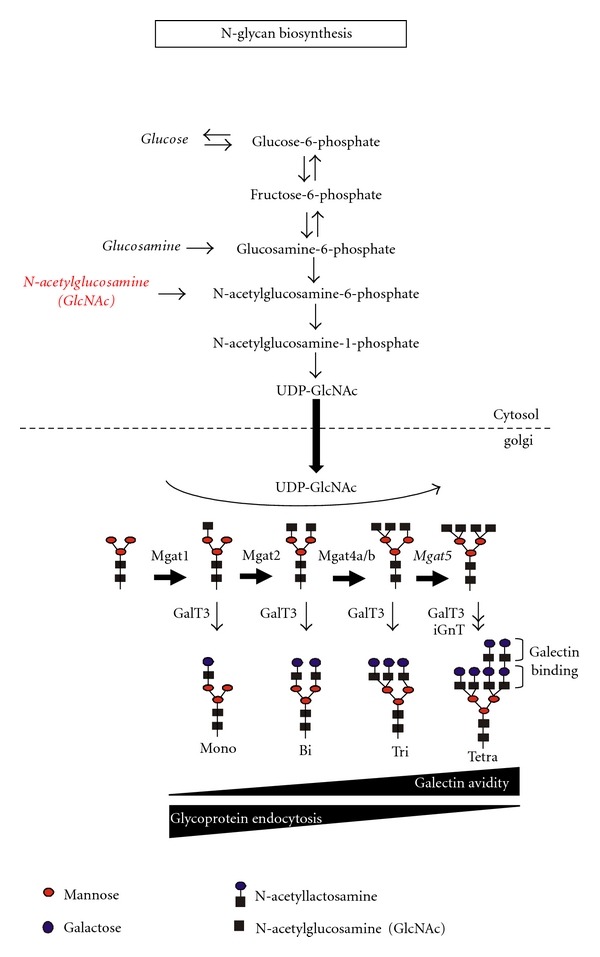
GlcNAc-branched N-glycan biosynthesis. UDP-GlcNAc is required by the N-acetylglucosaminyltransferases Mgat1, 2, 4, and 5 and iGnT. Cytosolic UDP-GlcNAc enters the Golgi via antiporter exchange with Golgi UMP, a reaction product of the N-acetylglucosaminyltransferases. Galectins bind N-acetyllactosamine, with avidity increasing in proportion to the number of N-acetyllactosamine units (i.e., branching). β-1,6-GlcNAc-branching by Mgat5 promotes poly-N-acetyllactosamine production, further enhancing avidity for galectins. GalT3, galactosyltransferase 3.
In T cells, Mgat5 branching and the galectin lattice inhibit basal and activation signaling, through the T-cell receptor (TCR) and CD45 in resting cells, promote growth arrest by cytotoxic T-lymphocyte antigen-4 (CTLA-4) in blasting cells and enhance TH2 over TH1/ TH17 differentiation [15, 16, 21–26]. These T-cell-specific phenotypes are consistent with enhanced susceptibility to demyelinating disease in Mgat5-deficient mice; however, they do not exclude disease promotion by non-T cells. Here we investigate whether Mgat5 branching deficiency in myelin-reactive T cells is sufficient to promote EAE.
2. Materials and Methods
2.1. Experimental Autoimmune Encephalomyelitis (EAE) Induction
Adoptive transfer EAE was induced by subcutaneous immunization of Mgat5−/− PL/J mice with 100 μg of bovine myelin basic protein (MBP) (Sigma) emulsified in complete Freund's adjuvant containing 4 mg/mL heat-inactivated Mycobacterium tuberculosis (H37RA; Difco) distributed over two spots on the hind flank. Splenocytes were harvested 10 days following immunization and stimulated in vitro with 50 μg/mL MBP. After 48 h of incubation, CD3+ T cells were purified by negative selection (R&D Systems). 2.7 million CD3+ T cells were injected intraperitoneally into naïve PL/J Mgat5+/+ (n = 7) and Mgat5−/− (n = 8) mice. Trypan blue exclusion determined <5% dead cells prior to injection. Mice were weighed and examined daily for clinical signs of EAE over the next 40 days with the observer blinded to experimental conditions. Mice were scored daily in a blinded fashion as follows: 0, no disease; 1, loss of tail tone; 2, hindlimb weakness; 3, hindlimb paralysis; 4, forelimb weakness or paralysis and hindlimb paralysis; 5, moribund or dead. All procedures and protocols with mice were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine, Calif, USA.
2.2. Cytokine Measurement
Supernatant from splenocyte cultures simulated with 50 μg/mL bovine MBP (Sigma) for 48 hours were tested for IFN-γ and TNF-α levels by a multiplexing immunoassay, a bead-based analyte detection system using flow cytometry (FlowCytomix; eBioscience).
2.3. Statistical Analysis
Statistical analysis and P values for EAE disease incidence was determined by Fisher's exact test. P values for EAE mean clinical score, disease duration, and the highest clinical score were determined by the Mann-Whitney test.
3. Results
To further investigate the effects of N-glycan processing deficiency in T-cell-dependent autoimmunity in vivo, we utilized an adoptive transfer model of EAE induction. Specifically, we wanted to examine whether N-glycan branching deficiency in self-reactive T cells is sufficient to enhance autoimmune demyelination. EAE may be induced by adoptive transfer of myelin antigen-specific T cells into naïve mice, leading to inflammatory demyelination of axons and progressive motor weakness. Splenocytes from myelin basic protein- (MBP-) immunized Mgat5−/− PL/J mice were restimulated in vitro with MBP, then purified T cells were transferred into Mgat5+/+ or Mgat5−/− PL/J mice for induction of EAE. Although the same myelin-reactive T cells were injected, the recipient mice deficient in Mgat5 displayed dramatically increased incidence and severity of EAE compared with wild-type mice (Figures 2(a) and 2(b)). At the peak of disease, less than 15% of the wild-type mice had disease, whereas greater than 60% of the Mgat5−/− mice displayed EAE (Figure 2(a)). Furthermore, the mean highest clinical score and disease duration were significantly increased in the Mgat5−/− mice (Figures 2(c) and 2(d)) (Table 1). The Mgat5−/− mice also appeared to have slightly greater weight loss, but this was not significantly different (Figure 2(e)). Splenocyte cultures from Mgat5−/− mice displayed increased levels of the proinflammatory cytokines IFN-γ and TNF-α when restimulated with MBP compared with wild-type mice (Figure 3). These results suggest that recipient mice lacking Mgat5-modified glycans are more susceptible to EAE autoimmune disease following adoptive transfer of encephalitogenic Mgat5−/− T cells, implicating cells of the host's own immune or central nervous system. This raises the possibility that Mgat5 deficiency in host, nonantigen-specific T cells and/or non-T cells (i.e., B cells, antigen-presenting cells, and nonimmune cells) contribute to increased susceptibility to inflammatory demyelination.
Figure 2.
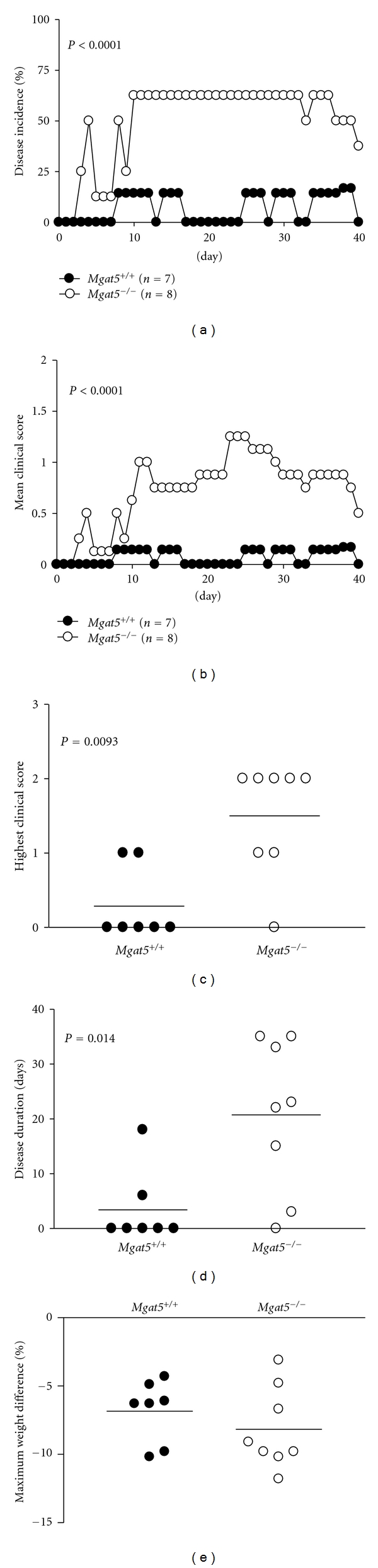
Mice deficient in Mgat5 are more susceptible to EAE. (a–e), Splenocytes were isolated from Mgat5−/− mice 10 days after immunization with MBP + complete Freund's adjuvant (CFA) and restimulated in vitro with MBP for two days. 2.7 million CD3+ T cells were injected into naïve Mgat5+/+ or Mgat5−/− mice and scored for clinical signs of EAE daily for 40 days. Mice were weighed daily throughout the duration of the experiment and used to determine maximum weight fluctuations. P value for EAE incidence was determined by Fisher's exact test. P values for EAE mean clinical score, disease duration, and the highest clinical score were determined by the Mann-Whitney test.
Table 1.
Clinical observations of adoptive transfer EAE.
| Genotype | n | Mean High Score | Incidence | Day of Onset | Mean Duration (Days) | |
|---|---|---|---|---|---|---|
| (Mean ± SEM) | (Day 20) | (Day 40) | (Mean ± SEM) | (Mean ± SEM) | ||
| P = 0.009 | P = 0.014 | |||||
| Mg at5+/+ | 7 | 0.3 ± 0.2 | 0% | 0% | 9.5 ± 1.5 | 3.4 ± 2.6 |
| Mg at5−/− | 8 | 1.5 ± 0.3 | 63% | 38% | 7.3 ± 1.8 | 20.8 ± 4.9 |
Mice were scored daily on a scale of 0–5 with: 0, no disease; 1: loss of tail tone; 2: hindlimb weakness; 3: hindlimb paralysis; 4: forelimb weakness or paralysis and hindlimb paralysis; 5: moribund or dead. P values for EAE mean high score and disease duration were determined by the Mann-Whitney test.
Figure 3.
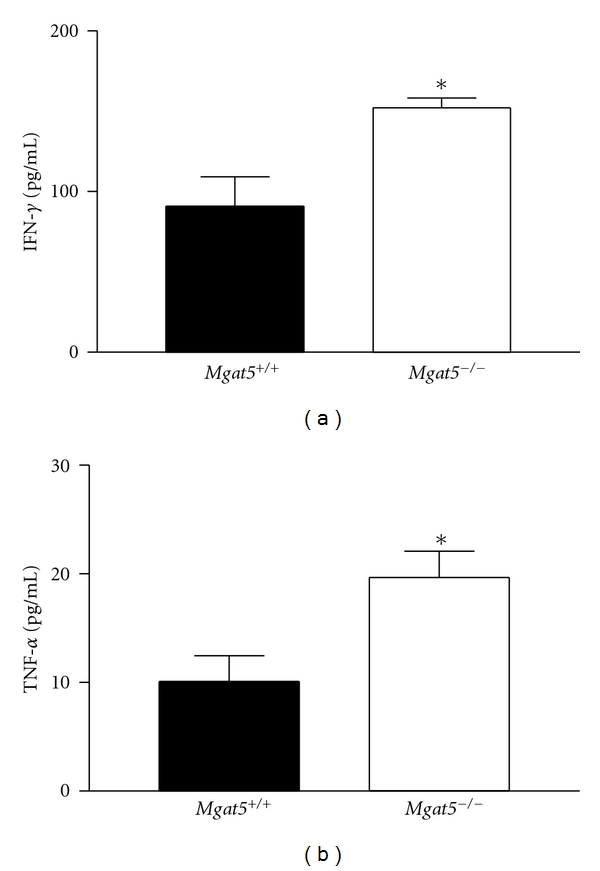
Mice deficient in Mgat5 have increased IFN-γ and TNF-α levels. Splenocytes were harvested from representative mice from both EAE groups and re-stimulated with 50 ug/mL MPB in vitro. Supernatants from splenocyte cultures were tested for IFN-γ and TNF-α levels by a bead-based analyte detection system using flow cytometry. *P ≤ 0.05. Error bars indicate the means ± S.E. of duplicate samples.
4. Discussion and Conclusions
During CNS inflammation, antigen-presenting cells (APC) such as invading macrophages and resident microglia can perpetuate the inflammatory milieu by secreting inflammatory factors and presenting myelin epitopes to autoreactive T cells. Endogenous presentation of myelin epitopes by APCs during acute inflammation can initiate epitope spreading and augment the progression of disease [27]. The differences in EAE observed in Mgat5+/+ versus Mgat5−/− mice following adoptive transfer of Mgat5−/− MBP-reactive T cells may result from a number of different mechanisms. Hyperactive endogenous T cells in Mgat5−/− recipients may respond more vigorously to epitope spreading, thereby enhancing disease. Mgat5−/− macrophages have impaired motility and phagocytosis, which may promote inflammatory demyelination and epitope spreading by inhibiting migration away from sites of demyelination and/or clearance of apoptotic cells [28]. Others have suggested that N-glycan branching deficiency in the kidney induced by Golgi α-mannosidase-II deficiency may trigger kidney autoimmunity via loss of self-recognition by the innate immune system [29], raising the possibility that reduced branching in oligodendrocytes may similarly activate innate immune cells. Galectin-1 has also been shown to regulate dendritic cell function by increasing tolerogenic signals to T cells and suppressing autoimmune neuroinflammation [30].
Multiple Sclerosis is also characterized by neurodegeneration. Although this may be triggered by inflammatory mediators and/or demyelination, deficiencies in N-glycan branching in neurons/axons may directly contribute to neurodegeneration independent from effects on inflammatory cells. The progressive MS-like disease that spontaneously develops in Mgat5−/− PL/J mice displays neuronal loss and axonal damage in both demyelinated areas and otherwise normal appearing white matter, the latter a hallmark of MS [17]. Moreover, neuron-specific deletion of Mgat1, a Golgi enzyme upstream of Mgat5 that eliminates all branching in N-glycans, results in apoptosis of adult neurons in vivo [31]. This confirms that N-glycan branching is required for neuronal viability in the adult CNS.
Restoration of neuronal integrity and regeneration of myelinated axons in the damaged CNS is another important mechanism to consider. Neural stem cells residing in the CNS are implicated in regenerating the damaged CNS, and, even within an acute inflammatory brain lesion, spontaneous remyelination occurs [32]. Recently, it has been shown that galectin-1 promotes proliferation of adult neural stem cells in the CNS through its carbohydrate-binding ability, suggesting that N-glycan branching deficiency may also limit regenerative mechanisms in MS and thereby promote progression [33]. Indeed, a polymorphism in Mgat5 strongly associates with disease severity in MS [10]. Future investigations are warranted to examine the many potential mechanisms by which deficiency of N-glycan branching by Mgat5 and other Golgi enzymes contribute to demyelinating disease initiation and progression.
Conflict of Interests
None of the authors declared conflict of interest.
Acknowledgments
Research was supported by the National Institutes of Health R01 AI053331 to M. Demetriou and F32AI081456 to A. Grigorian through the National Institute of Allergy and Infectious Disease as well as through a Collaborative Multiple Sclerosis Research Center Award to M. Demetriou.
References
- 1.Ebers GC, Sadovnick AD, Risch NJ, et al. A genetic basis for familial aggregation in multiple sclerosis. Nature. 1995;377(6545):150–151. doi: 10.1038/377150a0. [DOI] [PubMed] [Google Scholar]
- 2.Ebers GC, Bulman DE, Sadovnick AD. A population-based study of multiple sclerosis in twins. New England Journal of Medicine. 1986;315(26):1638–1642. doi: 10.1056/NEJM198612253152603. [DOI] [PubMed] [Google Scholar]
- 3.Baranzini SE, Mudge J, Van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464(7293):1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munger KL, Zhang SM, O’Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60–65. doi: 10.1212/01.wnl.0000101723.79681.38. [DOI] [PubMed] [Google Scholar]
- 5.Dyment DA, Sadovnick AD, Willer CJ, et al. An extended genome scan in 442 Canadian multiple sclerosis-affected sibships a report from the Canadian Collaborative Study Group. Human Molecular Genetics. 2004;13(10):1005–1015. doi: 10.1093/hmg/ddh123. [DOI] [PubMed] [Google Scholar]
- 6.Brynedal B, Duvefelt K, Jonasdottir G, et al. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PLoS ONE. 2007;2(7, article e664) doi: 10.1371/journal.pone.0000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nature Genetics. 2007;39(9):1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 8.Lundmark F, Duvefelt K, Iacobaeus E, et al. Variation in interleukin 7 receptor α chain (IL7R) influences risk of multiple sclerosis. Nature Genetics. 2007;39(9):1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- 9.Arthur AT, Armati PJ, Bye C, et al. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Medical Genetics. 2008;9, article 17 doi: 10.1186/1471-2350-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brynedal B, Wojcik J, Esposito F, et al. MGAT5 alters the severity of multiple sclerosis. Journal of Neuroimmunology. 2010;220(1-2):120–124. doi: 10.1016/j.jneuroim.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Schachter H. The “yellow brick road” to branched complex N-glycans. Glycobiology. 1991;1(5):453–461. doi: 10.1093/glycob/1.5.453. [DOI] [PubMed] [Google Scholar]
- 12.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 13.Mkhikian H, Grigorian A, Li CF, et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nature Communications. 2011;2, article 334 doi: 10.1038/ncomms1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grigorian A, Torossian S, Demetriou M. T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunological Reviews. 2009;230(1):232–246. doi: 10.1111/j.1600-065X.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau KS, Partridge EA, Grigorian A, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129(1):123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 16.Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409(6821):733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- 17.Lee SU, Grigorian A, Pawling J, et al. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. Journal of Biological Chemistry. 2007;282(46):33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- 18.Steinman L. Multiple sclerosis: two-stage disease. Nature Immunology. 2001;2(9):762–764. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- 19.Tranchant C, Bhatia KP, Marsden CD. Movement disorders in multiple sclerosis. Movement Disorders. 1995;10(4):418–423. doi: 10.1002/mds.870100403. [DOI] [PubMed] [Google Scholar]
- 20.Grigorian A, Lee SU, Tian W, et al. Control of T cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. Journal of Biological Chemistry. 2007;282(27):20027–20035. doi: 10.1074/jbc.M701890200. [DOI] [PubMed] [Google Scholar]
- 21.Calvert RC, Shabbir M, Thompson CS, Mikhailidis DP, Morgan RJ, Burnstock G. Immunocytochemical and pharmacological characterisation of P2-purinoceptor-mediated cell growth and death in PC-3 hormone refractory prostate cancer cells. Anticancer Research. 2004;24(5 A):2853–2859. [PubMed] [Google Scholar]
- 22.Toscano MA, Bianco GA, Ilarregui JM, et al. Differential glycosylation of T1, T2 and T-17 effector cells selectively regulates susceptibility to cell death. Nature Immunology. 2007;8(8):825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 23.Motran CC, Molinder KM, Liu SD, Poirier F, Miceli MC. Galectin-1 functions as a Th2 cytokine that selectively induces Th1 apoptosis and promotes Th2 function. European Journal of Immunology. 2008;38(11):3015–3027. doi: 10.1002/eji.200838295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nature Immunology. 2005;6(12):1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 25.Chen IJ, Chen HL, Demetriou M. Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling. Journal of Biological Chemistry. 2007;282(48):35361–35372. doi: 10.1074/jbc.M706923200. [DOI] [PubMed] [Google Scholar]
- 26.Chen HL, Li CF, Grigorian A, Tian W, Demetriou M. T cell receptor signaling co-regulates multiple golgi genes to enhance N-glycan branching. Journal of Biological Chemistry. 2009;284(47):32454–32461. doi: 10.1074/jbc.M109.023630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuohy VK, Yu M, Yin L, et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunological Reviews. 1998;164:93–100. doi: 10.1111/j.1600-065x.1998.tb01211.x. [DOI] [PubMed] [Google Scholar]
- 28.Partridge EA, Le Roy C, Di Guglielmo GM, et al. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science. 2004;306(5693):120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 29.Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth J. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity. 2007;27(2):308–320. doi: 10.1016/j.immuni.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Ilarregui JM, Croci DO, Bianco GA, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nature Immunology. 2009;10(9):981–991. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 31.Ye Z, Marth JD. N-glycan branching requirement in neuronal and postnatal viability. Glycobiology. 2004;14(6):547–558. doi: 10.1093/glycob/cwh069. [DOI] [PubMed] [Google Scholar]
- 32.Patani R, Balaratnam M, Vora A, Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathology and Applied Neurobiology. 2007;33(3):277–287. doi: 10.1111/j.1365-2990.2007.00805.x. [DOI] [PubMed] [Google Scholar]
- 33.Sakaguchi M, Shingo T, Shimazaki T, et al. A carbohydrate-binding protein, Galectin-1, promotes proliferation of adult neural stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):7112–7117. doi: 10.1073/pnas.0508793103. [DOI] [PMC free article] [PubMed] [Google Scholar]