

Abstract
Maturity onset diabetes of the young (MODY) is a monogenic form of diabetes inherited as an autosomal dominant trait. The second most common cause is GCK-MODY due to heterozygous mutations in the GCK gene which impair the glucokinase function through different mechanisms such as enzymatic activity, protein stability, and increased interaction with its receptor. The enzyme normally acts as a glucose sensor in the pancreatic beta cell and regulates insulin secretion. We report here a three-generation nonobese family diagnosed with diabetes. All affected family members presented with mild hyperglycemia and mostly slightly elevated hemoglobin A1c values. Genetic testing revealed a novel heterozygous T → C exchange in exon 8 of the GCK gene which resulted in a phenylalanine330 TTC → serine (TCC)/p.Phe330Ser/F330S substitution.
1. Introduction
MODY is a monogenic disease which accounts for 2–5% of all diabetes cases. The most frequent form is HNF-1α-MODY (MODY type 3), which is caused by mutations in the HNF1A gene encoding hepatic nuclear factor 1α. The second most frequent form is GCK-MODY (MODY type 2), which has been shown to be the result of mutations in the GCK gene [1]. The GCK gene maps to chromosome 7p15.3-p15.1 and consists of 12 exons that encode the 465-amino-acid protein glucokinase [2, 3], which is one of four members of the hexokinase family of enzymes. It catalyzes the phosphorylation of glucose as the first step of glycolysis. Glucokinase is exclusively expressed in mammalian liver and pancreatic islet beta cells. The enzyme plays an important regulatory role in glucose metabolism. As a glucose sensor, it regulates insulin secretion in the pancreatic β-cell by changing the glucose phosphorylation rate over a range of physiological glucose concentrations (4–15 mmoL/L; [4]). GCK gene mutations can cause both hypo- and hyperglycemia. Heterozygous inactivating mutations cause GCK-MODY, which mostly presents with mild hyperglycemia and is inherited in an autosomal dominant fashion [5, 6]. Usually, no diabetes-related complications such as nephropathy or retinopathy occur in patients with GCK-MODY. Homozygous or compound heterozygous inactivating GCK mutations result in a more severe phenotype presenting at birth as permanent neonatal diabetes mellitus [7–11]. Heterozygous activating GCK mutations, in contrast, cause persistent hyperinsulinemic hypoglycemia of infancy [12–16].
In 1992, GCK was the first MODY gene to be linked to disease in French and UK families [5, 6]. These linkage studies were quickly followed by the identification of the first genetic defects. Meanwhile, over 600 different GCK mutations have been described in many populations, the majority having been identified in Europe [17]. Missense, nonsense, frameshift, and splice site mutations have been reported, which are distributed throughout the 10 exons encoding the pancreatic β-cell isoform of the enzyme. There are no mutational hot spots. Over 250 mutations have been reported in more than one family [17]. All inactivating mutations are associated with mild fasting hyperglycemia while other specific symptoms are missing. Therefore, GCK-MODY is likely to be underdiagnosed, and no reliable data on its prevalence are available. In Caucasians, approximately 2% of the population are diagnosed as having gestational diabetes and approximately 2–5% of these patients will have GCK-MODY [18]. This would suggest a population prevalence of 0.04–0.1%.
The identification of a GCK gene mutation is important for the correct and definite diagnosis of GCK-MODY. It also helps the clinician to predict the clinical course of the disease and to advise appropriate therapy. Since only mild fasting hyperglycemia and no diabetes-related complications are usually present, diet is sufficient as a therapeutic approach in most cases [19].
We report a novel heterozygous inactivating GCK gene mutation in a nonobese family diagnosed with diabetes over three generations.
2. Case Report
A 17-year-old, nonobese (BMI 24.3 kg/m²) female was admitted for the evaluation of recurrent syncopes. A blood glucose profile was obtained, and she was found to be mildly hyperglycemic (blood glucose values between 6.7 and 10 mmoL/L). Hemoglobin A1c was slightly elevated with 6.5% (normal range 4.0–6.0%) or 48 mmoL/moL (normal range 20–42 mmoL/moL). A standard oral glucose tolerance test with 75 g of glucose equivalent was performed (Table 1) with a fasting glucose of 6.5 mmoL/L and a 2-hour glucose of 14.6 mmoL/L. The fasting insulin concentration was 3.6 μU/mL and 66.1 μU/mL after two hours.
Table 1.
Glucose and insulin concentrations during a standard oral glucose tolerance test with 75 g glucose equivalent.
| Time [min.] | Glucose [mg/dl] | Glucose [mmol/L] | Insulin [μU/mL] |
|---|---|---|---|
| 0 | 117 | 6.5 | 3.6 |
| 30 | 191 | 10.6 | 31.1 |
| 60 | 247 | 13.7 | 53.2 |
| 120 | 263 | 14.6 | 66.1 |
Her family history was strongly positive for diabetes. The patient's brother, her father, and the grandparents on her father's side were diagnosed with diabetes. Her grandmother was treated with oral antidiabetic medication (glimepiride). The index patient also reported that she had been diagnosed with mild hyperglycemia ten years ago, but this had no therapeutic consequences, and no followup analyses were performed.
This clinical presentation was highly suggestive of GCK-MODY. Therefore, a mutation analysis of the GCK gene was initiated. Genomic DNA of the patient was isolated, and PCR amplification of the pancreas-specific exon 1a as well as of exons 2–10 of the GCK gene was performed. Sequencing of the PCR products identified a novel phenylalanine330 (TTC) → serine (TCC)-/p.Phe330Ser-/F330S substitution encoded by exon 8. The index patient was also screened for type 1 diabetes-specific antibodies in the context of a Bavarian diabetes registry (DiMelli). Insulin autoantibodies (IAA), tyrosinephosphatase antibodies (IA2A), and zinc transporter antibodies (ZnT8c) were negative. Antibodies against glutamatedecarboxylase (GADA) were weakly positive.
Carrier screening of clinically affected family members revealed the same mutation in the pedigree with the exception of the grandfather, who has type 2 diabetes mellitus (Figure 1). The phenotype of the index patient's brother was comparable to that of the index patient (BMI 21.5 kg/m², hemoglobin A1c 6.2%, or 44 mmoL/moL). The father of the index patient was diagnosed with diabetes at 25 years of age. He also presented with very mild hyperglycemia and haemoglobin A1c at the upper end of the normal range (5.9% or 41 mmoL/moL). The father's BMI was 27.8 kg/m². Despite normal haemoglobin A1c, an antidiabetic treatment with metformin was started by the father's diabetologist. The index patient's grandmother was diagnosed with diabetes at the age of 62 years. Her BMI was 30.0 kg/m², and she had a haemoglobin A1c value of 7.0% or 53 mmoL/moL. She was treated with glimepiride. The grandfather was also diagnosed with diabetes but was not on any antidiabetic medication. His BMI was 29.8 kg/m² and his haemoglobin A1c never exceeded 7.0% or 53 mmoL/moL. No GCK mutation was found in him, so his final diagnosis is type 2 diabetes mellitus.
Figure 1.
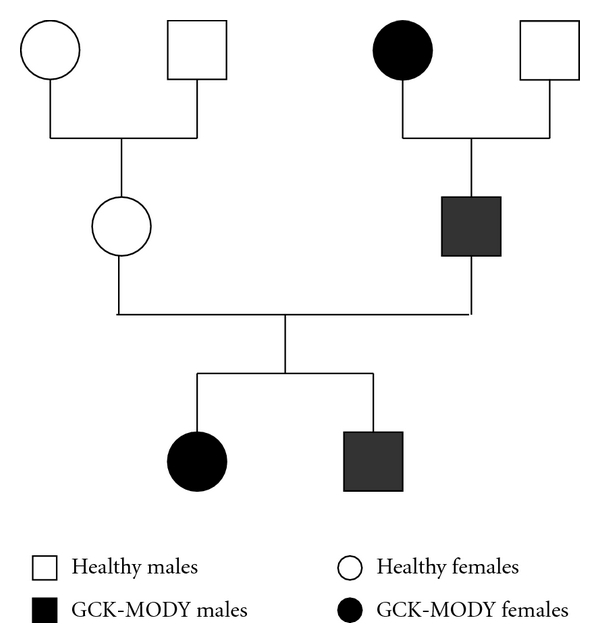
Circle: females, boxes: males, white: healthy, black: GCK-MODY.
Because of the typical clinical symptoms and the cosegregation of the phenotype with the genotype, we conclude that this novel mutation is a pathogenic one and not a polymorphism without an effect on protein function.
3. Discussion
So far, more than 620 GCK gene mutations have been reported in over 1400 patients with GCK-MODY (Table 2), permanent neonatal diabetes, and hyperinsulinemic hypoglycemia [17, 20, 21]. The mutations are spread over the ten exons of the gene which encode the pancreatic beta cell isoform of glucokinase (exons 1a and 2–10). Only few mutations have been detected in exon 1a, since most laboratories only screen for GCK mutations in exons 2–10 [18]. Missense, nonsense, frameshift, and splice site mutations have been reported. There are no mutation hot spots. Most of the mutations are private, although 255 mutations have been described in more than one family [18, 22]. We here report a novel heterozygous p.Phe330Ser mutation encoded by GCK exon 8 which results in the typical GCK-MODY phenotype. The mutation was found in three generations of a nonconsanguineous family, following an autosomal dominant inheritance pattern.
Table 2.
Published GCK mutations causing GCK-MODY (MODY 2).
| Region | Nucleotide and systematic name | Protein effect | Reference |
|---|---|---|---|
| Islet promoter | c.−71G>C | — | [25] |
| Exon 2 | c.106C>T | p.Arg36Trp | [26] |
| c.130G>A | p.Gly44Ser | [27] | |
| c.157G>T | p.Ala53Ser | [20] | |
| c.182A>C | p.Tyr61Ser | [28] | |
| c.185T>C | p.Val62Ala | [21] | |
| c.184G>A | p.Val62Met | [21, 29] | |
| c.208G>A | p.Glu70Lys | [20, 30] | |
| Exon 3 | c.214G>A | p.Gly72Arg | [29] |
| c.239G>C | p.Gly80Ala | [20] | |
| c.323A>G | p.Tyr108Cys | [21] | |
| Exon 4 | c.391T>C | p.Ser131Pro | [31] |
| c.410A>G | p.His137Arg | [20] | |
| c.437T>G | p.Leu146Arg | [21] | |
| c.480_482dupTAA | p.Asp160_Lys161 ins Asn | [32] | |
| Exon 5 | c.493C>T | p.Leu165Phe | [33] |
| c.502A>C | p.Thr168Pro | [20] | |
| c.523G>A | p.Gly175Arg | [20] | |
| c.524G>A | p.Gly175Glu | [20] | |
| c.544G>T | p.Val182Leu | [28] | |
| c.562G>A | p.Ala188Thr | [31] | |
| c.563C>A | p.Ala188Glu | [21] | |
| Exon 6 | c.608T>C | p.Val203Ala | [30] |
| c.617C>T | p.Thr206Met | [33] | |
| c.622G>A | p.Ala208Thr | [22] | |
| c.626C>T | p.Thr209Met | [26] | |
| c.629T>C | p.Met210Thr | [20] | |
| c.637T>C | p.Cys213Arg | [20] | |
| c.676G>A | p.Val226Met | [20] | |
| c.682A>G | p.Thr228Ala | [34] | |
| c.697T>C | p.Cys233Arg | [28] | |
| c.703A>G | p.Met235Val | [32] | |
| c.704T>C | p.Met235Thr | [21] | |
| c.755G>A | p.Cys252Tyr | [21] | |
| c.766G>A | p.Glu256Lys | [35] | |
| c.769T>C | p.Trp257Arg | [31] | |
| c.781G>A | p.Gly261Arg | [20] | |
| c.787T>C | p.Ser263Pro | [21] | |
| c.793G>A | p.Glu265Lys | [28] | |
| c.823C>T | p.Arg275Cys | [21] | |
| c.835G>C | p.Glu279Gln | [35] | |
| Exon 8 | c.893T>A | p.Met298Lys | [21] |
| c.895G>C | p.Gly299Arg | [35] | |
| c.898G>C | p.Glu300Gln | [35] | |
| c.898G>A | p.Glu300Lys | [30] | |
| c.922A>T | p.Arg308Trp | [32] | |
| c.926T>C | p.Leu309Pro | [20, 35] | |
| c.1007C>T | p.Ser336Leu | [20, 34] | |
| c.1016A>G | p.Glu339Gly | [21, 22] | |
| Exon 9 | c.1099G>A | p.Val367Met | [20, 26] |
| c.1129C>T | p.Arg377Cys | [21, 22] | |
| c.1136C>T | p.Ala379Val | [28] | |
| c.1148C>T | p.Ser383Leu | [21] | |
| c.1232C>T | p.Ser411Phe | [21] | |
| c.1240A>G | p.Lys414Glu | [20, 36] | |
| Exon 10 | c.1258A>G | p.Lys420Glu | [28] |
| c.1358C>T | p.Ser453Leu | [22] | |
| c.1364T>A | p.Val455Glu | [37] |
A GCK gene polymorphism with functional significance also has been reported in the literature [23, 24]. The G>A exchange at position −30 of the beta-cell-specific promoter (rs1799884) has been shown to be associated with increased fasting glucose levels in young adults and in pregnant women as well as an increased birth weight of their offspring [23].
The identification of a pathogenic GCK gene mutation is important for the correct and definite diagnosis of GCK-MODY and helps the clinician to predict the disease course and to initiate the appropriate therapy. Since only mild fasting hyperglycemia and usually no diabetes-related complications are present in patients with GCK-MODY, diet is sufficient as a therapeutic approach in most cases [19]. This may significantly improve the patient's quality of life.
References
- 1.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. The New England Journal of Medicine. 2001;345(13):971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 2.Stoffel M, Froguel P, Takeda J, et al. Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(16):7698–7702. doi: 10.1073/pnas.89.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoffel M, Patel P, Lo YMD, et al. Missense glucokinase mutation in maturity-onset diabetes of the young and mutation screening in late-onset diabetes. Nature Genetics. 1992;2(2):153–156. doi: 10.1038/ng1092-153. [DOI] [PubMed] [Google Scholar]
- 4.Matschinsky FM. Regulation of pancreatic β-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(3):S394–S404. doi: 10.2337/diabetes.51.2007.s394. [DOI] [PubMed] [Google Scholar]
- 5.Froguel P, Vaxillaire M, Sun F, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6365):162–164. doi: 10.1038/356162a0. [DOI] [PubMed] [Google Scholar]
- 6.Hattersley AT, Turner RC, Permutt MA, et al. Linkage of type 2 diabetes to the glucokinase gene. The Lancet. 1992;339(8805):1307–1310. doi: 10.1016/0140-6736(92)91958-b. [DOI] [PubMed] [Google Scholar]
- 7.Njølstad PR, Søvik O, Cuesta-Muñoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. The New England Journal of Medicine. 2001;344(21):1588–1592. doi: 10.1056/NEJM200105243442104. [DOI] [PubMed] [Google Scholar]
- 8.Njølstad PR, Sagen JV, Bjørkhaug L, et al. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52(11):2854–2860. doi: 10.2337/diabetes.52.11.2854. [DOI] [PubMed] [Google Scholar]
- 9.Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL. Permanent neonatal diabetes in an Asian infant. Journal of Pediatrics. 2005;146(1):131–133. doi: 10.1016/j.jpeds.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Rubio-Cabezas O, González FD, Aragonés A, Argente J, Campos-Barros A. Permanent neonatal diabetes caused by a homozygous nonsense mutation in the glucokinase gene. Pediatric Diabetes. 2008;9(3):245–249. doi: 10.1111/j.1399-5448.2007.00361.x. [DOI] [PubMed] [Google Scholar]
- 11.Turkkahraman D, Bircan I, Tribble ND, Akçurin S, Ellard S, Gloyn AL. Permanent neonatal diabetes mellitus caused by a novel homozygous(T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. Journal of Pediatrics. 2008;153(1):122–126. doi: 10.1016/j.jpeds.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 12.Christesen HBT, Jacobsen BB, Odili S, et al. The second activating glucokinase mutation (A456V): implications for glucose homeostasis and diabetes therapy. Diabetes. 2002;51(4):1240–1246. doi: 10.2337/diabetes.51.4.1240. [DOI] [PubMed] [Google Scholar]
- 13.Christesen HBT, Tribble ND, Molven A, et al. Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. European Journal of Endocrinology. 2008;159(1):27–34. doi: 10.1530/EJE-08-0203. [DOI] [PubMed] [Google Scholar]
- 14.Cuesta-Muñoz AL, Huopio H, Otonkoski T, et al. Severe persistent hyperinsulinemic hypoglycemic due to a de novo glucokinase mutation. Diabetes. 2004;53(8):2164–2168. doi: 10.2337/diabetes.53.8.2164. [DOI] [PubMed] [Google Scholar]
- 15.Glaser B, Kesavan P, Heyman M, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. The New England Journal of Medicine. 1998;338(4):226–230. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 16.Sayed S, Langdon DR, Odili S, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58(6):1419–1427. doi: 10.2337/db08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Human Mutation. 2009;30(11):1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- 18.Ellard S, Beards F, Allen LIS, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43(2):250–253. doi: 10.1007/s001250050038. [DOI] [PubMed] [Google Scholar]
- 19.Gill Carey OJSB, Colclough K, Ellard S, Hattersley AT. Finding a glucokinase mutation alters treatment. Diabetic Medicine. 2007;24:6–7. [Google Scholar]
- 20.Davis EA, Cuesta-Muñoz A, Raoul M, et al. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia. 1999;42(10):1175–1186. doi: 10.1007/s001250051289. [DOI] [PubMed] [Google Scholar]
- 21.Gloyn AL. Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Human Mutation. 2003;22(5):353–362. doi: 10.1002/humu.10277. [DOI] [PubMed] [Google Scholar]
- 22.Sagen JV, Bjørkhaug L, Molnes J, et al. Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY Registry. Pediatric Diabetes. 2008;9(5):442–449. doi: 10.1111/j.1399-5448.2008.00399.x. [DOI] [PubMed] [Google Scholar]
- 23.Weedon MN, Frayling TM, Shields B, et al. Genetic regulation of birth weight and fasting glucose by a common polymorphism in the islet cell promoter of the glucokinase gene. Diabetes. 2005;54(2):576–581. doi: 10.2337/diabetes.54.2.576. [DOI] [PubMed] [Google Scholar]
- 24.Weedon MN, Clark VJ, Qian Y, et al. A common haplotype of the glucokinase gene alters fasting glucose and birth weight: association in six studies and population-genetics analyses. American Journal of Human Genetics. 2006;79(6):991–1001. doi: 10.1086/509517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gašperíková D, Tribble ND, Staník J, et al. Identification of a novel β-cell glucokinase (GCK) promoter mutation (-71G>C) that modulates GCK gene expression through loss of allele-specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes. 2009;58(8):1929–1935. doi: 10.2337/db09-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller SP, Anand GR, Karschnia EJ, Bell GI, LaPorte DC, Lange AJ. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes. 1999;48(8):1645–1651. doi: 10.2337/diabetes.48.8.1645. [DOI] [PubMed] [Google Scholar]
- 27.Codner E, Rocha A, Deng L, et al. Mild fasting hyperglycemia in children: high rate of glucokinase mutations and some risk of developing type 1 diabetes mellitus. Pediatric Diabetes. 2009;10(6):382–388. doi: 10.1111/j.1399-5448.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estalella I, Rica I, De Nanclares GP, et al. Mutations in GCK and HNF-1α explain the majority of cases with clinical diagnosis of MODY in Spain. Clinical Endocrinology. 2007;67(4):538–546. doi: 10.1111/j.1365-2265.2007.02921.x. [DOI] [PubMed] [Google Scholar]
- 29.Arden C, Trainer A, de la Iglesia N, et al. Cell biology assessment of glucokinase mutations V62M and G72R in pancreatic β-cells: evidence for cellular instability of catalytic activity. Diabetes. 2007;56(7):1773–1782. doi: 10.2337/db06-1151. [DOI] [PubMed] [Google Scholar]
- 30.Burke CV, Buettger CW, Davis EA, McClane SJ, Matschinsky FM, Raper SE. Cell-biological assessment of human glucokinase mutants causing maturity-onset diabetes of the young type 2 (MODY-2) or glucokinase-linked hyperinsulinaemia (GK-HI) Biochemical Journal. 1999;342(2):345–352. [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda J, Gidh-Jain M, Xu LZ, et al. Structure/function studies of human β-cell glucokinase. Enzymatic properties of a sequence polymorphism, mutations associated with diabetes, and other site-directed mutants. Journal of Biological Chemistry. 1993;268(20):15200–15204. [PubMed] [Google Scholar]
- 32.García-Herrero CM, Galán M, Vincent O, et al. Functional analysis of human glucokinase gene mutations causing MODY2: exploring the regulatory mechanisms of glucokinase activity. Diabetologia. 2007;50(2):325–333. doi: 10.1007/s00125-006-0542-7. [DOI] [PubMed] [Google Scholar]
- 33.Galán M, Vincent O, Roncero I, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochemical Journal. 2006;393(1):389–396. doi: 10.1042/BJ20051137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marotta DE, Anand GR, Anderson TA, et al. Identification and characterization of the ATP-binding site in human pancreatic glucokinase. Archives of Biochemistry and Biophysics. 2005;436(1):23–31. doi: 10.1016/j.abb.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 35.Gidh-Jain M, Takeda J, Xu LZ, et al. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(5):1932–1936. doi: 10.1073/pnas.90.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pino MF, Kim KA, Shelton KD, et al. Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. Journal of Biological Chemistry. 2007;282(18):13906–13916. doi: 10.1074/jbc.M610094200. [DOI] [PubMed] [Google Scholar]
- 37.Gloyn AL, Tribble ND, van de Bunt M, Barrett A, Johnson PRV. Glucokinase (GCK) and other susceptibility genes for β-cell dysfunction: the candidate approach. Biochemical Society Transactions. 2008;36(3):306–311. doi: 10.1042/BST0360306. [DOI] [PubMed] [Google Scholar]