

Abstract
We have developed an animal model of degeneration of the nigrostriatal dopaminergic neurons, the neuronal system involved in Parkinson's disease (PD). The implication of neuroinflammation on this disease was originally established in 1988, when the presence of activated microglia in the substantia nigra (SN) of parkinsonians was reported by McGeer et al. Neuroinflammation could be involved in the progression of the disease or even has more direct implications. We injected 2 μg of the potent proinflammatory compound lipopolysaccharide (LPS) in different areas of the CNS, finding that SN displayed the highest inflammatory response and that dopaminergic (body) neurons showed a special and specific sensitivity to this process with the induction of selective dopaminergic degeneration. Neurodegeneration is induced by inflammation since it is prevented by anti-inflammatory compounds. The special sensitivity of dopaminergic neurons seems to be related to the endogenous dopaminergic content, since it is overcome by dopamine depletion. Compounds that activate microglia or induce inflammation have similar effects to LPS. This model suggest that inflammation is an important component of the degeneration of the nigrostriatal dopaminergic system, probably also in PD. Anti-inflammatory treatments could be useful to prevent or slow down the rate of dopaminergic degeneration in this disease.
1. Introduction
The pathological hallmark of Parkinson's disease (PD), an age-related neurodegenerative disorder, is the specific and progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) [1]. This results in extrapiramidal motor dysfunction accompanied by progressive impairment of autonomy, mood, and cognitive functions [1–3]. Although some genes have been identified as responsible for rare familial early onset PD [4], its etiology remains elusive and many distinct possibilities have been pointed out for that. The effects of exogenous agents, infection, inflammatory response, and selective oxidative stress in the SN have been suggested as the most probable causes of the dopaminergic degeneration in the SN [5]. Many exogenous compounds have been described to produce degeneration of dopaminergic neurons in the nigrostriatal system, as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone and many other pesticides [6], which suggest an energetic metabolism impairment as main cause of the disease. Other possibilities were also proposed, as head trauma, encephalitis, reduced expression of trophic factors, alterations of the ubiquitin-proteasome system and neuroinflammatory mechanisms that are thought to collaborate in the progressive demise of SNpc neurons [1, 3, 7–13].
The implication of neuroinflammation as a component of the neurodegeneration progress of the disease was originally established in 1988 when the presence of activated microglia in the SN of PD was reported (see [14]; for review see [15, 16]). In that moment, the fact that many cases of PD were accompanied with a general brain inflammation along with the neurodegeneration was pointed out [14, 17, 18]. The dramatic proliferation of reactive amoeboid macrophages and microglia seen in the SN of parkinsonian brains, together with oxidative stress, was highly compatible with the existence of an inflammatory process similar to that previously described for Alzheimer's disease (AD) [19] and multiple sclerosis [20].
All these data allowed suggesting the implication of inflammation in the neurodegeneration of nigrostriatal dopaminergic neurons; however, its importance was not clear. Is this process exclusively involved in the progress of degeneration or could it have other implications? Is it able to induce the neurodegeneration of dopaminergic neurons?
To address these questions, we took advantage of lipopolysaccaride (LPS), a component of the Gram-negative bacteria cell wall. LPS is a potent inducer of inflammation and has diverse effects on cells of the immune system [21]. It was described that the intracerebral injection of LPS activated glial cells in vivo [22–26]; these microglial cells could contribute to the neurotoxicity by secreting neurotoxins [27–32] including inflammatory cytokines, N-methyl-D-aspartate (NMDA) receptor agonists, oxygen free radicals, and nitric oxide (NO). Besides, in vitro studies [33] showed that dopaminergic neurons were twice as sensitive to the toxic effects of LPS as tyrosine hydroxylase (TH) negative neurons. All these data suggested that the injection of LPS in different areas of the brain could be a good experimental model to ascertain the importance of inflammation in such different areas and neurons.
2. Nigral Dopaminergic Neurons Degeneration (Loss) Produced by the Inflammatory Processes Induced by the Intranigral Injection of LPS
We injected 2 μL of a solution containing 1 mg of LPS/mL and 1% Monastral blue inert tracer in phosphate-buffered saline [34] in the left SN of female rats previously anesthetized. The results showed a significant inflammation with activation of microglia, a typical characteristic of brain inflammation. This effect was accompanied by the loss of dopaminergic neurons (Figure 1) and the decrease of the intracellular content of dopamine (DA) and its metabolites in SN and striatum. These effects suggested the loss of dopaminergic neurons of SN along with its terminals in striatum. This was corroborated by the loss of TH activity, mainly in striatum. The highest loss was found 15 days after the lesion and maintained during the 21 days of the experiments. The average loss of the dopaminergic system was around 35%; at this level, rats showed no obvious motor disturbances. Another interesting result came from serotonin (5-HT); although 5-HT levels in the SN reached a minimum 15 days after the lesion, they recovered to the initial levels by day 21. These results obviously suggested that the degenerative process induced by inflammation (LPS) in the SN was specific for dopaminergic neurons and allowed us to suggest that the injection of a single dose of LPS within the SN could be an interesting model for studying the selective effects of inflammatory reactions on dopaminergic system, and potentially for studying PD. Moreover, taking into account that Andersson et al. [23] had shown a loss of pyramidal cells in the CA1 field in mice following an acute intrahippocampal infusion of 2 μg of LPS, we used this dose to study the specificity of the LPS effect in different areas and neurons of the brain. Specificity would obviously increase the importance of the effect.
Figure 1.

Effect of LPS on glial cells and dopaminergic neurons. (a) Injection of vehicle within the SN; (b) injection of LPS. OX-6 is a commercial antibody directed against a monomorphic determinant of the rat major histocompatibility complex (MHC) class II antigens, expressed by activated microglia but not for the resting cells. LPS increases OX-6 immunoreactivity around the injection track, filling the area of activated microglia characterized by its round morphology. On the contrary, there is an area lacking GFAP immunoreactivity, a marker of astroglia, around the injection site of LPS. As hallmark of this model, LPS induces the loss of dopaminergic (TH positive) neurons in the SN. Scale bar: 500 μm. (c) Represents the average values of some parameters in the SN (as percentage of controls) after the single injection of 2 μg of LPS: DA/TH/DAT, dopamine content, neurons expressing tyrosine hydroxylase, and dopamine transporter; OX-42/OX-6, density of activated microglial cells; amounts of the proinflammatory cytokines TNF-α and IL-1β, the adhesion molecule ICAM-1, the inducible nitric oxide synthase (iNOS), and the heat shock protein (HSP)-70; the phosphorylated (active) forms of the MAP kinases p38 (associated with promotion of apoptosis) and Akt (cell surviving signal). Alterations on the expression of GFAP and the endothelial barrier antigen (EBA), as area lacking expression (in mm2), are also shown.
3. The SN Is the Most Sensitive Area of the Brain to the Inflammation Induced by the Injection of LPS, Which Produced a Selective Degeneration of Dopaminergic Neurons
We injected 2 μL of the LPS solution described before into four different locations: the left SN, the medial forebrain bundle (MFB), the striatum, and the dorsal raphe nucleus (DR) [35]. LPS induced a strong macrophage/microglial reaction in SN, with a characteristic clustering of macrophage cells around blood vessels. These macrophage cells contained blue particles inside (from Monastral blue), indicating phagocytic activity. Fifteen days after injection, the number of lipid-laden cells began to decrease gradually and 1 year after injection most of the OX-42-positive cells showed ramified morphology. The SN was far more sensitive than the striatum and the other areas studied to the inflammatory stimulus. Moreover, only the dopaminergic neurons of the SN were affected, with no detectable damage to either the GABAergic or the serotoninergic neurons. The damage to the DA neurons in the SN was permanent, as observed 1 year after injection. Moreover, the injection of LPS into MFB did not produce any significant effect on the inflammation process, nor neuronal degeneration. Similar results were found when injections were made within the striatum or the DR. Therefore, these results seem to support that the injection of a single dose of LPS within the SN produced a selective effect on the dopaminergic neurons. Moreover, this work pointed out that inflammation was able to induce the degeneration of nigral dopaminergic neurons; the induction of inflammatory processes could be involved in the degeneration of dopaminergic neurons after injury (as boxing) or infection; it could also be involved in the progression of neuronal degeneration induced by other causes. Kim et al. [36] published the same conclusions. They also studied the effect of LPS injection within hippocampus and found no significant induction of inflammation or neuronal degeneration. Neurotoxicity after intraparenchymal injection of LPS has also been reported by other authors [37, 38].
4. Protective Effect Produced by the Treatment with Anti-Inflammatory Compounds on This Neurodegenerative Model
Obviously, the possible protective effect of an anti-inflammatory compound would support this model along with the importance of the inflammatory response in the LPS-induced degeneration of the nigral dopaminergic neurons. Inflammation is an attractive pharmacological target since it progresses over several days after injury and because intervention with anti-inflammatory agents may not result in intolerable side-effects [39].
4.1. Dexamethasone
Thus, we used dexamethasone, a potent anti-inflammatory drug. It was known that glucocorticoids were potent anti-inflammatory drugs that have long been used in clinical neurology for the treatment of brain inflammation [40, 41] and spinal cord injury [42]. They are potent inhibitors of the IFN-induced activation of microglia and macrophages in vitro [43] and downregulates the expression of the major histocompatibility complex (MHC) class II molecules on macrophages, both in vivo and in vitro [44]. Dexamethasone prevents the induction of cyclo-oxygenase (COX)-2 mRNA and prostaglandins in the lumbar spinal cord following intraplantar injection of Freund's complete adjuvant, in parallel with inhibition of edema [45]. Moreover, dexamethasone down-regulates the expression of MHC class II on rat microglia [43, 46] and reversibly inhibits the microglial proliferation in vitro [47] induced by axotomy and IFN-γ.
When animals were treated with dexamethasone [48], we found a significant protection against the damage caused by the intranigral injection of LPS. The treatment with dexamethasone interferes with many of the features characterizing proinflammatory glial activation, proliferation and also the upregulated expression of the MHC class II produced by LPS. We found that dexamethasone inhibited not only the number of OX-42-positive cells but also the number of microglia/macrophages expressing MHC class II antigens as revealed by staining with OX-6. These observations were in agreement with previous works reporting that glucocorticoids downregulate MHC class II [43, 44, 46].
Dexamethasone treatment was also able to prevent the loss of DA and its metabolites in SN and striatum. Similarly, it also prevented the loss of TH (measured by its activity and by TH immunostaining) in SN and striatum. These results confirmed that inflammatory response was implicated in LPS-induced neurodegeneration and open the possibility to use anti-inflammatory drugs to slow down the progress of the disease.
4.2. Minocycline
We have used other anti-inflammatory compounds; minocycline is a semisynthetic tetracycline derivative that is able to cross the BBB, reaching the cerebrospinal fluid [49]. Moreover, this new antibiotic exerts anti-inflammatory effects that are completely separated and distinct from its antimicrobial origin [50–52]; its use in neurodegenerative diseases has been suggested. Minocycline has been shown to protect hippocampal neurons against global ischemia [53] and to reduce cortical infarction volume in a rat model of focal brain ischemia [54]. It is also a very efficient neuroprotective agent in animal models of traumatic brain injury [55], multiple sclerosis [56], Huntington's disease [57], and amyotrophic lateral sclerosis [58]. Moreover, other studies had also demonstrated a neuroprotective effect of minocycline over the dopaminergic system in response to either MPTP [59, 60] or 6-OHDA [61].
When we evaluated the potential neuroprotective activity of minocycline in our animal model of Parkinson's disease induced by the intranigral injection of LPS [62], we found it was very effective preventing different inflammatory features along with the loss of nigral dopaminergic neurons. Minocycline treatment highly prevented the activation of reactive microglia as visualized by OX-42 and OX-6 immunohistochemistry, as well as the induction of interleukin (IL)-1α and tumor necrosis factor (TNF)-α mRNAs. Moreover, minocycline treatment also partially prevented the loss of dopaminergic neurons (12% against 50%) produced by LPS.
4.3. Simvastatin
At this time, some experimental and clinical evidence indicated that statins—extensively used in medical practice as effective lipid-lowering agents through the inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase—had other cholesterol independent effects, as improving endothelial function, decreasing oxidative stress, inhibiting the thrombogenic response in the vascular wall, immunomodulatory and, anti-inflammatory properties [63–67]. Thus, we investigated the influence of simvastatin on the degenerative process of the dopaminergic neurons in our animal model of Parkinson's disease [68, 69]. In these works, we found that simvastatin treatment prevented the inflammatory process induced by LPS. Simvastatin prevented the bulk microglial activation found after LPS injection, reducing the number of microglia/macrophages expressing MHC class II antigens (40% of LPS group) as revealed by immunostaining with OX-6; it also inhibited the activation of IL-1β, TNF-α, and inducible nitric oxide synthase (iNOS) produced by LPS. These effects could be, at least, one of the causes of the prevention of dopaminergic neuronal loss induced by LPS produced by the simvastatin treatment. However, simvastatin showed another important effect that could help to the survival of dopaminergic neurons: simvastatin produced the activation of the neurotrophic factor BDNF, along with the prevention of the oxidative damage to proteins, conditions in which BDNF has an important protective effect. Moreover, it also prevents the main changes produced by LPS on different mitogen-activated protein kinases, featured as increases of P-c-Jun N-terminal protein kinase, P-extracellular signal-regulated kinase, p-38, and P-glycogen synthase kinase and the decrease of the promotion of cell survival signals such as cAMP response element-binding protein and Akt. Our results suggest that statins could delay the progression of dopaminergic degeneration in disorders involving inflammatory processes.
Many other anti-inflammatory compounds have been described to act as neuroprotectors in this and other models of Parkinson's disease. Nalaxone, an opiate receptor antagonist, and its inactive isomer (+)-nalaxone, inhibited both the activation of microglia and the loss of dopaminergic neurons induced by LPS in the SN [37, 38]. This effect was improved when nalaxone was combined with indomethacin [70]. Triptolide, another anti-inflammatory compound, also prevents the dopaminergic degeneration induced by LPS intranigral injection [71]. A similar effect was also produced by silymarin [72] and catalpol [73], other anti-inflammatory compounds.
5. Why Does the SN Have a Special Sensitivity to the Inflammation Induced by the Injection of LPS?
As described above, our results showed that the inflammatory reaction was stronger in SN than in striatum and other areas studied [34–36]. This was also clear in the characteristic disappearance of the GFAP immunoreactivity at the LPS injection site. We suggested at that moment that the differences could be due to specific structural differences between the SN and other areas. Regarding this, it was thought that SN had the highest concentration of microglia in the brain [74] and consequently a high concentration of the inflammation-related factors produced by these cells, such as TNF-α and NO. This proposal is supported by the fact that many anti-inflammatory compounds protect dopaminergic neurons. However, other authors have reported more recently that in intact brain, the densities of CD11b+ microglía are similar in SNpc and cortex, although LPS injection enhanced the number of CD11b+ cells in the former but not in the latter [75].
Other possibilities have been pointed out, as the increase in vascularization induced by inflammation in the SN, also described in a MPTP model of PD that is accompanied by the increase in the vascular endothelial growth factor (VEGF) [76]. One of the effects of the change could be the increase in the BBB permeability that could have some influence in the sensitivity, since the intranigral injection of VEGF also induce the degeneration of the dopaminergic system [77]. Another important consequence could be the infiltration of peripheral monocytes/macrophages, which could act as protector or neurotoxic [78]. These circumstances could increase the sensitivity of SN to inflammation.
6. Why Are the Dopaminergic Neurons of the SN Especially Affected in Our Inflammation Model?
We had described that dopaminergic neurons were especially sensitive to inflammation, and this did not occur when LPS was delivered within the MFB (dopaminergic axons) or the striatum (dopaminergic terminals). The main suggestion was that SN was highly vulnerable to oxidative damage [79–82], taking into account its reduced antioxidative capability along with its high content on iron and DA [83]. However, this rationale did not account for the special sensitivity of the DA neurons with respect to other non-DA neurons within the SN, such as the GABAergic ones. Although unexplained, this is very interesting for Parkinson's disease, in which the dopaminergic neurons are especially sensitive, probably not only due to the inflammatory process.
To answer this question, we studied the possible influence of DA. There was a general agreement on the toxic capability of DA, which generates redox metabolites including semiquinone, quinone, zwitterionic 5,6-hydroxyindoles, and possibly oxygen free radicals. Theoretically, this neurotoxicity also accelerates autooxidation of the released DA, which results in the generation of free radicals in vivo (Figure 2). Enhanced DA autoxidation and oxygen free radicals may initiate a cascade of oxidant stress, leading to injury and loss of SNc neurons in Parkinson's disease. This possibility is supported by increased basal malonaldehyde formation (lipid peroxidation) and iron content in surviving pigmented SN neurons [80, 84, 85].
Figure 2.
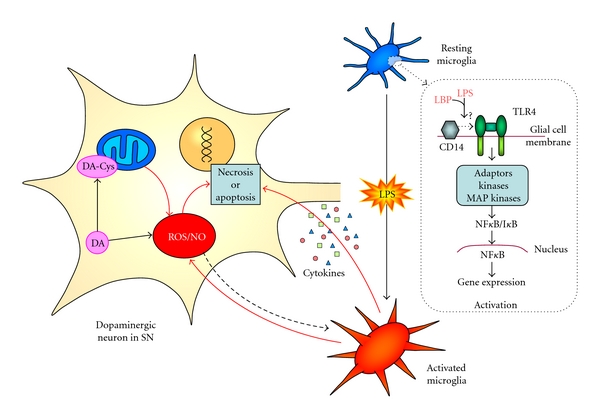
Interaction between glia and neurons may lead to neuron damage and death. LPS activates microglia from a resting state to an activated one, probably through the CD14 and the TLR4 receptors; this signalling pathway, mediated by different molecular adaptors, kinases, and MAP kinases, activates NF-κB with the consequent transcription of specific genes leading to microglial activation. Then, activated microglial cells release several compounds, as proinflammatory cytokines, radical oxygen species (ROS), and NO that may eventually lead to neuronal death. Within dopaminergic neurons, the mitochondrial respiratory chain can be affected by several substances, leading to energy failure, production of ROS, and reduction of the neurons viability. ROS can act as signal for the activation of microglia, indicating that neurons are not healthy. DA can exert a toxic action through the ROS formed in its oxidative metabolism; it may also forms complexes with cysteine, inhibiting the respiratory chain and producing more ROS. The reduction/elimination of microglial induction could ameliorate neuronal damage.
To test the involvement of DA in the degeneration induced by LPS, we treated albino Wistar rats with different concentrations of α-methyl-p-tyrosine (α-MPT), an inhibitor of TH activity that blocks synthesis of cytoplasmic DA [86, 87]. Pretreatment with α-MPT reduces DA content by 79% [88]. Results showed that α-MPT prevented, in a dose-dependent manner, the inflammatory features induced by LPS: activation of microglial cells (characterized by a change of morphology from resting, ramified cells with thin processes, to reactive microglial cells that eventually become phagocytic), loss of the astroglial population accompanied by the appearance of GFAP immunoreactive debris from hypertrophic, degenerating astrocytes, loss of TH immunostaining, and expression of mRNA for TH and DA transporter (DAT). These protective effects resulted from inhibition of TH and the consequent decrease in DA concentration, since cotreatment with L-DOPA/benserazide, which bypasses TH inhibition increasing the extracellular levels of DA in the SN approximately fourfold with respect to basal levels [89], avoided the protective effects induced by inhibition of TH produced by α-MPT and restored the degenerative effects induced by LPS on dopaminergic neurons as well as the loss of astrocytes. All these results strongly supported the implication of DA in the dopaminergic neurodegeneration induced by LPS in the SN. Moreover, not only did DA seem to be involved in dopaminergic degeneration of the SN but it could also be responsible for the special sensitivity of the SN to the inflammation induced by LPS. The SN inflammatory response to LPS injection after TH inhibition was similar to that described for other brain areas, where LPS had little or no effect on neuron degeneration or the glial inflammatory process [35, 36]. However, the treatment with LPS + α-MPT + L-DOPA/benserazide did not produce activation of microglial cells, although it induced damage to astroglia. This result is quite intriguing and could be involved in the inflammation process in SN. These results point out the important contribution of DA to the vulnerability and degeneration of dopaminergic neurons of the SN. Further knowledge about the involvement of DA in this process may lead to the possibility of new protection strategies against this important degenerative process.
7. Other Natural and Endogenous Compounds Able to Induce the Inflammatory Process along with the Degeneration of Dopaminergic Neurons by Intranigral Injection
LPS is a compound derived from de bacterial cell wall; thus, it could be considered an exogenous substance to the nervous system; other natural and endogenous compounds have been described to have similar properties on this process.
7.1. Histamine
We have evaluated the effects of a direct infusion of histamine (HA) in SN, striatum, medial septum, and medial lemniscus. There were many reasons for carrying out these experiments. Histamine is an important component of mast cells of the brain and participates in the enhancement of microvascular permeability, leukocyte rolling, adhesion, and extravasations of inflammatory cells into the brain and spinal cord. These events are important in many inflammatory diseases of the CNS, such as encephalomyelitis and multiple sclerosis [90]. It is also released after trauma, ischemia, seizures, and inflammation [91]. Moreover, the brain also has histaminergic neurons located in the tuberomammillary nucleus of the posterior hypothalamus nucleus that project to almost all regions of the brain from the olfactory bulb to the spinal cord [92]. This neuronal HA acts as neuromodulator or neurotransmitter [93, 94], participating also in many physiological functions [93–95]. It also interacts with some neurotransmitter as DA [96–101]. This relationship could be involved in the interaction between central HA neurons with the extrapyramidal system. It is known that HA induces catalepsy, which has been suggested as an animal model of Parkinson's disease [102]. HA antagonists are used as antiparkinsonian drugs [103].
HA has been shown to be involved in neuronal degeneration [104] and neurotoxicity [105]. Wernicke's encephalopathy is a disorder characterized by selective pathologic damage to the midline thalamus, mammillary bodies, and certain brainstem nuclei. Thiamine deficiency is a critical factor in the etiology of this disorder. Langlais et al. [104] using a rat model of Wernicke's encephalopathy induced by an acute bout of pyrithiamine-induced thiamine deficiency (PTD), pointed out that HA mediated neuronal death in this model.
In our study [106], the injection of histamine in SN produced an acute inflammatory response manifested by an activation of microglia, where OX-42-immunoreactive microglia exhibited typical features of phagocytic cells. In addition, a marked induction of MHC class II antigen expression and a characteristic disappearance of the GFAP immunoreactivity were produced in the same area. These effects were accompanied by a selective damage in dopaminergic neurons, evidenced by the loss of tyrosine hydroxylase mRNA-expressing cells, tyrosine hydroxylase-immunolabeled-positive cell bodies, and a decrease of DA and 3,4-dihydroxyphenylacetic acid levels. However, HA injection had little effect in other areas; only high doses produced an evident dopaminergic terminal degeneration. Moreover, similarly to that described for LPS, HA produced the specific degeneration of DA neurons of SN; HA did not affect GABAergic neurons, alter the pattern of ChAT mRNA-expressing cells, or change the pattern of 5-HT-immunolabeled cells when it was injected in SN, septum, or the medial lemniscus, respectively. We conclude that, similarly to LPS, HA is a selective neurotoxin for dopaminergic cells.
HA effect must be related to its inflammatory capability along with the rapid and reversible increase in the BBB permeability, producing a disturbance of the normal brain homeostasis. Moreover, a direct neurotoxic effect of HA over the nigral dopaminergic population should never be discarded, since there are reports on the induction of neuronal death by HA [104, 105]. In spite of the strict and remarkable selective neurotoxic action of HA on the dopaminergic system, HA may be taken into account in relationship to its involvement in the etiopathology of PD. So, higher blood HA levels have been found in patients with PD than in healthy controls [107]. However, central histaminergic systems are not affected in PD [108]. It has been shown that mast cells can rapidly penetrate brain blood vessels from extraneuronal sources [109]. It is interesting to note that therapies used in PD (L-DOPA + carbidopa) decrease the blood HA concentration [107], probably due to the decrease in its formation [110]. In addition, HA antagonists have long been used as antiparkinsonian drugs [103]. Patients with idiopathic PD treated with famotidine, an HA H2 antagonist, demonstrated improvement in their motor symptoms [111]. In addition, some HA metabolites and derivatives have been correlated with the degree of severity in PD patients. Levels of pros-methylimidazolacetic acid in cerebrospinal fluid showed a high positive correlation with the severity of PD in a group of not medicated, mildly to moderately affected patients [112, 113]. Further indications supporting the involvement of HA in the etiopathology of PD come from the observation that an increase in HA concentration was associated with the appearance or worsening of parkinsonian symptoms. Thus, acute tacrine administration produced tremulous jaw movements that shared some characteristics with parkinsonian tremor [114–116]. Tacrine is an inhibitor of brain histamine N-methyltransferase, which increases brain HA concentration [117]. Combined treatment with tacrine and haloperidol increases the parkinsonian-like symptoms [118, 119]. Secondary Parkinsonism has been described in neurocysticercosis [120, 121], characterized by a diffuse brain edema [122] generally accompanied by an increase of HA levels.
In conclusion, we showed for the first time that HA was able to produce a specific degeneration of dopaminergic neurons in SN along with a highly inflammatory process. The dose of HA needed to produce this effect was remarkably lower than that described in other studies. Besides, many of the relationships between elevated HA concentration and parkinsonism-like symptoms were pointed out. All these data strongly suggested an important relationship between elevated concentrations of HA and degeneration of the nigrostriatal dopaminergic system, which requires further investigation.
7.2. Thrombin
Thrombin is a multifunctional serine protease, best known for its role in the blood coagulation cascade. It is derived from its zymogen, prothrombin, and converts fibrinogen into fibrin. It has many functions: activates platelets, stimulating the proliferation of vascular smooth muscle cells [123–125]; it is involved in the chemotaxis and adhesion in monocytes, macrophages, and neutrophils (for review, see Grand et al. [126]); it has specific effects in cells of the CNS, regulating neurite outgrowth in neuronal cells [127] and inducing the proliferation of astrocytes [128]; it is also able to induce the activation and proliferation of cultured rodent microglia along with the production of nitric oxide (NO); it evokes the release of cytokines and chemokines [129] and induces the proliferation of astrocytes [128, 130]. The latter effects could be important in relationship to inflammatory processes. It is also interesting that brain contains this and some other proteases, along with protease-activated receptors (PARs) that serve key roles in blood coagulation and wound healing. These proteases have actually been found in the brain in situations in which the BBB is compromised, with a possible role in the neuropathological triggering of this situation (for review, see Gingrich and Traynelis [131]). Taking into account all these properties, along with the possible effect of thrombin in stroke, we studied thrombin effects on the nigrostriatal dopaminergic system.
Following the experimental procedure carried out in our previous works, we injected different amounts of thrombin into the nigrostriatal pathway [132]. Seven days after injection, we found a strong inflammatory response in the SN with a strong macrophage/microglial reaction pointed out by immunostaining (using OX-42 and OX-6 antibodies) and the induction of iNOS, IL-1α, Il-1β, and TNF-α. These results showed that the infusion of thrombin induced an inflammatory process in a similar way to other proinflammatory compounds such as LPS, probably by the activation of microglial cells, as described for microglial cultures [129]. The thrombin-induced inflammation was produced by its biological activity, maybe through the protease-activated receptors- (PAR-) receptor, since the injection of heat denatured thrombin or thrombin plus α-NAPAP (a specific inhibitor of thrombin) did not induce these processes, or they were strongly reduced. Moreover, selective damage to dopaminergic neurons was produced after thrombin injection, evidenced by loss of TH immunostaining and TH mRNA expressing cell bodies, and the unaltered transcription of glutamic acid decarboxylase (GAD) mRNA in the SN and striatum. These effects were also produced by its biological activity since they almost disappeared when thrombin was heat-inactivated or injected with its inhibitor α-NAPAP.
These thrombin effects were even more interesting if we remember that thrombin is activated from prothrombin in the coagulation cascade and also that it enters interstitial fluid during penetrating head wounds, hemorrhagic stroke, rupture of cerebral aneurysms, and arteriovenous malformations. In addition, BBB breakdown associated with cerebrovascular insults reflects a largely nonselective increase in the permeability of brain capillaries and tight junctions to high molecular weight proteins. Moreover, taking into account that prothrombin circulates in blood at high concentrations (1 μM) [133] and vascular injury triggers its rapid conversion to thrombin, direct entry of thrombin into the interstitial space with the consequence of a significant increase in its concentration is plausible [134]. Moreover, the elevation of thrombin levels in cerebrospinal fluid from 100 pM to 25 nM for a period of more than a week has been reported in subdural hematoma [135]. This suggests that appreciable amounts of thrombin can be generated and persist at sites of cerebrovascular injury. When bleeding occurs directly within brain tissue, active thrombin, and other proteases will freely penetrate the interneuronal spaces by diffusion until clotting closes the injured vessels and thrombin becomes depleted from the clot. High concentrations of thrombin, such as those that can be produced by a cerebral hemorrhage, appear to cause brain damage [133, 136] and may also contribute to damage in Alzheimer's disease and vascular dementia [137–139]. Vascular Parkinsonism has also been reported [140]; moreover, features of Parkinsonism have been seen in up to 38% of lacunar stroke patients at one year [141].
All these data provided evidence that thrombin activates microglia along with other inflammatory features in the SN, resulting in specific degeneration of dopaminergic neurons. These results suggest that thrombin, as an endogenous neurotoxin, could be implicated in dopaminergic neuron degeneration. Besides PD, these features could also be of relevance for a wide range of neurological diseases which present pathological signs of inflammation, such as cerebrovascular diseases and CNS trauma.
Similar results were described by Choi et al. [142]. It is interesting to note that the inflammatory process and the degenerative induction produced by intranigral injection of thrombin are not overcome by the treatment with dexamethasone as anti-inflammatory agent [143]. In fact, when we studied the possible protection exerted by dexamethasone in the thrombin paradigm, we found unexpected results. Dexamethasone was able to prevent partially the loss of astrocytes but was unable to overcome other inflammatory features, as the microglial proliferation induced by thrombin. Moreover, this treatment resulted in a greater loss of dopaminergic neurons that was accompanied by the accumulation of α-synuclein in dopaminergic neurons, probably produced by the diminution of its degradation, an effect described to be produced by dexamethasone on the beta-amyloid [144]. Dexamethasone produced other effects, as the consequent aggravation of the ischemic neuronal damage in striatum [145] and hippocampal CA1 area [146] as the extracellular accumulation of glutamate increased and thereby the overload of cellular calcium [147].
We also found that dexamethasone produced a significant decrease in PAR-1, which was increased by thrombin plus dexamethasone; nexin-1, the most abundant, potent endogenous thrombin inhibitor in brain [148], was not also affected by dexamethasone alone but was increased by thrombin; the effect was greater and in the same direction when thrombin and dexamethasone were used together. Dexamethasone produced a greater amount of oxidative stress (measured as carbonyl groups) than thrombin, in a similar extension that in animals treated with thrombin plus dexamethasone. We suggest that this increase of oxidative stress could be the cause of the great damage produced by dexamethasone. The cause of the oxidative stress is the increase of DA metabolism produced by the release of DA along with its metabolism through MAO enzymes. It is known that MAO-A is induced by glucocorticoids in tissues as human skeletal muscle [149] or bovine adrenal endothelial cells [150]. Dexamethasone increased the expression of MAO-B in rat astrocytes, which was inhibited by the glucocorticoids receptor inhibitor RU486 [151]. Moreover, the relationship between MAO-dependent H2O2 overproduction and degenerative processes had been suggested in PD [152]. These data suggested that H2O2 produced by MAO played an important role in the induction of apoptosis under some conditions. In our experimental design, both MAO A and B increased in animals treated with dexamethasone. Moreover, when we blocked MAO activity with tranylcypromine, the death of dopaminergic neurons caused by the combined treatment with thrombin and dexamethasone was prevented. This supports that induction of MAO activity by dexamethasone might play an important role in the lack of protection against thrombin observed in the animals treated with this glucocorticoid. This result is also in agreement with Tazik et al. [153], who recently described the protective effect of rasagiline, an inhibitor of MAO B, on dexamethasone-induced brain apoptosis. Increase of DA metabolism produced by dexamethasone through the induction of MAOs could be important in dopaminergic neurons, since DA seems to play an active role in their degeneration [154].
7.3. Tissue Plasminogen Activator (tPA)
tPA is a highly specific serine proteinase and one of the two principal plasminogen activators. It is widely distributed in all brain regions across the neuroaxis, being more elevated in the spinal cord and the SN [155, 156]. Different functions have been suggested for tPA in the CNS; for our work, the most important was the neurotoxicity effect [157–162]. Different animal models had demonstrated that the genetic deficiency of tPA [159, 163] and its inhibition with the natural tPA inhibitor neuroserpin [164, 165] were associated with a decrease in infarct volume and significant neuronal survival compared with control animals. Moreover, tPA plays important roles in blood coagulation and fibrinolysis outside the CNS. By cleaving the precursor molecule plasminogen, plasminogen activators produce the active enzyme plasmin, which dissolves fibrin-based clots in focal cerebral ischemia. Clinical trials have demonstrated that treatment with plasminogen activators in selected patients can improve outcome after acute ischemic stroke [166]. Moreover, tPA is the only drug approved for the treatment of thromboembolic stroke, but it might lead to some neurotoxic side effects. Some of these effects could be related with the fact that tPA, as thrombin, is able to induce microglia [167], probably with the consequent activation of an inflammatory process that could be responsible for some of the neurotoxic effects reported after its pharmacological use.
We have used the intranigral injection of tPA, which let us to describe its effects under standardized conditions. We found that injection in the SN led to the induction of inflammatory process, microglial activation, loss of astroglia, and increase in the expression of nNOS, iNOS, and aquaporin 4, along with the disruption of the BBB integrity. This latter result is in agreement with Yepes et al. [168] and Goto et al. [169] which showed that recombinant tPA promotes acute direct neurotoxicity with disruption of the BBB and increased formation of edema. Moreover, we also found a dose-dependent loss of dopaminergic neurons, pointed out by TH-immunoreactivity and changes in the expression of TH and DAT mRNAs. The toxic effect of tPA seemed to be specific on dopaminergic neurons of the SN since the GABAergic population of the ventral midbrain was not affected.
These results could be related to—and could also justify—the possible neurotoxic effect produced by tPA in those pathological conditions which result in a significant increase of its production and release. It is known that endogenous tPA activity increases significantly by ischemia [159]; this effect is prevented in animals with genetic deficiency of tPA [159, 163] and also by the treatment with the natural tPA-inhibitor neuroserpin [164, 165]. Ischemia should be produced early in the SN to observe the degeneration of dopaminergic neurons induced for tPA. However, we must note that we carried out other experiments in which we injected tPA intravenously at nearly clinical concentration, along with the injection of saline solution in the SN in order to disrupt the BBB; we did not find a significant degeneration of the dopaminergic system, suggesting that the damage observed in our model should be produced by the increase in the endogenous concentration of tPA and its extravasations by isquemia.
7.4. Trisialoganglioside
The intranigral injection of Trisialoganglioside GT1b, one of the major brain gangliosides, induced a great inflammatory response along with the degeneration of dopaminergic neurons [170]. This is also relevant since gangliosides, a component of membranes, are also involved in some neurodegenerative processes in which they accumulate. The ability of these compounds to induce inflammation could be important.
8. Peripheral Inflammation Enhanced the Inflammation Produced by the Intranigral Injection of LPS and, Consequently, the Degeneration of Dopaminergic Neurons
As we have described above, evidence suggesting that inflammation may play a central role in the cell loss seen in PD has been accumulating during years (see [14]; for review see [15, 16]). Moreover, some relationship between central inflammation and peripheral could be possible. It is known that the chance of developing AD is increased twice in aged persons exposed to systemic infections [171]. Furthermore, Strang [172] described the increased prevalence of peptic ulcer prodromal to idiopathic Parkinsonism [173]. Thus, we were interested in using our animal model of PD to study the implication of peripheral inflammation [174]. To achieve peripheral inflammation, we used a model of ulcerative colitis (UC) induced by dextran sulphate sodium (DSS) ingestion [175]. We described an increase in the levels of inflammatory markers from serum (TNF-α, IL-1β, IL-6, and the acute phase protein C-reactive protein). Moreover, it also increased the inflammatory parameters in SN (TNF-α, IL-1β, IL-6, iNOS, intercellular adhesion molecule-1 (ICAM-1), and microglial and astroglial populations) caused by the intranigral injection of vehicle in animals with UC. Consequently, peripheral inflammation induced by UC increased all the LPS-induced inflammatory markers examined in the ventral mesencephalon, demonstrating that SN becomes more sensitive to central inflammatory stimulus under conditions of peripheral inflammation. The inflammatory response of SN was associated to increased vulnerability of its dopaminergic population. These results suggest that inflammation produced in a peripheral organ (intestine) could induce loss of dopaminergic neurons in the SN, enhancing the inflammation and the dopaminergic degeneration induced in the SN by a previous inflammation (LPS). Moreover, we also studied the possibility that leukocyte infiltration through an impaired BBB could be involved in the deleterious effects of systemic inflammation on nigral dopaminergic population under conditions of brain inflammation. Brochard et al. [176] had shown lymphocyte T infiltration into brain parenchyma in MPTP-intoxicated mice showing induction of ICAM-1 but no BBB disruption. Our analysis demonstrated that UC induced ICAM-1 levels in the ventral mesencephalon; flow cytometry analyses showed that the amount of circulating monocytes was higher in animals from the LPS and UC groups (around fourfold) than in the control animals. Infiltration in the UC + LPS group increased nearly eightfold. Moreover, using the intravenous injection of clodronate encapsulated in liposomes (ClodLip), which produced a peripheral macrophage depletion lasting 5 days in blood, liver and spleen of normal rats and mice [177–179], we found the reversion of the deleterious effect of peripheral inflammation on microglial activation, BBB disruption, astrocytes loss, and degeneration of nigral dopaminergic neurons induced by LPS. Taken together, our results demonstrate that peripheral inflammation induced by UC contributes to dopaminergic degeneration. Activation of macrophages seems a decisive factor, since destruction of this peripheral leukocyte type by ClodLip abolishes the nocive effects on the ventral mesencephalon associated with UC. This study shows that BBB disruption may increase brain susceptibility to subsequent exposure to circulating leukocytes. This study may also shed light into previous observations; it described the existence of an increased prevalence of peptic ulcer prodromal to idiopathic Parkinsonism [172, 180]. This has prompted some authors to suggest a prominent role of inflammation in gastrointestinal tract in the etiology and pathogenesis of idiopathic Parkinsonism, including a possible role for Helicobacter pylori infections [173, 181]. This infective process is the most prevalent in the world, affecting approximately 50% of the population [182], and it is considered the causative agent of many gastrointestinal and extradigestive conditions. Colonization of gastric mucosa by H. pylori is accompanied by an inflammatory response associated with gastric mucosal damage through the activation of polymorphonuclear neutrophil leukocytes [183], inflammatory infiltration of lymphocytes, plasma cells and macrophages in the stomach tissue [184–186], and the production of pro-inflammatory factors such as IL-8, IL-1, and TNF-α [187–189].
9. Conclusions
On 1998, we proposed an animal model of PD based on the inflammatory process triggered by the intranigral injection of LPS. Afterwards, this model was further developed and corroborated that the SN had the highest response to inflammation process induced by LPS with respect to other brain areas studied. In addition, one of the responses to inflammation was the specific degeneration of dopaminergic neurons. The implication of inflammation in the degeneration of dopaminergic neurons was also supported by the protection produced by many different anti-inflammatory compounds. The interest of this model is to point out the fact that inflammation is the inductor of the degeneration of dopaminergic neurons, probably in cases as injury, boxing, infection, and others; it could be important in the progression of the disease at the same time that it could enhance the damage induced by the main (unknown) cause of the disease. Moreover, the interest of the scientific community in inflammation in PD has increased significantly afterwards. In all cases, the anti-inflammatory pharmacology could be important in the disease, and this model could be also very useful to test anti-inflammatory treatments. Moreover, the model has allowed pointing out that any endogenous substance able to induced inflammation or activation of microglia produced similar effects to LPS. Many endogenous compounds have been used in this way, as histamine, thrombin, or tPA among others. Some of them could have a direct effect in this disease, as stroke or infection. With respect to the special sensitivity of the SN to inflammation, there are no clear answers, but its knowledge would be of interest for the treatment of PD. The special sensitivity of dopaminergic neurons to inflammation is also of special interest; in this case, however, the increase of free radicals production through MAO enzymes and the effect of DA seem clear. Our model make possible to study other processes, as peripheral inflammation, which could enhance the effects produced by central inflammation.
Acknowledgments
This work was supported by Grant no. SAF2008-04366. M. Sarmiento thanks Junta de Andalucía for a Beca de Proyecto de Excelencia; M. J. Delgado-Cortés and R. Mauriño thank the Spanish Ministerio de Educación for a FPU and a FPI fellowships, respectively. This work is in the memorial of J. Cano.
References
- 1.Olanow CW, Schapira AHV, Agid Y. Neuroprotection for Parkinson’s disease: prospects and promises. Annals of Neurology. 2003;53(3):S1–S2. doi: 10.1002/ana.10566. [DOI] [PubMed] [Google Scholar]
- 2.Di Monte DA, Langston JW. Idiopathic and 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York, NY, USA: Oxford UP; 1995. pp. 989–997. [Google Scholar]
- 3.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 4.Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Human Molecular Genetics. 2009;18(1):R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 5.Jankovic J. Theories on the etiology and pathogenesis of Parkinson’s disease. Neurology. 1993;43(1, part 2):S21–S23. [Google Scholar]
- 6.Arif IA, Khan HA. Environmental toxins and Parkinson’s disease: putative roles of impaired electron transport chain and oxidative stress. Toxicology and Industrial Health. 2010;26(2):121–128. doi: 10.1177/0748233710362382. [DOI] [PubMed] [Google Scholar]
- 7.Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson’s disease. Trends in Pharmacological Sciences. 2003;24(8):395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- 8.Jenner P. Oxidative stress in Parkinson’s disease. Annals of Neurology. 2003;53(3):S26–S38. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 9.McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism and Related Disorders. 2004;10(1):S3–S7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Marchetti B, Abbracchio MP. To be or not to be (inflamed)—is that the question in anti-inflammatory drug therapy of neurodegenerative disorders? Trends in Pharmacological Sciences. 2005;26(10):517–525. doi: 10.1016/j.tips.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Marchetti B, Serra PA, L’Episcopo F, et al. Hormones are key actors in gene X environment interactions programming the vulnerability to Parkinson’s disease: glia as a common final pathway. Annals of the New York Academy of Sciences. 2005;1057:296–318. doi: 10.1196/annals.1356.023. [DOI] [PubMed] [Google Scholar]
- 12.Marchetti B, Serra PA, Tirolo C, et al. Glucocorticoid receptor-nitric oxide crosstalk and vulnerability to experimental parkinsonism: pivotal role for glia-neuron interactions. Brain Research Reviews. 2005;48(2):302–321. doi: 10.1016/j.brainresrev.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 13.Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson’s disease. Annals of Neurology. 2003;53(3):S49–S60. doi: 10.1002/ana.10481. [DOI] [PubMed] [Google Scholar]
- 14.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 15.McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Movement Disorders. 2008;23(4):474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 16.Klegeris A, McGeer EG, McGeer PL. Therapeutic approaches to inflammation in neurodegenerative disease. Current Opinion in Neurology. 2007;20(3):351–357. doi: 10.1097/WCO.0b013e3280adc943. [DOI] [PubMed] [Google Scholar]
- 17.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Annals of Neurology. 1988;24(4):574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama H, McGeer PL. Microglial response to 6-hydroxydopamine-induced substantia nigra lesions. Brain Research. 1989;489(2):247–253. doi: 10.1016/0006-8993(89)90857-3. [DOI] [PubMed] [Google Scholar]
- 19.Rogers J, Civin WH, Styren SD, McGeer PL. Immune-related mechanisms of Alzheimer‘s disease pathogenesis. In: Khachatunan ZS, Blass JP, editors. Alzheimer‘s Disease, New Treatment Strategies. New York, NY, USA: Marcel Dekker; 1992. pp. 147–163. [Google Scholar]
- 20.Compston A. Inflammation and the brain. Molecular and Chemical Neuropathology. 1993;19(1-2):47–64. doi: 10.1007/BF03160168. [DOI] [PubMed] [Google Scholar]
- 21.Burrell R. Immunomodulation by bacterial endotoxin. Critical Reviews in Microbiology. 1990;17(3):189–208. doi: 10.3109/10408419009105725. [DOI] [PubMed] [Google Scholar]
- 22.Bourdiol F, Toulmond S, Serrano A, Benavides J, Scatton B. Increase in ω 3 (peripheral type benzodiazepine) binding sites in the rat cortex and striatum after local injection of interleukin-1, tumour necrosis factor-α and lipopolysaccharide. Brain Research. 1991;543(2):194–200. doi: 10.1016/0006-8993(91)90028-t. [DOI] [PubMed] [Google Scholar]
- 23.Andersson PB, Perry VH, Gordon S. The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience. 1992;48(1):169–186. doi: 10.1016/0306-4522(92)90347-5. [DOI] [PubMed] [Google Scholar]
- 24.Montero-Menei CN, Sindji L, Pouplard-Barthelaix A, Jehan F, Denechaud L, Darcy F. Lipopolysaccharide intracerebral administration induces minimal inflammatory reaction in rat brain. Brain Research. 1994;653(1-2):101–111. doi: 10.1016/0006-8993(94)90377-8. [DOI] [PubMed] [Google Scholar]
- 25.Montero-Menei CN, Sindji L, Garcion E, et al. Early events of the inflammatory reaction induced in rat brain by lipopolysaccharide intracerebral injection: relative contribution of peripheral monocytes and activated microglia. Brain Research. 1996;724(1):55–66. doi: 10.1016/0006-8993(96)00268-5. [DOI] [PubMed] [Google Scholar]
- 26.Szczepanik AM, Fishkin RJ, Rush DK, Wilmot CA. Effects of chronic intrahippocampal infusion of lipopolysaccharide in the rat. Neuroscience. 1996;70(1):57–65. doi: 10.1016/0306-4522(95)00296-u. [DOI] [PubMed] [Google Scholar]
- 27.Piani D, Frei K, Do KQ, Cuenod M, Fontana A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neuroscience Letters. 1991;133(2):159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- 28.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. Journal of Immunology. 1992;149(8):2736–2741. [PubMed] [Google Scholar]
- 29.Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7(1):111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 30.Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia. 1993;7(1):102–110. doi: 10.1002/glia.440070116. [DOI] [PubMed] [Google Scholar]
- 31.Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. Journal of Neuroscience. 1993;13(1):29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lees GJ. The possible contribution of microglia and macrophages to delayed neuronal death after ischemia. Journal of the Neurological Sciences. 1993;114(2):119–122. doi: 10.1016/0022-510x(93)90285-7. [DOI] [PubMed] [Google Scholar]
- 33.Bronstein DM, Perez-Otano I, Sun V, et al. Glia-dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Research. 1995;704(1):112–116. doi: 10.1016/0006-8993(95)01189-7. [DOI] [PubMed] [Google Scholar]
- 34.Castaño A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. Journal of Neurochemistry. 1998;70(4):1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 35.Herrera AJ, Castaño A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiology of Disease. 2000;7(4):429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 36.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. Journal of Neuroscience. 2000;20(16):6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu B, Jiang JW, Wilson BC, et al. Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. Journal of Pharmacology and Experimental Therapeutics. 2000;295(1):125–132. [PubMed] [Google Scholar]
- 38.Lu X, Bing G, Hagg T. Naloxone prevents microglia-induced degeneration of dopaminergic substantia nigra neurons in adult rats. Neuroscience. 2000;97(2):285–291. doi: 10.1016/s0306-4522(00)00033-6. [DOI] [PubMed] [Google Scholar]
- 39.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. Journal of Cerebral Blood Flow and Metabolism. 1999;19(8):819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Anderson DC, Cranford RE. Corticosteroids in ischemic stroke. Stroke. 1979;10(1):68–71. doi: 10.1161/01.str.10.1.68. [DOI] [PubMed] [Google Scholar]
- 41.Norris JW, Hachinski VC. High dose steroid treatment in cerebral infarction. British Medical Journal. 1986;292(6512):21–23. doi: 10.1136/bmj.292.6512.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. New England Journal of Medicine. 1990;322(20):1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- 43.Loughlin AJ, Woodroofe MN, Cuzner ML. Modulation of interferon-γ-induced major histocompatibility complex class II and Fc receptor expression on isolated microglia by transforming growth factor-β1, interleukin-4, noradrenaline and glucocorticoids. Immunology. 1993;79(1):125–130. [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder DS, Unanue ER. Corticosteroids inhibit murine macrophage Ia expression and interleukin 1 production. Journal of Immunology. 1982;129(5):1803–1805. [PubMed] [Google Scholar]
- 45.Hay CH, De Belleroche JS. Dexamethasone prevents the induction of COX-2 mRNA and prostaglandins in the lumbar spinal cord following intraplantar FCA in parallel with inhibition of oedema. Neuropharmacology. 1998;37(6):739–744. doi: 10.1016/s0028-3908(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 46.Kiefer R, Kreutzberg GW. Effects of dexamethasone on microglial activation in vivo: selective downregulation of major histocompatibility complex class II expression in regenerating facial nucleus. Journal of Neuroimmunology. 1991;34(2-3):99–108. doi: 10.1016/0165-5728(91)90119-r. [DOI] [PubMed] [Google Scholar]
- 47.Ganter S, Northoff H, Mannel D, Gebicke-Harter PJ. Growth control of cultured microglia. Journal of Neuroscience Research. 1992;33(2):218–230. doi: 10.1002/jnr.490330205. [DOI] [PubMed] [Google Scholar]
- 48.Castaño A, Herrera AJ, Cano J, Machado A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-α IL-1β IFN-γ . Journal of Neurochemistry. 2002;81(1):150–157. doi: 10.1046/j.1471-4159.2002.00799.x. [DOI] [PubMed] [Google Scholar]
- 49.O'Neil MJ. Merck Manual of Diagnosis and Therapy. 17th edition. Whitehouse Station, NJ, USA: Merck; 1999. [Google Scholar]
- 50.Amin AR, Attur MG, Thakker GD, et al. A novel mechanism of action of tetracyclines: Effects on nitric oxide synthases. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne J, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Advances in dental research. 1998;12(2):12–26. doi: 10.1177/08959374980120010501. [DOI] [PubMed] [Google Scholar]
- 52.Rifkin BR, Vernillo AT, Golub LM, Ramamurthy NS. Modulation of bone resorption by tetracyclines. Annals of the New York Academy of Sciences. 1994;732:165–180. doi: 10.1111/j.1749-6632.1994.tb24733.x. [DOI] [PubMed] [Google Scholar]
- 53.Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanchez-Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48(6):1393–1401. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 56.Popovic N, Schubart A, Goetz BD, Zhang SUC, Linington C, Duncan D. Inhibition of autoimmune encephalomyelitis by a tetracycline. Annals of Neurology. 2002;51(2):215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- 57.Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nature Medicine. 2000;6(7):797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 58.Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 59.Du Y, Ma Z, Lin S, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(25):14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu DC, Jackson-Lewis V, Vila M, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. Journal of Neuroscience. 2002;22(5):1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He YI, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Research. 2001;909(1-2):187–193. doi: 10.1016/s0006-8993(01)02681-6. [DOI] [PubMed] [Google Scholar]
- 62.Tomás-Camardiel M, Rite I, Herrera AJ, et al. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiology of Disease. 2004;16(1):190–201. doi: 10.1016/j.nbd.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 63.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(11):1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 64.Takemoto M, Node K, Nakagami H, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. Journal of Clinical Investigation. 2001;108(10):1429–1437. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liao JK. Beyond lipid lowering: the role of statins in vascular protection. International Journal of Cardiology. 2002;86(1):5–18. doi: 10.1016/s0167-5273(02)00195-x. [DOI] [PubMed] [Google Scholar]
- 66.Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blanco-Colio LM, Muñoz-García B, Martín-Ventura JL, et al. 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors decrease Fas ligand expression and cytotoxicity in activated human T lymphocytes. Circulation. 2003;108(12):1506–1513. doi: 10.1161/01.CIR.0000089086.48617.2B. [DOI] [PubMed] [Google Scholar]
- 68.Hernández-Romero MDC, Argüelles S, Villarán RF, et al. Simvastatin prevents the inflammatory process and the dopaminergic degeneration induced by the intranigral injection of lipopolysaccharide. Journal of Neurochemistry. 2008;105(2):445–459. doi: 10.1111/j.1471-4159.2007.05148.x. [DOI] [PubMed] [Google Scholar]
- 69.Santiago M, Hernández-Romero MC, Machado A, Cano J. Zocor Forte® (simvastatin) has a neuroprotective effect against LPS striatal dopaminergic terminals injury, whereas against MPP+ does not. European Journal of Pharmacology. 2009;609(1–3):58–64. doi: 10.1016/j.ejphar.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 70.Wang V, Chia LG, Ni DR, et al. Effects of the combined treatment of naloxone and indomethacin on catecholamines and behavior after intranigral lipopolysaccharide injection. Neurochemical Research. 2004;29(2):341–346. doi: 10.1023/b:nere.0000013736.80749.4b. [DOI] [PubMed] [Google Scholar]
- 71.Zhou HF, Liu XY, Niu DB, Li FQ, He QH, Wang XM. Triptolide protects dopaminergic neurons from inflammation-mediated damage induced by lipopolysaccharide intranigral injection. Neurobiology of Disease. 2005;18(3):441–449. doi: 10.1016/j.nbd.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 72.Wang MJ, Lin WW, Chen HL, et al. Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. European Journal of Neuroscience. 2002;16(11):2103–2112. doi: 10.1046/j.1460-9568.2002.02290.x. [DOI] [PubMed] [Google Scholar]
- 73.Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B. Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sciences. 2006;80(3):193–199. doi: 10.1016/j.lfs.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 74.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 75.Ji KA, Eu MY, Kang SH, Gwag BJ, Jou I, Joe EH. Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. GLIA. 2008;56(10):1039–1047. doi: 10.1002/glia.20677. [DOI] [PubMed] [Google Scholar]
- 76.Barcia C, Bautista V, Sánchez-Bahillo A, et al. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. Journal of Neural Transmission. 2005;112(9):1237–1248. doi: 10.1007/s00702-004-0256-2. [DOI] [PubMed] [Google Scholar]
- 77.Rite I, Machado A, Cano J, Venero JL. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. Journal of Neurochemistry. 2007;101(6):1567–1582. doi: 10.1111/j.1471-4159.2007.04567.x. [DOI] [PubMed] [Google Scholar]
- 78.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? Journal of Neuroimmune Pharmacology. 2009;4(4):462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ben-Shachar D, Riederer P, Youdim MBH. Iron-melanin interaction and lipid peroxidation: implications for Parkinson’s disease. Journal of Neurochemistry. 1991;57(5):1609–1614. doi: 10.1111/j.1471-4159.1991.tb06358.x. [DOI] [PubMed] [Google Scholar]
- 80.Gerlach M, Ben-Shachar D, Riederer P, Youdim MBH. Altered brain metabolism of iron as a cause of neurodegenerative diseases? Journal of Neurochemistry. 1994;63(3):793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- 81.Duffy S, So A, Murphy TH. Activation of endogenous antioxidant defenses in neuronal cells prevents free radical-mediated damage. Journal of Neurochemistry. 1998;71(1):69–77. doi: 10.1046/j.1471-4159.1998.71010069.x. [DOI] [PubMed] [Google Scholar]
- 82.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. Journal of Neurochemistry. 1998;70(1):268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 83.Jenner P, Olanow CW. Understanding cell death in Parkinson’s disease. Annals of Neurology. 1998;44(3):S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 84.Dexter DT, Carter CJ, Wells FR, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. Journal of Neurochemistry. 1989;52(2):381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 85.Gotz ME, Kunig G, Riederer P, Youdim MBH. Oxidative stress: free radical production in neural degeneration. Pharmacology and Therapeutics. 1994;63(1):37–122. doi: 10.1016/0163-7258(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 86.Spector S, Sjoerdsma A, Udenfriend S. Blockade of endogenous norepinephrine synthesis by α-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. The Journal of Pharmacology And Experimental Therapeutics. 1965;147:86–95. [PubMed] [Google Scholar]
- 87.Weissman A, Koe BK, Tenen SS. Antiamphetamine effects following inhibition of tyrosine hydroxylase. Journal of Pharmacology and Experimental Therapeutics. 1966;151(3):339–352. [PubMed] [Google Scholar]
- 88.White FJ, Bednarz LM, Wachtel SR, Hjorth S, Brooderson RJ. Is stimulation of both D1 and D2 receptors necessary for the expression of dopamine-mediated behaviors? Pharmacology Biochemistry and Behavior. 1988;30(1):189–193. doi: 10.1016/0091-3057(88)90442-x. [DOI] [PubMed] [Google Scholar]
- 89.Jonkers N, Sarre S, Ebinger G, Michotte Y. MK801 influences L-DOPA-induced dopamine release in intact and hemi-parkinson rats. European Journal of Pharmacology. 2000;407(3):281–291. doi: 10.1016/s0014-2999(00)00753-6. [DOI] [PubMed] [Google Scholar]
- 90.Bebo BF, Jr., Yong T, Orr EL, Linthicum DS. Hypothesis: a possible role for mast cells and their inflammatory mediators in the pathogenesis of autoimmune encephalomyelitis. Journal of Neuroscience Research. 1996;45(4):340–348. doi: 10.1002/(SICI)1097-4547(19960815)45:4<340::AID-JNR3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 91.Wahl M, Schilling L. Regulation of cerebral blood flow—a brief review. Acta Neurochirurgica, Supplement. 1993;59:3–10. doi: 10.1007/978-3-7091-9302-0_1. [DOI] [PubMed] [Google Scholar]
- 92.Watanabe T, Taguchi Y, Shiosaka S, et al. Distribution of the histaminergic neuron system in the central nervous system of rats: a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Research. 1984;295(1):13–25. doi: 10.1016/0006-8993(84)90811-4. [DOI] [PubMed] [Google Scholar]
- 93.Prell GD, Green JP. Histamine as a neuroregulator. Annual Review of Neuroscience. 1986;9:209–254. doi: 10.1146/annurev.ne.09.030186.001233. [DOI] [PubMed] [Google Scholar]
- 94.Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiological Reviews. 1991;71(1):1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- 95.Wada H, Inagaki N, Yamatodani A, Watanabe T. Is the histaminergic neuron system a regulatory center for whole-brain activity? Trends in Neurosciences. 1991;14(9):415–418. doi: 10.1016/0166-2236(91)90034-r. [DOI] [PubMed] [Google Scholar]
- 96.Subramanian N, Mulder AH. Modulation by histamine of the efflux of radiolabeled catecholamines from rat brain slices. European Journal of Pharmacology. 1977;43(2):143–152. doi: 10.1016/0014-2999(77)90126-1. [DOI] [PubMed] [Google Scholar]
- 97.Onodera K, Yamatodani A, Watanabe T. Effects of α-fluoromethylhistidine on locomotor activity, brain histamine and catecholamine contents in rats. Methods and Findings in Experimental and Clinical Pharmacology. 1992;14(2):97–105. [PubMed] [Google Scholar]
- 98.Fleckenstein AE, Lookingland KJ, Moore KE. Differential role of histamine in mediating stress-induced changes in central dopaminergic neuronal activity in the rat. Brain Research. 1994;653(1-2):267–272. doi: 10.1016/0006-8993(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki T, Takamori K, Misawa M, Onodera K. Effects of the histaminergic system on the morphine-induced conditioned place preference in mice. Brain Research. 1995;675(1-2):195–202. doi: 10.1016/0006-8993(95)00064-w. [DOI] [PubMed] [Google Scholar]
- 100.Prast H, Heistracher M, Philippu A. Modulation by dopamine receptors of the histamine release in the rat hypothalamus. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1993;347(3):301–305. doi: 10.1007/BF00167449. [DOI] [PubMed] [Google Scholar]
- 101.Ito C, Onodera K, Sakurai E, Sato M, Watanabe T. Effects of dopamine antagonists on neuronal histamine release in the striatum of rats subjected to acute and chronic treatments with methamphetamine. Journal of Pharmacology and Experimental Therapeutics. 1996;279(1):271–276. [PubMed] [Google Scholar]
- 102.Onodera K, Yamatodani A, Watanabe T, Wada H. Neuropharmacology of the histaminergic neuron system in the brain and its relationship with behavioral disorders. Progress in Neurobiology. 1994;42(6):685–702. doi: 10.1016/0301-0082(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 103.Oertel WH, Dodel RC. International guide to drugs for Parkinson’s disease. Movement Disorders. 1995;10(2):121–131. doi: 10.1002/mds.870100202. [DOI] [PubMed] [Google Scholar]
- 104.Langlais PJ, Zhang SX, Weilersbacher G, Hough LB, Barke KE. Histamine-mediated neuronal death in a rat model of Wernicke’s encephalopathy. Journal of Neuroscience Research. 1994;38(5):565–574. doi: 10.1002/jnr.490380509. [DOI] [PubMed] [Google Scholar]
- 105.Thoburn KK, Hough LB, Nalwalk JW, Mischler SA. Histamine-induced modulation of nociceptive responses. Pain. 1994;58(1):29–37. doi: 10.1016/0304-3959(94)90182-1. [DOI] [PubMed] [Google Scholar]
- 106.Vizuete ML, Merino M, Venero JL, Santiago M, Cano J, Machado A. Histamine infusion induces a selective dopaminergic neuronal death along with an inflammatory reaction in rat substantia nigra. Journal of Neurochemistry. 2000;75(2):540–552. doi: 10.1046/j.1471-4159.2000.0750540.x. [DOI] [PubMed] [Google Scholar]
- 107.Coelho MH, Silva IJ, Azevedo MS, Manso CF. Decrease in blood histamine in drug-treated Parkinsonian patients. Molecular and Chemical Neuropathology. 1991;14(2):77–85. doi: 10.1007/BF03159928. [DOI] [PubMed] [Google Scholar]
- 108.Cumming P, Jakubovic A, Vincent SR. Cerebral histamine levels are unaffected by MPTP administration in the mouse. European Journal of Pharmacology. 1989;166(2):299–301. doi: 10.1016/0014-2999(89)90071-x. [DOI] [PubMed] [Google Scholar]
- 109.Silverman AJ, Sutherland AK, Wilhelm M, Silver R. Mast cells migrate from blood to brain. Journal of Neuroscience. 2000;20(1):401–408. doi: 10.1523/JNEUROSCI.20-01-00401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharpless NS, Muenter MD, Tyce GM. Effect of L Dopa on endogenous histamine metabolism. Medical Biology. 1975;53(2):85–92. [PubMed] [Google Scholar]
- 111.Molinari SP, Kaminski R, Di Rocco A, Yahr MD. The use of famotidine in the treatment of Parkinson’s disease: A pilot study. Journal of Neural Transmission. 1995;9(2-3):243–247. doi: 10.1007/BF02259665. [DOI] [PubMed] [Google Scholar]
- 112.Prell GD, Green JP. Histamine metabolites and prosmethylimidazole acetic acid in human cerebrospinal fluid. Agents and Actions. Supplements. 1991;33:343–363. doi: 10.1007/978-3-0348-7309-3_25. [DOI] [PubMed] [Google Scholar]
- 113.Prell GD, Khandelwal JK, Burns RS, Blandina P, Morrishow AM, Green JP. Levels of pros-methylimidazoleacetic acid: correlation with severity of Parkinson’s disease in CSF of patients and with the depletion of striatal dopamine and its metabolites in MPTP-treated mice. Journal of Neural Transmission. 1991;3(2):109–125. doi: 10.1007/BF02260886. [DOI] [PubMed] [Google Scholar]
- 114.Carriero DL, Outslay G, Mayorga AJ, Aberman J, Gianutsos G, Salamone JD. Motor dysfunction produced by tacrine administration in rats. Pharmacology Biochemistry and Behavior. 1997;58(4):851–858. doi: 10.1016/s0091-3057(97)98986-3. [DOI] [PubMed] [Google Scholar]
- 115.Mayorga AJ, Carriero DL, Cousins MS, Gianutsos G, Salamone JD. Tremulous jaw movements produced by acute tacrine administration: possible relation to parkinsonian side effects. Pharmacology Biochemistry and Behavior. 1997;56(2):273–279. doi: 10.1016/s0091-3057(96)00225-0. [DOI] [PubMed] [Google Scholar]
- 116.Trevitt JT, Lyons M, Aberman J, Carriero D, Finn M, Salamone JD. Effects of clozapine, thioridazine, risperidone and haloperidol on behaviovar tests related to extrapyramidal motor function. Psychopharmacology. 1997;132(1):74–81. doi: 10.1007/s002130050322. [DOI] [PubMed] [Google Scholar]
- 117.Cumming P, Reiner PB, Vincent SR. Inhibition of rat brain histamine-N-methyltransferase by 9-amino-1,2,3,4-tetrahydracridine (THA) Biochemical Pharmacology. 1990;40(6):1345–1350. doi: 10.1016/0006-2952(90)90402-7. [DOI] [PubMed] [Google Scholar]
- 118.McSwain ML, Forman LM. Severe parkinsonian symptom development on combination treatment with tacrine and haloperidol. Journal of Clinical Psychopharmacology. 1995;15(4):p. 284. doi: 10.1097/00004714-199508000-00008. [DOI] [PubMed] [Google Scholar]
- 119.Maany I. Adverse interaction of tacrine and haloperidol. American Journal of Psychiatry. 1996;153(11):p. 1504. doi: 10.1176/ajp.153.11.1504a. [DOI] [PubMed] [Google Scholar]
- 120.Keane JR, Horwitz NH, Grossman RG. Tremor as the result of shunt obstruction: four patients with cysticercosis and secondary Parkinsonism: report of four cases. Neurosurgery. 1995;37(3):520–522. doi: 10.1227/00006123-199509000-00023. [DOI] [PubMed] [Google Scholar]
- 121.Verma A, Berger JR, Bowen BC, Sanchez-Ramos J. Reversible Parkinsonian syndrome complicating cysticercus midbrain encephalitis. Movement Disorders. 1995;10(2):215–219. doi: 10.1002/mds.870100214. [DOI] [PubMed] [Google Scholar]
- 122.Agapejev S, Yela DA, Gomes AE. Chronic brain oedema in neurocysticercosis. Arquivos de Neuro-Psiquiatria. 1998;56(3 B):569–576. doi: 10.1590/s0004-282x1998000400009. [DOI] [PubMed] [Google Scholar]
- 123.Davey MG, Lüscher EF. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature. 1967;216(5118):857–858. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 124.Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry. 1991;30(43):10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 125.McNamara CA, Sarembock IJ, Gimple LW, II, Fenton JW, Coughlin SR, Owens GK. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. Journal of Clinical Investigation. 1993;91(1):94–98. doi: 10.1172/JCI116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grand RJA, Turnell AS, Grabham PW. Cellular consequences of thrombin-receptor activation. Biochemical Journal. 1996;313(2):353–368. doi: 10.1042/bj3130353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gurwitz D, Cunningham DD. Thrombin modulates and reverses neuroblastoma neurite outgrowth. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(10):3440–3444. doi: 10.1073/pnas.85.10.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grabham P, Cunningham DD. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: involvement of tyrosine phosphorylation. Journal of Neurochemistry. 1995;64(2):583–591. doi: 10.1046/j.1471-4159.1995.64020583.x. [DOI] [PubMed] [Google Scholar]
- 129.Möller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. Journal of Neurochemistry. 2000;75(4):1539–1547. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- 130.Cavanaugh KP, Gurwitz D, Cunningham DD, Bradshaw RA. Reciprocal modulation of astrocyte stellation by thrombin and protease nexin-1. Journal of Neurochemistry. 1990;54(5):1735–1743. doi: 10.1111/j.1471-4159.1990.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 131.Gingrich MB, Traynelis SF. Serine proteases and brain damage—is there a link? Trends in Neurosciences. 2000;23(9):399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- 132.Carreño-Müller E, Herrera AJ, De Pablos RM, et al. Thrombin induces in vivo degeneration of nigral dopaminergic neurones along with the activation of microglia. Journal of Neurochemistry. 2003;84(5):1201–1214. doi: 10.1046/j.1471-4159.2003.01634.x. [DOI] [PubMed] [Google Scholar]
- 133.Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. Journal of Neurosurgery. 1996;84(1):91–96. doi: 10.3171/jns.1996.84.1.0091. [DOI] [PubMed] [Google Scholar]
- 134.Vu TKH, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64(6):1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 135.Suzuki M, Ogawa A, Sakurai Y, et al. Thrombin activity in cerebrospinal fluid after subarachnoid hemorrhage. Stroke. 1992;23(8):1181–1182. doi: 10.1161/01.str.23.8.1181. [DOI] [PubMed] [Google Scholar]
- 136.Lee KR, Betz AL, Keep RF, Chenevert TL, Kim S, Hoff JT. Intracerebral infusion of thrombin as a cause of brain edema. Journal of Neurosurgery. 1995;83(6):1045–1050. doi: 10.3171/jns.1995.83.6.1045. [DOI] [PubMed] [Google Scholar]
- 137.Akiyama H, Ikeda K, Kondo H, McGeer PL. Thrombin accumulation in brains of patients with Alzheimer’s disease. Neuroscience Letters. 1992;146(2):152–154. doi: 10.1016/0304-3940(92)90065-f. [DOI] [PubMed] [Google Scholar]
- 138.Smith MA, Perry G. Alzheimer disease: an imbalance of proteolytic. Medical Hypotheses. 1994;42(4):277–279. doi: 10.1016/0306-9877(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 139.Kario K, Matsuo T, Hoshide S, Umeda Y, Shimada K. Effect of thrombin inhibition in vascular dementia and silent cerebrovascular disease: an MR spectroscopy study. Stroke. 1999;30(5):1033–1037. doi: 10.1161/01.str.30.5.1033. [DOI] [PubMed] [Google Scholar]
- 140.Ondo WG, Chan LL, Levy JK. Vascular parkinsonism: clinical correlates predicting motor improvement after lumbar puncture. Movement Disorders. 2002;17(1):91–97. doi: 10.1002/mds.1270. [DOI] [PubMed] [Google Scholar]
- 141.Foltynie T, Barker R, Brayne C. Vascular parkinsonism: a review of the precision and frequency of the diagnosis. Neuroepidemiology. 2002;21(1):1–7. doi: 10.1159/000048607. [DOI] [PubMed] [Google Scholar]
- 142.Choi SH, Joe EH, Kim SU, Jin BK. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. Journal of Neuroscience. 2003;23(13):5877–5886. doi: 10.1523/JNEUROSCI.23-13-05877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Argüelles S, Herrera AJ, Carreño-Müller E, et al. Degeneration of dopaminergic neurons induced by thrombin injection in the substantia nigra of the rat is enhanced by dexamethasone: role of monoamine oxidase enzyme. NeuroToxicology. 2010;31(1):55–66. doi: 10.1016/j.neuro.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 144.Harris-White ME, Chu T, Miller SA, et al. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and Aβ clearance in vitro and in vivo. Neurochemistry International. 2001;39(5-6):435–448. doi: 10.1016/s0197-0186(01)00051-1. [DOI] [PubMed] [Google Scholar]
- 145.Mitsuyo T, Adachi N, Yorozuya T, Tabo E, Nagaro T, Arai T. Facilitation of ischemia-induced release of dopamine and neuronal damage by dexamethasone in the rat striatum. European Journal of Pharmacology. 2003;465(3):267–274. doi: 10.1016/s0014-2999(03)01534-6. [DOI] [PubMed] [Google Scholar]
- 146.Chen J, Adachi N, Tsubota S, Nagaro T, Arai T. Dexamethasone augments ischemia-induced extracellular accumulation of glutamate in gerbil hippocampus. European Journal of Pharmacology. 1998;347(1):67–70. doi: 10.1016/s0014-2999(98)00198-8. [DOI] [PubMed] [Google Scholar]
- 147.Adachi N, Chen J, Liu K, Tsubota S, Arai T. Dexamethasone aggravates ischemia-induced neuronal damage by facilitating the onset of anoxic depolarization and the increase in the intracellular Ca2+ concentration in gerbil hippocampus. Journal of Cerebral Blood Flow and Metabolism. 1998;18(3):274–280. doi: 10.1097/00004647-199803000-00005. [DOI] [PubMed] [Google Scholar]
- 148.Guenther J, Nick H, Monard D. A glia-derived neurite-promoting factor with protease inhibitory activity. EMBO Journal. 1985;4(8):1963–1966. doi: 10.1002/j.1460-2075.1985.tb03878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Manoli I, Le H, Alesci S, et al. Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB Journal. 2005;19(10):1359–1361. doi: 10.1096/fj.04-3660fje. [DOI] [PubMed] [Google Scholar]
- 150.Youdim MBH, Banerjee DK, Kelner K, Offutt L, Pollard HB. Steroid regulation of monoamine oxidase activity in the adrenal medulla. FASEB Journal. 1989;3(6):1753–1759. doi: 10.1096/fasebj.3.6.2495232. [DOI] [PubMed] [Google Scholar]
- 151.Carlo P, Violani E, Del Rio M, et al. Monoamine oxidase B expression is selectively regulated by dexamethasone in cultured rat astrocytes. Brain Research. 1996;711(1-2):175–183. doi: 10.1016/0006-8993(95)01353-9. [DOI] [PubMed] [Google Scholar]
- 152.Cohen G, Farooqui R, Kesler N. Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(10):4890–4894. doi: 10.1073/pnas.94.10.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tazik S, Johnson S, Lu D, et al. Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone-induced brain cell apoptosis. Neurotoxicity Research. 2009;15(3):284–290. doi: 10.1007/s12640-009-9030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.De Pablos RM, Herrera AJ, Villarán RF, Cano J, Machado A. Dopamine-dependent neurotoxicity of lipopolysaccharide in substantia nigra. FASEB Journal. 2005;19(3):407–409. doi: 10.1096/fj.04-2153fje. [DOI] [PubMed] [Google Scholar]
- 155.Hayden SM, Seeds NW. Modulated expression of plasminogen activator system components in cultured cells from dissociated mouse dorsal root ganglia. Journal of Neuroscience. 1996;16(7):2307–2317. doi: 10.1523/JNEUROSCI.16-07-02307.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Scarisbrick IA, Isackson PJ, Ciric B, Windebank AJ, Rodriguez M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. Journal of Comparative Neurology. 2001;431(3):347–361. [PubMed] [Google Scholar]
- 157.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361(6411):453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 158.Seeds NW, Williams BL, Bickford PC. Tissue plasminogen activator induction in purkinje neurons after cerebellar motor learning. Science. 1995;270(5244):1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- 159.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nature Medicine. 1998;4(2):228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 160.Kim YH, Park JH, Hong SH, Koh JY. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999;284(5414):647–650. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- 161.Mataga N, Nagai N, Hensch TK. Permissive proteolytic activity for visual cortical plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(11):7717–7721. doi: 10.1073/pnas.102088899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yepes M, Sandkvist M, Coleman TA, et al. Regulation of seizure spreading by neuroserpin and tissue-type plasminogen activator is plasminogen-independent. Journal of Clinical Investigation. 2002;109(12):1571–1578. doi: 10.1172/JCI14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99(18):2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- 164.Yepes M, Sandkvist M, Wong MKK, et al. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood. 2000;96(2):569–576. [PubMed] [Google Scholar]
- 165.Cinelli P, Madani R, Tsuzuki N, et al. Neuroserpin, a neuroprotective factor in focal ischemic stroke. Molecular and Cellular Neuroscience. 2001;18(5):443–457. doi: 10.1006/mcne.2001.1028. [DOI] [PubMed] [Google Scholar]
- 166.Marler JR. Tissue plasminogen activator for acute ischemic stroke. New England Journal of Medicine. 1995;333(24):1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 167.Gravanis I, Tsirka SE. Tissue plasminogen activator and glial function. Glia. 2005;49(2):177–183. doi: 10.1002/glia.20115. [DOI] [PubMed] [Google Scholar]
- 168.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. Journal of Clinical Investigation. 2003;112(10):1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Goto H, Fujisawa H, Oka F, et al. Neurotoxic effects of exogenous recombinant tissue-type plasminogen activator on the normal rat brain. Journal of Neurotrauma. 2007;24(4):745–752. doi: 10.1089/neu.2006.0183. [DOI] [PubMed] [Google Scholar]
- 170.Ryu JK, Shin WH, Kim J, et al. Trisialoganglioside GT1b induces in vivo degeneration of nigral dopaminergic neurons: role of microglia. Glia. 2002;38(1):15–23. doi: 10.1002/glia.10047. [DOI] [PubMed] [Google Scholar]
- 171.Tilvis RS, Kähönen-Väre MH, Jolkkonen J, Valvanne J, Pitkala KH, Strandberg TE. Predictors of cognitive decline and mortality of aged people over a 10-year period. Journals of Gerontology. Series A. 2004;59(3):268–274. doi: 10.1093/gerona/59.3.m268. [DOI] [PubMed] [Google Scholar]
- 172.Strang RR. The association of gastro-duodenal ulceration with Parkinson’s disease. The Medical journal of Australia. 1965;310:842–843. doi: 10.5694/j.1326-5377.1965.tb72277.x. [DOI] [PubMed] [Google Scholar]
- 173.Dobbs RJ, Dobbs SM, Weller C, et al. Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 1: eradication of Helicobacter in the cachexia of idiopathic parkinsonism. Helicobacter. 2005;10(4):267–275. doi: 10.1111/j.1523-5378.2005.00331.x. [DOI] [PubMed] [Google Scholar]
- 174.Villarán RF, Espinosa-Oliva AM, Sarmiento M, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson's disease. Journal of Neurochemistry. 2010;114(6):1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 175.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98(3):694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 176.Brochard V, Combadière B, Prigent A, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. Journal of Clinical Investigation. 2009;119(1):182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Van Rooijen N. The liposome-mediated macrophage ’suicide’ technique. Journal of Immunological Methods. 1989;124(1):1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- 178.Van Rooijen N, Sanders A, Van Den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. Journal of Immunological Methods. 1996;193(1):93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 179.Nieuwenhuijzen GAP, Knapen MFCM, Hendriks T, Van Rooijen N, Goris RJA. Elimination of various subpopulations of macrophages and the development of multiple-organ dysfunction syndrome in mice. Archives of Surgery. 1997;132(5):533–539. doi: 10.1001/archsurg.1997.01430290079016. [DOI] [PubMed] [Google Scholar]
- 180.Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1(8336):1273–1275. [PubMed] [Google Scholar]
- 181.Weller C, Oxlade N, Dobbs SM, Dobbs RJ, Charlett A, Bjarnason IT. Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunology and Medical Microbiology. 2005;44(2):129–135. doi: 10.1016/j.femsim.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 182.Megraud F. Epidemiology of Helicobacter pylori infection. Gastroenterology Clinics of North America. 1993;22(1):73–88. [PubMed] [Google Scholar]
- 183.Ernst P. Review article: the role of inflammation in the pathogenesis of gastric cancer. Alimentary Pharmacology and Therapeutics, Supplement. 1999;13(1):13–18. doi: 10.1046/j.1365-2036.1999.00003.x. [DOI] [PubMed] [Google Scholar]
- 184.Shimada T, Terano A. Chemokine expression in Helicobacter pylori-infected gastric mucosa. Journal of Gastroenterology. 1998;33(5):613–617. doi: 10.1007/s005350050146. [DOI] [PubMed] [Google Scholar]
- 185.Peek RM. Helicobacter pylori strain-specific activation of signal transduction cascades related to gastric inflammation. American Journal of Physiology. 2001;280(4):G525–G530. doi: 10.1152/ajpgi.2001.280.4.G525. [DOI] [PubMed] [Google Scholar]
- 186.Aydemir S, Tekin IO, Numanoglu G, Borazan A, Ustundag Y. Eosinophil infiltration, gastric juice and serum eosinophil cationic protein levels in Helicobacter pylori-associated chronic gastritis and gastric ulcer. Mediators of Inflammation. 2004;13(5-6):369–372. doi: 10.1155/S0962935104000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Supajatura V, Ushio H, Wada A, et al. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. Journal of Immunology. 2002;168(6):2603–2607. doi: 10.4049/jimmunol.168.6.2603. [DOI] [PubMed] [Google Scholar]
- 188.Brandt S, Kwok T, Hartig R, König W, Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao Y, Yokota K, Ayada K, et al. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR)2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. Journal of Medical Microbiology. 2007;56(2):154–164. doi: 10.1099/jmm.0.46882-0. [DOI] [PubMed] [Google Scholar]