

Abstract
Cell and tissue movement are essential processes at various stages in the life cycle of most organisms. The early development of multi-cellular organisms involves individual and collective cell movement; leukocytes must migrate towards sites of infection as part of the immune response; and in cancer, directed movement is involved in invasion and metastasis. The forces needed to drive movement arise from actin polymerization, molecular motors and other processes, but understanding the cell- or tissue-level organization of these processes that is needed to produce the forces necessary for directed movement at the appropriate point in the cell or tissue is a major challenge. In this paper, we present three models that deal with the mechanics of cells and tissues: a model of an arbitrarily deformable single cell, a discrete model of the onset of tumour growth in which each cell is treated individually, and a hybrid continuum–discrete model of the later stages of tumour growth. While the models are different in scope, their underlying mechanical and mathematical principles are similar and can be applied to a variety of biological systems.
Keywords: multi-scale models, cell motility, stress effects, tumour dynamics
1. Introduction
(a). Movement of single cells
Cell locomotion in multi-cellular organisms plays an essential role during embryonic development, in tissue movement and regeneration, in the immune response, during cancer metastasis and in wound healing. Movement is a very complex process that involves the spatio-temporal control and integration of a number of subprocesses, including the transduction of chemical or mechanical signals from the environment, intracellular biochemical responses, and translation of the intra- and extracellular signals into a mechanical response (Mitchison & Cramer 1996; Sheetz et al. 1999). Individual cells detect extracellular chemical and mechanical signals via membrane receptors, and this initiates signal transduction cascades that produce intracellular signals. Directed motion in response to signals requires that cells orient properly, and this frequently involves amplification of small differences in the extracellular signal over the cell into large end-to-end intracellular signal differences (Chung et al. 2001). These signals control the motile machinery of the cell and thereby determine the spatial localization of contact sites with the substrate and the sites of force generation needed to produce directed motion. As cell movement is fundamentally a mechanical process, one cannot understand movement without an integrated model that can predict the effects of all the component processes involved in the mechanical response.
The mechanical response of a single cell is often characterized by four major subprocesses. (i) Extension of directed protrusions (lamellipodia, filopodia, or pseudopodia) at the leading edge. The force for this results from localized polymerization of monomeric actin into cross-linked networks of actin filaments (F-actin) in lamellipodia, or bundles of filaments in filopodia or pseudopodia. (ii) Anchoring of protrusions to the substrate or the extracellular matrix via adhesive complexes, which serve as sites for force transmission to the substrate (Small et al. 2002). In cells such as fibroblasts, these complexes, which are called focal complexes, can mature into larger focal adhesions that serve as ‘traction pads’ over which the cell body moves (Small 1989; Friedl & Wolf 2003). (iii) Next, depending on the cell type, actomyosin filaments (complexes of F-actin and the motor protein myosin II) contract at the front, in the perinuclear region or at the rear, to move the cell body forward. (iv) Finally, cells release attachments at the rear (Pollard et al. 2000). Of course, these processes usually occur simultaneously in different parts of a cell.
Numerous pathways are involved in transducing signals from surface receptors to downstream effectors such as the Arp2/3 complex, which associates with actin to nucleate new filaments and initiate branches on existing filaments (Dumontier et al. 2000). Behind the leading edge there is a region of actin disassembly, where filaments are disassembled, cross-links broken and the actin monomers resulting from disassembly freed to diffuse to the site of active polymerization (Alberts et al. 2002). Protrusions are stabilized by formation of adhesive complexes, which serve as sites for molecular signalling and also transmit mechanical force to the substrate. During migration, the small nascent adhesive complexes may mature into focal adhesions.
At a higher level of abstraction, one can identify three essential processes involved in movement: (i) controlled spatio-temporal remodelling of the actin network; (ii) generation of traction forces to move the cell body; and (iii) the construction and destruction of either focal complexes or focal adhesions (Mitra et al. 2005). Four actin subnetworks have been identified in different motile cells: (i) the lamellipodium, a region of rapid actin turnover that extends 3–5 μm from the leading edge of the cell; (ii) the lamellum, a contractile network that extends from just behind the leading edge to the interior of the cell; (iii) the convergence zone, where the retrograde flow in the lamellipodium meets the anterograde flow in the cell body; and (iv) the actin network in the cell body, which contains the major organelles (Vallotton et al. 2004; Ponti et al. 2005). The lamellipodium and lamellum have distinct network structures, different turnover rates and are driven by distinct forces, viz. actin assembly and disassembly in the lamellipodium, and contractile forces in the lamellum (Ponti et al. 2004). In order to produce directed cell movement, the interaction of the actin subnetworks and force transmission to the substrate must be properly integrated in space and time. In addition, how much force a cell exerts is a function of the substrate: on a rigid, adhesive substrate they can generate large contractile forces, but on a pliable substrate they exhibit less organized actin and smaller, weaker adhesion complexes (Lo et al. 2000). One method of probing the mechanical response of a cell is to measure the traction patterns that a cell exerts on a deformable substrate (Burton et al. 1999; Munevar et al. 2001; Saez et al. 2007; Rabodzey et al. 2008), and results from studies in keratocytes (Lee et al. 1994; Oliver et al. 1999; Doyle et al. 2004) will serve as a test of our model of cell motility.
Understanding the coherent interplay between cellular subprocesses and extracellular properties that produces movement requires a mathematical model that links molecular-level behaviour with macroscopic observations on forces exerted, cell shape and cell speed, but how to formulate a multi-scale model that integrates the microscopic steps into a macroscopic model is poorly understood. Numerous models that address components of individual cell motility have been proposed, and the interested reader is referred to Lim et al. (2006), Mogilner (2006) and Flaherty et al. (2007) for reviews. The model for cell motility that is proposed here differs from previous work in several essential ways. It accounts for arbitrary three-dimensional large deformations that cells undergo as they move, and it incorporates experimentally observed viscoelastic material properties (Bausch et al. 1998; Feneberg & Westphal 2001). This model is a first step towards formulating a multi-scale model of cell motility; further steps will be the inclusion of signal transduction, control of network formation and dynamic cell–substrate adhesions.
(b). Tissue growth and movement
Cell growth and movement, the latter both individually and collectively as a tissue, play important roles in angiogenesis, morphogenesis and tumour development. Growth produces stresses that can deform neighbouring cells, and, via mechanical signal transduction through adhesion sites, can affect growth and gene expression in those cells. Morphogenesis comprises processes such as pattern formation and cell or tissue movement that produce the complex shapes of adults from the simple ball of cells that results from division of the fertilized egg. Collective motion of cells as a tissue involves coordinated migration in which cells maintain their adhesive contacts while moving. One striking example of this occurs in the early morphogenetic movements in Xenopus (Keller 2005).
To understand movement exhibited by cellular aggregates, one must understand how local interactions between moving cells affect the collective motion. A well-studied system in this context is the cellular slime mould Dictyostelium discoideum (Dd). In early aggregation the cells move autonomously, but in late aggregation they form connected streams that migrate towards the pacemaker cell. These streams converge in a loose mound of cells that usually topples over to form a cigar-shaped mass called the slug. How individual cell behaviour produces the collective tissue-like motion during aggregation is only partially understood. A cell-based model described later leads to the conclusion that the force exerted by a slug on a substrate scales with the number of actively moving cells in contact with the substrate, not with the volume of the slug (Dallon & Othmer 2004). This conclusion is, of course, not only applicable to the Dd slug, but wherever tissue movements are involved.
In the following section we introduce a new continuum model for a single cell that incorporates many of the mechanical processes involved in cell motility, and apply it to keratocyte motion. In §3 we discuss cell-based and hybrid models for growth and movement in tissues and tissue-like cellular aggregates, and apply these models to the context of tumour growth.
2. A continuum model for single cells
(a). The patterns of movement in model systems
Fragments of motile fish epithelial keratocytes have a flat, crescent-shaped bilaterally symmetric structure, whereas stationary fragments or whole cells are disc-shaped and radially symmetric (Verkhovsky et al. 1999; Keren et al. 2008). Application of a localized mechanical stimulus to a stationary fragment induces global reorganization of the actin network and initiates movement (Henson et al. 1999; Yam et al. 2007), but random perturbations can also initiate movement in approximately 30 per cent of the cells per hour (Yam et al. 2007). The leading edge of a fragment is a brush-like network, which merges into a region of longer, sparsely bundled F-actin in the centre of the fragment, connected to a highly bundled transverse network at the rear (Svitkina et al. 1997). Labelled actin in moving keratocytes exhibits slow retrograde flow relative to the substrate (Jurado et al. 2004; Vallotton et al. 2004), whereas in stationary cells it flows centripetally from the cell edge to the cell body at a rate of approximately 25–60 nm s−1. It has recently been shown that the dynamics of the actin network coupled to the tension in the keratocyte membrane couples spatially distinct processes in the cell (such as extension and contraction), thereby determining the shape of the cell (Keren et al. 2008).
Keratocyte motion is smooth, but other cell types move in a more pulsatile manner. For example, Giannone et al. (2007) show that extension of the lamellipodium is periodic in mouse embryonic fibroblasts. In each cycle, the adhesion strength at the front of the lamellipodium increases due to concentration of integrin clusters. They suggest that, once the adhesion sites are initiated, the weakest link of the fibronectin–integrin–actin connection is the interaction with actin, and this results in periodic tearing of the network from the adhesion sites with a period of about 25 s. Uchida et al. (2003) measured the rate of the displacement of the leading edge and the rear of Dd cells, and found that both rates as well as the total area of the cell–substrate contact region vary periodically. Koehl & McNally (2002) found that myosin II at the rear of Dd undergoes an oscillatory ‘C-to-spot’ redistribution, which is required for the periodic contractility of the rear. Thus, the overall motion in these cells is quite different from the steady gliding motion seen in keratocytes.
In the remainder of this section we describe a computational model designed to shed light on the diverse patterns of movement described earlier, and we present some preliminary results.
(b). The description of active and passive deformation
Cell motility involves large deformations and a free boundary, the latter because the spatial position of the boundary of a deforming cell has to be determined. We adopt a Lagrangian description in which the boundary comprises material points and moves under the deformation. We denote by 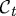 the set of points in space occupied by the cell at time t, we label points in
the set of points in space occupied by the cell at time t, we label points in 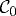 by X, and points in
by X, and points in 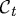 for t>0 are given by the mapping
for t>0 are given by the mapping
| 2.1 |
which satisfies x(X,0)=X. The gradient of this mapping,
| 2.2 |
which governs infinitesimal stretching and rotation via the relation dx=F dX, is a second-rank tensor called the deformation gradient. Here, dX is a vector connecting two nearby material points in the initial configuration, and x is a vector connecting the images of these points in the current configuration at time t>0. The displacement vector is defined as the difference u=x−X, and leads to an alternative definition of the deformation gradient, viz.
| 2.3 |
The deformation of the cell is the result of active and passive processes, the former stemming from actin polymerization that deforms the leading edge and the action of actomyosin contractile forces in the interior. The passive component stems from the passive material properties embodied in the viscoelastic description described later. Together, these produce local deformations in the form of extensions and contractions, and a passive resistance to these forces and external forces. Even in the absence of external forces, a typical cell is under residual stress due to local internal deformations, and interactions with other cells or a substrate add to these stresses.
Active processes that produce extension and contraction, which we term active deformation, are incorporated into the model by postulating a multiplicative decomposition of the total deformation gradient F into a passive part FP and an active part FA, such that
| 2.4 |
As shown in figure 1, the factorization postulates a two-step deformation process in which FA first maps the original configuration into a fictitious, intermediate local configuration that by definition is assumed to be stress-free. It is not necessarily true that the partial deformation generated by the active component produces a smooth configuration; discontinuities may result from incompatibilities present in the active part of the deformation gradient, which, unlike F itself, does not result from a displacement field but must be explicitly defined. In other words, if the configuration is to remain locally stress-free, adjacent material segments may have to separate as they deform. FP enforces compatibility of the deformations and accounts for body forces and surface tractions that may be acting on the cell. The fact that F results from a continuous displacement field implies that FP is incompatible whenever FA is.
Figure 1.

Decomposition of deformation gradient.
A multiplicative decomposition of the deformation gradient was introduced in Lee & Liu (1967) to model elastoplasticity, and has subsequently been used to describe elastoplasticity and thermoelasticity. It has also been used to describe growth in biological systems such as the heart, blood vessels and cancerous tumours (Skalak 1981; Rodriguez et al. 1998; Taber & Perucchio 2000; Ambrosi & Mollica 2002). To date, there has been no derivation of this form from molecular-level descriptions of a material, but it can be shown that, in the small strain limit, the multiplicative decomposition of F reduces to an additive decomposition of the strain.
(c). The equations of change and the constitutive equation
We assume throughout that the system is isothermal and that the stress tensor is symmetric, and therefore we only require equations of change for mass and linear momentum. We assume that the cytoplasm is incompressible (Wilkes & Athanasiou 1996; Vasiev & Weijer 2003), and therefore the mass balance equation is given by
| 2.5 |
where v=du/dt is the velocity. Since cell motion is slow, we neglect inertial forces, but this assumption may not be valid for all cell types, especially those that exhibit relatively rapid recoil at the leading edge. In addition, we neglect external fields such as gravity, and therefore the momentum balance equation is given by
| 2.6 |
where σ is the Cauchy stress tensor, and p is a pressure term that is used to satisfy the incompressibility constraint. The symmetry of the stress tensor σ guarantees conservation of angular momentum.
To complete the formulation, we have to specify constitutive equations and the boundary conditions. Given the displacement field, one can calculate the full deformation gradient according to equation (2.3). One can show that its rate of change is related to the velocity field via
| 2.7 |
where ∇ is the gradient taken with respect to spatial coordinates, and 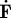 is the material time derivative of F (Holzapfel 2000). The active component of the deformation captures extension and contraction of the cell, and, as a result, should depend on local filamentous actin and myosin II concentrations. In this section, we are only concerned with the mechanics of motion, not the biochemistry, and we compute the active deformation gradient by assuming that
is the material time derivative of F (Holzapfel 2000). The active component of the deformation captures extension and contraction of the cell, and, as a result, should depend on local filamentous actin and myosin II concentrations. In this section, we are only concerned with the mechanics of motion, not the biochemistry, and we compute the active deformation gradient by assuming that
| 2.8 |
and specifying LA. The tensor LA defines the rate and direction of extension and contraction, and, in effect, we later specify LA via a constitutive relation. To satisfy the incompressibility condition, LA must have zero trace (Tr(LA)=0), and to obtain FA for a given LA, one must solve the nine-dimensional system of differential equations (2.8) with the initial conditions FA(0)=I.
The passive component of the response is determined by the cytoplasm, which in many cell types has been characterized as a viscoelastic material comprising actin filaments, intermediate filaments and microtubules, collectively termed the cytoskeleton (Janmey 1991). The elastic modulus of actin solutions is concentration dependent (MacKintosh 1998), and these solutions exhibit strain hardening (Xu et al. 2000), a property that may be important in tissue movement. The cytoplasm in Dd has been characterized as an ‘active viscoplastic’ material (Feneberg & Westphal 2001), because it exhibits viscoelastic behaviour above a yield stress, but little deformation below this level. The cytoplasm of other cell types has similar properties: it is viscoelastic in leukocytes and neutrophils (Evans & Yeung 1989; Bausch et al. 1998; Heidemann et al. 1999), but large regional variations in elasticity and viscosity coefficients are found within a cell (Yanai et al. 1999; Laurent et al. 2005).
We shall use a viscoelastic material model for the passive response, and the simplest constitutive equation that captures some of the essential characteristics is termed the standard solid model. For small strains in one dimension, this is described by the following relation between stresses (σ), strains (ε) and their rates of change:
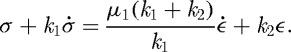 |
2.9 |
Here k1, k2 and μ1 are elastic constants and the viscosity coefficient, respectively. For our purposes, this must be extended to an equation that is appropriate for an actively deforming body that undergoes large strains in three space dimensions. It is known that incompatible active deformation strains lead to residual stresses, but if the active deformation is compatible, as happens if the extension or contraction rate is spatially uniform and there are no applied loads, the stresses should vanish throughout. This stems from the fact that the local, intermediate, stress-free configuration will form a continuous configuration globally, and there exist displacements 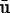 such that
such that 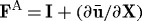 . If no external forces are present, then the displacement in equation (2.3) satisfies
. If no external forces are present, then the displacement in equation (2.3) satisfies 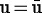 , and it follows that F=FA, FP=I, and the stress field vanishes. These requirements can be met if at every point of the cell the constitutive equation relates the local value of the stress to the local value of the passive component of the deformation gradient FP.
, and it follows that F=FA, FP=I, and the stress field vanishes. These requirements can be met if at every point of the cell the constitutive equation relates the local value of the stress to the local value of the passive component of the deformation gradient FP.
The constitutive equation used here reflects this property and properly incorporates the effects of active deformation. It is a large-strain, three-dimensional equivalent of equation (2.9) that has the following form:
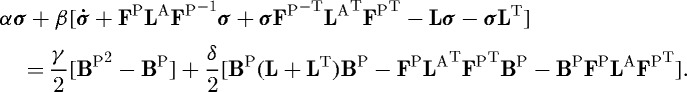 |
2.10 |
Here,
| 2.11 |
is the Finger tensor (Holzapfel 2000) for the passive part of the deformation, and α, β, γ and δ are material constants. One can show that this equation transforms properly under rigid body motion (Holzapfel 2000); details will be provided elsewhere.
(d). Boundary and interface conditions
In the remainder of this section we consider cell movement on a deformable substrate. If no displacement boundary conditions are specified on a surface, that surface is assumed to be traction-free. All displacements and forces transmitted across the cell–substrate interface are continuous at the attachment sites that lie on this interface. All normal displacements are fixed on the boundaries of the substrate, except for the top boundary, which is traction-free outside of the zones where the cell and substrate are attached. Thus, there are no interaction forces between the two surfaces except at the contact sites. To account for forces that may arise when the two surfaces are in contact at sites other than adhesion sites, the current interface conditions must be modified into ‘contact conditions’, as will be discussed later. The boundary and cell–substrate interface conditions we use are illustrated in figure 2a and are summarized in the following:
| 2.12 |
| 2.13 |
| 2.14 |
| 2.15 |
and
| 2.16 |
Here n, σ and p (nsub, σsub and psub) are the unit outer normal, the stress and the pressure in the cell (substrate), ∂Ωa represents any part of the cell boundary that is attached to the substrate, ∂Ωb is the bottom surface of the cell that is not an attachment site, ∂Ωsub refers to the portion of the substrate on which normal displacements are fixed and ∂Ωt is the remainder of the boundary. The union of the individual components constitutes the boundary of the cell and substrate, and thus 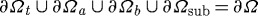 .
.
Figure 2.

(a) Displacement boundary conditions for cell motility on a deformable substrate. (b) One set of attachment sites under consideration in the keratocyte model. (c) The second set of attachment sites used for the numerical experiments of the keratocyte.
It should be noted that, because we account for active stresses in the factorization of the deformation gradient, they enter into the momentum balance equation via the stress tensor, and volume-based stress automatically translates into boundary surface forces via the divergence theorem. Thus, if one integrates the momentum equation (2.6) over the volume of the cell one obtains
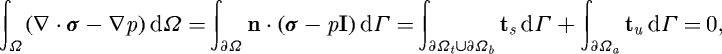 |
2.17 |
where ts is the applied surface traction and tu is the resultant traction that occurs at regions of attachment to the substrate. As indicated in equation (2.12), all applied surface tractions are zero (ts=0), and it is clear that the centre of mass of the cell cannot move if it cannot transmit force to the substrate, i.e. if tu≡0. Thus, the formulation of active stress is self-consistent; no further constraints are needed, unlike models in which active forces are specified in non-divergence form.
(e). Computational results
Next, we apply the foregoing model to a keratocyte in the initial stages of movement over a deformable substrate. In experiments done by Lee et al. (1994), Oliver et al. (1999) and Doyle et al. (2004), keratocytes are placed on a deformable substrate into which microbeads were implanted, and traction forces exerted by a cell on the substrate are determined from the bead displacements. Lee et al. (1994) and Oliver et al. (1999) observe that the displacements at the leading edge of the keratocyte are in the direction of motion, rather than in the opposite direction, as was observed for other cell types (Munevar et al. 2001). At the lateral edges of the cell, the beads are displaced perpendicular to the direction of motion, and these displacements are significantly larger than those at the leading edge. However, in Doyle et al. (2004) it is shown that the beads at the leading edge of the motile keratocyte are displaced towards the interior of the cell, opposite to the direction of motion. It has not been established what accounts for these differences, but they may stem from differences in the attachment of keratocytes to the substrate (Doyle et al. 2004), which is gelatin in Doyle et al. (2004) and silicon in Lee et al. (1994) and Oliver et al. (1999). Our goal in this section is to use the model to shed light on differences between the two sets of experimental results and to use the most recent experimental results in Doyle et al. (2004) to test the validity of the model.
The observed traction patterns in all three of the foregoing experiments lead to the conclusion that the strongest actomyosin contractility is at the rear of the cell, and that the extending actin network at the leading edge shows significantly less contractility. These conclusions are further supported by the fact that the actin network in a keratocyte has a bundled structure with associated myosin II filaments in the transition zone between the lamellipodium and the cell body, leading to significantly larger contractility there than at the leading edge of the cell (Svitkina et al. 1997). Experiments by Burton et al. (1999) and additional experiments described in Oliver et al. (1999) demonstrated similar keratocyte traction patterns measured by substrate wrinkling.
The adhesions in rapidly moving cells such as keratocytes are generally much weaker than those of slowly moving cells such as fibroblasts. Lee & Jacobson (1997) observe dynamic close contacts in a narrow rim at the leading edge and in small foci throughout the lamellipodium of a keratocyte. Focal adhesions, which are significantly stronger attachments than close contacts, were observed at the tips of actin stress fibres located at the rear of the cell. Anderson & Cross (2000) also demonstrate that the strongest cell–substrate adhesions in keratocytes are found at the rear of the cell.
To apply the model to keratocyte motility we must specify the rate and localization of extension and contraction, the location of the cell–substrate adhesions, the initial cell shape and the material properties. We base the form of LA used later on the experimental observations described earlier, and assume that extension occurs at the cell’s leading edge in a direction perpendicular to the leading edge, and that contraction occurs at the rear in the direction perpendicular to the lateral sides and the rear edge. We also incorporate the effect of increased contractility at the leading edge of the cell on the traction patterns occurring in the substrate. The specific form of the rate of change of the active component of the deformation gradient used here is
| 2.18 |
Here, NXY is the unit vector in the xy-plane at the periphery of the contact area between cell and substrate in the initial configuration, k is a unit vector orthogonal to the xy-plane, and N⊥ is the unit vector in the direction 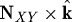 . The amplitude A1,2 is given by one of two functions,
. The amplitude A1,2 is given by one of two functions,
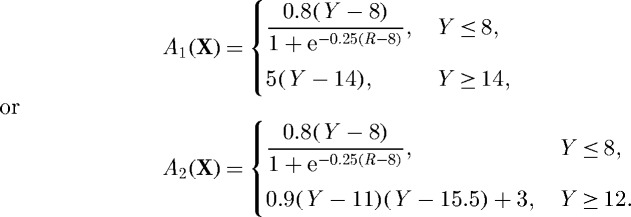 |
2.19 |
Here 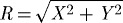 and X and Y are coordinates of the cell in its initial configuration. The maximum and minimum values of A1,2(X) are chosen so that the speed of the cell compares to an average experimentally determined value of approximately 30 μm per min (Doyle et al. 2004). From the rear to its leading edge, the domain of the cell is given by −5≤Y ≤18. Therefore, the function A1 represents the case in which the contractility at the leading edge is negligible compared with that at the rear, and primary extension due to actin polymerization occurs in the region in front of leading edge adhesion sites. In the function A2, we consider the case in which contractility at the leading edge is significant. In this case, we assume that the cell has a thin contractile band directly behind the zone in which actin polymerization, and therefore extension, is occurring.
and X and Y are coordinates of the cell in its initial configuration. The maximum and minimum values of A1,2(X) are chosen so that the speed of the cell compares to an average experimentally determined value of approximately 30 μm per min (Doyle et al. 2004). From the rear to its leading edge, the domain of the cell is given by −5≤Y ≤18. Therefore, the function A1 represents the case in which the contractility at the leading edge is negligible compared with that at the rear, and primary extension due to actin polymerization occurs in the region in front of leading edge adhesion sites. In the function A2, we consider the case in which contractility at the leading edge is significant. In this case, we assume that the cell has a thin contractile band directly behind the zone in which actin polymerization, and therefore extension, is occurring.
To test the effect of adhesions on substrate traction patterns, we consider two configurations. In one configuration, we assume that the largest adhesions are localized to the lamellipodium–cell body transition zone, and that smaller adhesions are also found near the cell’s leading edge (cf. figure 2b). In the second configuration, we consider only attachments at the lamellipodium–cell body transition zone (cf. figure 2c). In this second configuration we assume, in effect, that any attachments at the leading edge are significantly weaker than those at the rear. As a simplification, we further assume that all adhesions are circular patches, as illustrated in figure 2b,c, and that the cell is attached to a deformable substrate there. We assume that the adhesions are fixed and only consider the movement that is possible for the given set of adhesions. This results in a translocation of approximately 2–2.5 μm over the course of 5.7 s. Incorporating dynamic adhesions into a model of cell–substrate interaction, which will allow us to investigate cell motility over longer spatial and time scales, is part of our future work.
It has been shown that there is a gradient of rigidity in the lamellipodium of keratocytes (Laurent et al. 2005), and we therefore assume that the leading edge of the cell is stiffer than the rear, and that the elastic modulus is given by
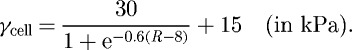 |
2.20 |
The rheological model used for the cell is also used to describe the mechanics of the substrate, but with a different elastic modulus and without active deformation. We use the elastic modulus γsub=2 kPa for the substrate, as reported in Doyle et al. (2004). We assume that both the cell and the substrate are slightly viscous, and for both we set the remaining parameters as α=1 (unitless), β=0.1 in min and δ=0.1 in kPa min.
Figure 3a shows the computational domain at t=0. The cell is shown in light grey, and the substrate to which it is attached is shown in dark grey. Figure 3b shows a view from above of the original position, outlined in black, and the final location, at t=5.7 s. This particular deformed configuration is obtained via the extension–contraction function A1 and the full set of adhesions illustrated in figure 2b. Based on the displacement fields measured in Lee et al. (1994), we assume that at this time the cell must detach from the substrate in certain regions (presumably at the rear) and form new adhesions in other regions. We note that the lamellipodium of the simulated cell is significantly thicker than it is in reality (5 μm versus <1–2 μm). Performing three-dimensional finite-element simulations on a lamellipodium of a more realistic thickness would present significant challenges, but the thickness we use nevertheless allows us to predict accurately the tractions at the cell–substrate interface.
Figure 3.

(a) Initial position of the keratocyte cell (light grey) and the substrate (dark grey). (b) The final position of the cell, viewed from above, for a given set of attachments. The initial position of the cell is outlined in black.
In figure 4, we exhibit the qualitative displacement patterns resulting from three combinations for the location of the extension–contraction/adhesion sites. In figure 4a the displacement patterns are obtained by applying the extension–contraction function A1 to a cell that is attached at the full set of adhesion sites shown in figure 2b. For this choice, the contractility at the leading edge is negligible compared with both the extension there and the contractility at the rear, and we assume that the attachments at the front are relatively strong. The displacement field in figure 4b is obtained by applying extension–contraction function A1 to a cell that is only attached towards its rear, as illustrated in figure 2c. In essence, in this case we assume that leading edge adhesions are very weak. Finally, in figure 4c we show the displacement patterns obtained using the function A2 and the full set of leading edge and rear adhesions. In this case, contractility at the leading edge is relatively strong. Because the displacements at the lateral edges of the cell are significantly larger than the displacements at the front of the cell, we have normalized the displacement vectors and scaled these normalized vectors by a factor of four to visualize better the qualitative nature of the displacement patterns. In all three cases, the black outline corresponds to the position of the deformed cell.
Figure 4.

(a) Displacement pattern resulting from extension–contraction function A1 and the full set of adhesion sites shown in figure 2b. (b) Displacement pattern for function A1 and only rear adhesions, as in figure 2c. (c) Displacement pattern for function A2 and the full set of adhesion sites. In all three figures the displacement arrows are uniform in length and do not reflect the magnitudes of the displacements.
The displacement patterns obtained under the three cases considered are qualitatively different in nature. When the contractility at the leading edge is negligible and the full set of adhesions are used, the displacements throughout much of the cell are oriented towards the interior of the cell (figure 4a). This displacement pattern is most similar to those observed in Doyle et al. (2004). When the contractility at the leading edge is negligible but the attachments there are released, the displacement field at the leading edge is oriented towards the direction of motion, while some displacements towards the rear of the cell are oriented in the opposite direction (cf. figure 4b). When contractility at the leading edge is significant, the entire displacement field beneath the cell is oriented towards the direction of motion (cf. figure 4c). These displacement patterns in figure 4b,c are more similar in nature to those observed in Lee et al. (1994) and Oliver et al. (1999).
As discussed later, these results suggest that the discrepancy between the experimentally measured traction patterns in the keratocyte stems either from a difference between the adhesion properties of a keratocyte adhering to a gelatin substrate versus adhering to a silicon substrate, or from a difference in contractile properties of the cell on these substrates. Furthermore, we note that, in all the cases considered here, the displacements in front of the leading edge are oriented in the direction of motion. This indicates that, regardless of the extension–contraction pattern at the leading edge, most of the force required for keratocyte motion is generated by contraction of the rear and lateral edges of the cell; in essence, keratocytes push themselves forwards. The traction patterns here are unlike those that we observed in numerical simulations of a fibroblast. In that case, the cell pulls itself forwards by strong contraction of the region directly behind the extending leading edge, and this type of mechanism results in a rearward displacement pattern in front of the leading edge (data not shown). This has been described as the ‘frontal-towing’ model of movement (Munevar et al. 2001).
To test the model quantitatively we compare the numerically computed traction patterns with experimentally determined traction patterns from Doyle et al. (2004). Figure 5 illustrates the direction (a) and magnitude (b) of the numerically computed traction field acting on the substrate at the cell–substrate interface at the final time. These traction patterns are obtained by applying extension–contraction function A1 and the full set of leading edge and rear adhesions. This combination was chosen because the qualitative displacement patterns for this case are most similar to those observed in Doyle et al. (2004). The experimentally determined maximum traction field from Doyle et al. (2004) is shown in figure 5c(i,ii). The magnitude of the largest experimental traction is approximately 4 kPa, and that of the largest numerical traction is approximately 1 kPa. While the maximum traction magnitudes compare in order of magnitude, we speculate that the primary reason for the discrepancy between the numerical and experimental results is the fact that the current model does not incorporate cell–substrate attachment dynamics. The time course of experimentally measured shear traction magnitudes (Doyle et al. 2004) is illustrated in figure 5d(i–iv). As noted in Doyle et al. (2004), the magnitudes and localization of cell–substrate surface tractions depend on calcium transients and intracellular tension, and, over the time course considered, the maximum shear tractions vary from approximately 1 kPa to approximately 6 kPa. The future incorporation of dynamic cell–substrate attachments into the model should result in an even more accurate comparison with experimental data.
Figure 5.

(a) Numerically computed shear traction vectors (tshear=〈σxz,σyz〉) magnified four-fold. (b) Magnitude of the numerically computed shear tractions (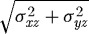 in kPa) acting on the substrate at the cell–substrate interface for a substrate stiffness of 2 kPa. (c) Experimentally determined maximum shear traction vectors (i) and magnitudes (ii) from Doyle et al. (2004). (d) Time course of traction magnitudes as measured in Doyle et al. (2004). (1 kPa=1×104 dyne cm−2.)
in kPa) acting on the substrate at the cell–substrate interface for a substrate stiffness of 2 kPa. (c) Experimentally determined maximum shear traction vectors (i) and magnitudes (ii) from Doyle et al. (2004). (d) Time course of traction magnitudes as measured in Doyle et al. (2004). (1 kPa=1×104 dyne cm−2.)
3. Tissue-level models of growth and movement
At present, it is computationally prohibitive to use the detailed cell-based model described in the previous section for a growing, deforming tissue. However, it is desirable to incorporate some cell-level detail into a model for such tissues, and in this section we present some applications of a previously developed model to this problem.
(a). Cell growth in a tissue
As elaborated in Kim et al. (2007), three factors needed to describe individual cells in a growing tissue are (i) how an individual cell reacts to forces on it, (ii) how cells interact mechanically with their surroundings, and (iii) how growth and division are described, and how stress affects growth.
The mechanical behaviour of individual cells is based on the model developed by Dallon & Othmer (2004) (hereafter the paper and model are denoted DO), and the growth and division are modelled as in Kim et al. (2007) (hereafter the model and paper are referred to as KSO). In general, the cells are treated as oriented ellipsoids whose cytoplasm is an incompressible, viscoelastic solid. Growth is included in series with the active response and the passive forces (cf. §3b), which at the continuum level leads to a factorization of F like that used in the previous section. Here, we do not consider chemotaxis driven by active motile forces, but comment later on some consequences if it is included. When cells do not grow, their volume is constant under all deformations, but when there is growth this constraint does not apply. We define V0 as the volume cells attain immediately after division, and we assume here that this is the same for all cells.
In the KSO model, stress and nutrient levels affect the growth rate, and we assume that the effect of stress is isotropic. In the absence of nutrient or stress limitations, cells grow to 2V0 and then instantly divide into two equal daughter cells. In the presence of extracellular forces, the orientation of cell division is determined by the direction of the net force exerted on the cell, as others have assumed (Galle et al. 2005). Cells in a growing tissue interact indirectly via the nutrient pool, and this may lead to non-uniform growth in the population and an increased cell-cycle time. Without adequate nutrients cells may enter the quiescent phase, and if the nutrient level drops too low they die or undergo apoptosis. In a tumour this leads to the necrotic core. We also assume that growth halts if the stress is too large in magnitude, and in that case the passive response to the applied force is the same as in the DO model. However, if it is within a certain range, displacement of the solid-growth element increases and asymptotes to the linear growth profile that results from the growth component. A more detailed description can be found in KSO.
It is shown in KSO that the governing equations of the length of the ith axis, i=a,b,c, of a cell are
| 3.1 |
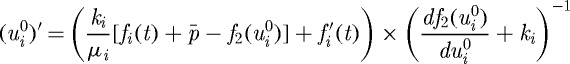 |
3.2 |
and
| 3.3 |
where ui is the change in the length of the ith axis, 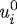 (
(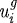 ) the change in the length of the ith axis due to a change in the passive (growth) element, f2 the nonlinear spring force from the spring in parallel (cf. §3b), fi the magnitude of the force applied at each end, μi the viscous coefficient of the dashpot, ki the spring constant for the spring in the Maxwell element,
) the change in the length of the ith axis due to a change in the passive (growth) element, f2 the nonlinear spring force from the spring in parallel (cf. §3b), fi the magnitude of the force applied at each end, μi the viscous coefficient of the dashpot, ki the spring constant for the spring in the Maxwell element, 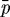 the pressure force and f the growth function given in equation (3.4). The specific form of the function f2 and details of how these equations are established are given in DO. To simplify the later computations of tumour growth, we hereafter consider only two-dimensional cells, which are ellipses, rather than ellipsoids; results on a full three-dimensional version will be reported elsewhere.
the pressure force and f the growth function given in equation (3.4). The specific form of the function f2 and details of how these equations are established are given in DO. To simplify the later computations of tumour growth, we hereafter consider only two-dimensional cells, which are ellipses, rather than ellipsoids; results on a full three-dimensional version will be reported elsewhere.
The effect of stress on growth is described by the relation 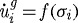 , for i=a,b,c, where σ is the axial component of the force applied along the ith axis. Here, we use a piecewise linear function and include the effect of tensile as well as compressive stresses (cf. figure 6).
, for i=a,b,c, where σ is the axial component of the force applied along the ith axis. Here, we use a piecewise linear function and include the effect of tensile as well as compressive stresses (cf. figure 6).
Figure 6.

The growth rate function f(σ). From Kim et al. (2007), with permission.
Thus f(⋅) is given by
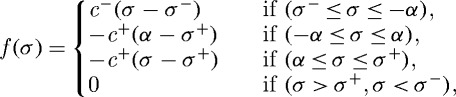 |
3.4 |
where c+,c− are positive constants, σ+>0, σ−<0, [σ−,σ+] is the interval of positive growth, and c+(α−σ+)=−c−(−α−σ−). Finally, Newton’s law for the ith cell reduces to
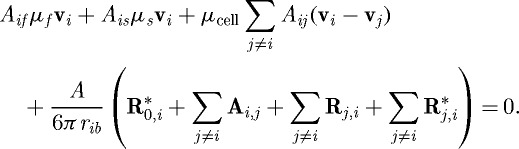 |
3.5 |
Here A=A(t) is the total area of an undeformed cell; Aij=Aij(t), Aif=Aif(t) and Ais=Ais(t) are the lengths of contact regions between cell i and cell j, cell i and the extracellular fluid, and cell i and the substrate at time t, respectively; μcell (respectively μs,μf) is the degree of adhesiveness between the cells (respectively, between the substrate and the cells, the fluid viscosity); and rib=ub+b0 where b0 is the initial length of the b axis and vi is the velocity of the ith cell. The capital boldface terms represent reactive cell–substrate and cell–cell forces (R*), static attachment forces between cells (A) and forces due to deformation of a cell by neighbours (R). The base parameters that characterize the cells are given in table 1; those that are changed later will be noted. Details as to how the various terms in equation (3.5) are computed can be found in DO.
Table 1.
Parameters for the cell-based component of the model. From Kim et al. (2007), with permission.
| parameter | description | value | refs |
|---|---|---|---|
| adhesion parameters | |||
| μcell | cell–cell adhesiveness | 27.0 dyne cm−1 | Dallon & Othmer (2004) |
| μs | cell–substrate adhesiveness | 27.0 dyne cm−1 | Dallon & Othmer (2004) |
| μf | fluid viscosity | 2.7 dyne cm−1 | Dallon & Othmer (2004) |
| rheological parameters | |||
| c+ | growth function parameter | 5.16089×10−9 mm (min nN)−1 | |
| σ+ | growth function parameter | 800 nN | |
| σ− | growth function parameter | −4 nN | |
| α | growth function parameter | 0.0 nN | |
| ka | standard solid parameter | 163.8 dyne cm−1 | Dallon & Othmer (2004) |
| k2 | standard solid parameter | 147.5 dyne cm−1, | Dallon & Othmer (2004) |
| μa | standard solid parameter | 123 dyne min cm−1 | Dallon & Othmer (2004) |
The cell-based KSO model facilitates a variety of computational experiments that probe the effects of variations in cell parameters and allows the tracking of lineages of specific cells. We first examine various behaviours of cells in a two-dimensional layer supplied with adequate nutrients. In figure 7a–f we show how clones evolve and how their spatial localization changes with time. One sees there how the competition for space affects the size of clones: cells in the interior of an aggregate grow slowly compared with those on the outer boundary because they are compressed by surrounding cells (see also figure 7g) even in the absence of constraints at the edge of the tissue. The simulation reveals an asymmetric pattern of clones and irregular boundaries between them, which are dictated in part by the initial conditions. The dramatic effect of different levels of the compression parameter σ− on the total number of cells is shown in figure 7h, where one sees that cells grow faster for smaller (more negative) values of σ−.
Figure 7.

Tumour growth in the presence of adequate nutrients. (a–f) Tumour growth. Parameters σ−=−4.0 nN, σ+=800.0 nN, c+=5.16089× 10−9 mm (min nN)−1 were used. (a) t=0 h, 7 cells; (b) t=60 h, 28 cells; (c) t=132 h, 179 cells; (d) t=168 h, 343 cells; (e) t=204 h, 650 cells; (f) t=276 h, 1614 cells. Diameter inside each figure is in the unit of 10 μm. (g) The occupancy (%) for each clone corresponding to (a–f). Notice that the occupancy by cells in the central clone (black cells) decreases significantly compared to other types due to the stress effect on growth. (h) Growth kinetics for different level of the compression parameter σ−=−400 nN (red circles), −4 nN (blue squares) and −0.04 nN (green crosses).
In a different vein, Dorie et al. (1982) investigated the internalization of cells in tumour cell spheroids by adhering cell-size microspheres on the surface of tumour spheroids. They found that these microspheres were internalized during growth, which demonstrated that cell sorting and cell movement occur during tumour spheroid growth. From a comparison of the movement of microspheres and labelled cells, they concluded that tumour growth involves active cell movement and cell–cell interaction rather than just passive movement, as for the microspheres. Here we test our model by tracking adherent discs, which were assumed to have the same internal mechanical properties as cells, but do not grow and have very weak adhesion to other cells. This can be compared with the labelled cells and microspheres seeded on the surface of the spheroid (cf. figure 8). As Dorie et al. observed in their experiments, we observe that the internalization pattern of these labelled cells (figure 8a) is different from that of the microspheres (figure 8b). Many labelled cells are located in the rim of the spheroid, rather than relocating towards the spheroid centre (cf. figure 8c). In contrast, we see significant internalization of discs during growth (figure 8f,d), while labelled cells tend to stay in the outer rim of the spheroid (figure 8e). These results indicate that the different behaviour of labelled cells and microspheres in Dorie’s experiments may be because of passive engulfing of the former, and only passive movement because of growth for the latter; active movement does not seem to be essential.
Figure 8.

Comparison of simulation results with experiments. (a) H3-labelled cells on the surface of EMT6 spheroids at 24 h (Dorie et al. 1982). (b) Cells remain at the surface in the simulations as observed experimentally in (a). (c) Internalized microspheres in spheroids at 48 h (Dorie et al. 1982). (d) Simulation results showing internalized discs. (e–f) Simulation results showing frequency of distance from the edge for (e) labelled cells at 0, 54, 114, 167 h and (f) discs (microspheres) at 0, 84, 198, 270 h. In the simulations, the labelled cells and discs were placed on the surface initially. ((a,c) Reprinted from Dorie et al. (1982), with permission.)
(b). A hybrid model for tissue growth and tumour development
The next step is to incorporate the mechanical interaction of a growing tissue with its microenvironment in vitro, which enables us to study the effect of external stresses on growth. There are up to four geometrically distinct regions in the hybrid model: the extracellular matrix (ECM) or agarose gel surrounding the tumour, a shell of actively proliferating cells at the outer edge of the tumour, a quiescent zone bordering the actively proliferating region, and a necrotic core (cf. figure 9). We denote these  and
and  , respectively. The actively proliferating region comprises a layer 3–5 cells thick in the radial direction. We assume that the outer gel, the quiescent region and the necrotic region are homogeneous materials, and in the model we represent them as continua. These regions have the same rheological properties, but different material parameters are used in
, respectively. The actively proliferating region comprises a layer 3–5 cells thick in the radial direction. We assume that the outer gel, the quiescent region and the necrotic region are homogeneous materials, and in the model we represent them as continua. These regions have the same rheological properties, but different material parameters are used in  and
and  . The irregular boundaries between the cell-based region
. The irregular boundaries between the cell-based region  and the continuum regions
and the continuum regions  and
and  are represented by two artificial boundaries across which the forces are transmitted, as shown in fig. 3 in KSO. The necrotic region
are represented by two artificial boundaries across which the forces are transmitted, as shown in fig. 3 in KSO. The necrotic region  is defined as a subdomain of the tumour interior in which the appropriate nutrient levels are below specified thresholds. Different diffusion coefficients are used in each region and the nutrient uptake function is taken into consideration only in tumour regions.
is defined as a subdomain of the tumour interior in which the appropriate nutrient levels are below specified thresholds. Different diffusion coefficients are used in each region and the nutrient uptake function is taken into consideration only in tumour regions.
Figure 9.

A schematic showing the notation used for the subdomains, the representation of cells in the proliferating zone as ellipsoids, and the representation of the standard solid and growth elements that characterize the internal rheology of each cell in  . (Reprinted from Kim et al. (2007), with permission.)
. (Reprinted from Kim et al. (2007), with permission.)
The proliferating zone  comprises a few hundred cells that grow and divide as dictated by nutrient conditions, and whose shape changes are governed by their internal rheology and the forces acting on them. As discussed in the previous section, we assume that cells grow as long as σ∈[σ−,σ+] and they have adequate nutrients. Some of the cells in
comprises a few hundred cells that grow and divide as dictated by nutrient conditions, and whose shape changes are governed by their internal rheology and the forces acting on them. As discussed in the previous section, we assume that cells grow as long as σ∈[σ−,σ+] and they have adequate nutrients. Some of the cells in 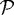 become quiescent when the level of nutrients drops below the threshold, and since the quiescent region
become quiescent when the level of nutrients drops below the threshold, and since the quiescent region  is represented as a continuum, this requires that those cells be transformed into the continuum region
is represented as a continuum, this requires that those cells be transformed into the continuum region  . The displacements of these transformed cells and the forces acting on them are converted into displacements and stress fields in this newly formed continuum material in
. The displacements of these transformed cells and the forces acting on them are converted into displacements and stress fields in this newly formed continuum material in  . To preserve mass during the transformation, it is also assumed that the ECM between cells that are converted into continuum is converted as well.
. To preserve mass during the transformation, it is also assumed that the ECM between cells that are converted into continuum is converted as well.
The outer gel (Ω0) and the inner region (Ωm,m=1,2) are treated as linear viscoelastic materials with different material properties 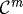 and
and 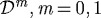 , and therefore the constitutive equations and the momentum equation, neglecting inertial effects, are
, and therefore the constitutive equations and the momentum equation, neglecting inertial effects, are
| 3.6 |
and
| 3.7 |
with boundary conditions u0=0 on Γ0×(0,T), σ0⋅n=q0 on Γc0×(0,T), and σ1⋅n=q1 on Γc1×(0,T). Here  and
and 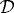 are second-order tensors with entries described in KSO; ε is the strain; Γ0 is the fixed outer boundary; Γc0 is the interface between
are second-order tensors with entries described in KSO; ε is the strain; Γ0 is the fixed outer boundary; Γc0 is the interface between  and
and  ; Γc1 is the interface between
; Γc1 is the interface between  and
and  ; u0 is the displacement field on
; u0 is the displacement field on  ; σ0 and σ1 are the stress fields on Ω0 and Ω1, respectively; and q0 and q1 are boundary forces acting on Γc0 and Γc1, respectively; these are calculated from the cell-based component as indicated in figure 10. These equations are solved using first- or second-order elements in a finite-element discretization. The parameters used in the computations are given in table 2 in KSO.
; σ0 and σ1 are the stress fields on Ω0 and Ω1, respectively; and q0 and q1 are boundary forces acting on Γc0 and Γc1, respectively; these are calculated from the cell-based component as indicated in figure 10. These equations are solved using first- or second-order elements in a finite-element discretization. The parameters used in the computations are given in table 2 in KSO.
Figure 10.

An illustration of how force is transmitted between cells and the continua. From Kim et al. (2007), with permission, wherein details of the algorithm are given.
The nutrients considered here are oxygen and glucose, and we assume that their consumption is described by Michaelis–Menten kinetics. The governing equations for the evolution of the nutrients, assuming Dirichlet boundary conditions, are
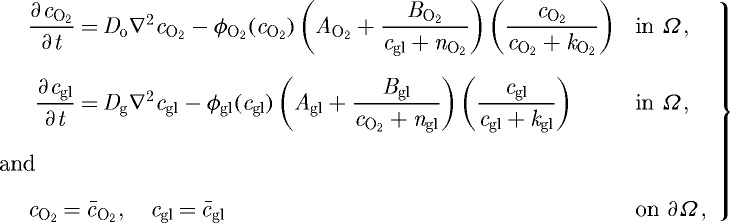 |
3.8 |
where cO2 (cgl) is the molar concentration of oxygen (glucose), the second term of each equation is a function describing the consumption of oxygen (glucose) by the tumour, Do (Dg) is the space-dependent (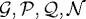 ) diffusion coefficient of oxygen (glucose), AO2, Agl, BO2, Bgl, kO2, kgl, nO2 and ngl are empirically determined parameters, and ϕO2(cO2), ϕgl(cgl) are the cell consumption indicator functions, which give 1 in
) diffusion coefficient of oxygen (glucose), AO2, Agl, BO2, Bgl, kO2, kgl, nO2 and ngl are empirically determined parameters, and ϕO2(cO2), ϕgl(cgl) are the cell consumption indicator functions, which give 1 in  and
and  and 0 otherwise. The parameter values for the reaction–diffusion equations are given in table 3 in KSO. The reaction–diffusion equations (3.8) are solved on a regular grid using an alternating-direction implicit (ADI) scheme and the package nksol (which has been superseded by nitsol) for nonlinear algebraic systems. Details of the computational algorithm can be found in KSO.
and 0 otherwise. The parameter values for the reaction–diffusion equations are given in table 3 in KSO. The reaction–diffusion equations (3.8) are solved on a regular grid using an alternating-direction implicit (ADI) scheme and the package nksol (which has been superseded by nitsol) for nonlinear algebraic systems. Details of the computational algorithm can be found in KSO.
(c). Computational results
In KSO we showed that (i) the shape of a tumour spheroid embedded in an agarose gel is relatively symmetric and much more regular than the shape of a tumour growing in free suspension, and (ii) tumours maintain a viable rim of relatively constant thickness regardless of the stiffness of the surrounding gel. Here, we show that the inhibition of tumour growth increases as the stiffness of the gel increases, i.e. a stiffer gel inhibits tumour growth more effectively, as shown in figure 11a. The diameters of the various regions were determined by taking the average distance from the centre to the nodes on each contact boundary (the  , the
, the 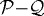 and the
and the 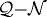 interfaces). Figure 12 shows the linear relationship between the tumour diameter and the diameter of the necrotic core for tumours embedded in an agarose gel of different stiffnesses. This linear relationship has also been found experimentally (Groebe & Mueller-Klieser 1996). One also observes a similar linear relationship between the spheroid diameter and the diameter of the quiescent region. Here, we assumed that the initial tumour diameter is 300 μm, which already includes a necrotic core, but in general the necrotic core will form once nutrient levels go below critical levels.
interfaces). Figure 12 shows the linear relationship between the tumour diameter and the diameter of the necrotic core for tumours embedded in an agarose gel of different stiffnesses. This linear relationship has also been found experimentally (Groebe & Mueller-Klieser 1996). One also observes a similar linear relationship between the spheroid diameter and the diameter of the quiescent region. Here, we assumed that the initial tumour diameter is 300 μm, which already includes a necrotic core, but in general the necrotic core will form once nutrient levels go below critical levels.
Figure 11.

(a) The effect of gel stiffness on tumour growth. Curves 1, 2, 3, 4 correspond to different Young’s modulus Ea of agarose gel, 10, 20, 80 and 200 MPa, respectively, while other parameters are fixed. The diameter of the tumour was defined as 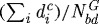 , where
, where 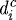 is the distance from the ith node point on the
is the distance from the ith node point on the 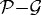 interface to the tumour centre and
interface to the tumour centre and 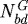 is the number of nodes on the
is the number of nodes on the 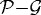 interface. (b) The effect of gel stiffness on packing density: packing density at 137 h, computed as given in the text. (c) The average cell area Ac(t) (normalized) in the
interface. (b) The effect of gel stiffness on packing density: packing density at 137 h, computed as given in the text. (c) The average cell area Ac(t) (normalized) in the 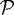 region for each case:
region for each case: 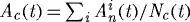 , where
, where 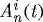 is the normalized cell area and Nc(t) is the number of cells at time t. Curves as in (a). (d) Area distribution of proliferating cells corresponding to cases 1 (light grey), 3 (dark grey) and 4 (black) in (c) at 137 h.
is the normalized cell area and Nc(t) is the number of cells at time t. Curves as in (a). (d) Area distribution of proliferating cells corresponding to cases 1 (light grey), 3 (dark grey) and 4 (black) in (c) at 137 h.
Figure 12.

The linear relationship between the spheroid diameter and the diameter of the necrotic core for tumours in gels of increasing stiffness. (a–d) Young’s modulus Ea of the gel of 10, 20, 80 and 200 MPa, respectively, while other parameters are as in the tables in Kim et al. (2007). The solid grey line in each panel represents the best linear relation between the diameter of the necrotic core and the spheroid diameter (square boxes). The circles are for the  region. The steps in the computational results arise from the manner in which cells are converted to continuum (Kim et al. 2007).
region. The steps in the computational results arise from the manner in which cells are converted to continuum (Kim et al. 2007).
We also investigated the effect of gel stiffness on the packing density of the cells in the proliferating region. Helmlinger et al. (1997, fig. 3c) found experimentally that the packing density increases as the gel stiffness increases, and our computational results reproduce this (cf. figure 11b). The packing density in the simulations is determined from the total area PA of region 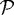 and the area CA covered by the cells in
and the area CA covered by the cells in 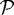 . It is given by 100×CA/PA where PA=total domain area − (
. It is given by 100×CA/PA where PA=total domain area − (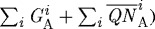 , where
, where 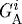 and
and 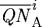 are the areas of the ith finite element in regions
are the areas of the ith finite element in regions 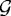 and
and 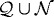 , respectively. The force required to deform a stiffer outer gel is large, and as a result the cells in region
, respectively. The force required to deform a stiffer outer gel is large, and as a result the cells in region 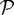 tend to rearrange themselves to fill a more constrained area, which leads to a larger packing density. The cells can deform a more compliant outer gel more easily, which leads to a lower packing density and more irregular interfaces at the gel and quiescent zone interfaces.
tend to rearrange themselves to fill a more constrained area, which leads to a larger packing density. The cells can deform a more compliant outer gel more easily, which leads to a lower packing density and more irregular interfaces at the gel and quiescent zone interfaces.
Also, the average cell area converges towards a limiting value after an initial fluctuation due to an initial massive growth of the tumour before transformation happens (cf. figure 11c). In our model, the cell area indicate the cells’ phase at a given time, and the distribution of the area of cells shows that more cells are staying in the early phase of the cell cycle (cf. figure 11d). Here, the cell area was normalized using the intrinsic cell area that a cell adopts immediately after cell division, V0.
4. Discussion
A cell must deform in order to move, and the single-cell model developed here reproduces the experimentally observed large strain and viscoelastic behaviour of the cytoplasm, and allows tracking of arbitrary deformations of the cell and the substrate on which it moves. In the model, a cell generates an active deformation that is incorporated via a multiplicative decomposition of the total deformation gradient F into its active (FA) and passive (FP) parts. The locally controlled active deformation, coupled to regions of cell–substrate adhesion, allows the cell to generate the force required to move. The same mechanical principles that were used in modelling the cell are also essential for describing a collection of cells and tissues, as was illustrated in the hybrid model of tumour growth.
In the time frame considered, the numerical simulations of the cell model applied to the motion of a keratocyte agree with experimental results. In addition, the model provides some insight into the various hypotheses arising from the experimental systems. As noted in Doyle et al. (2004), the resolution allowed by a gelatin substrate certainly plays a role in the accuracy of their measured traction patterns as compared with those measured in earlier experiments (Lee et al. 1994; Oliver et al. 1999). However, when one carefully considers the traction patterns illustrated in Doyle et al. (2004), in the region where the cell is in contact with the substrate, all the traction vectors are oriented towards the cell interior. This indicates that increased resolution in experimental technique is probably not the only explanation for the different traction patterns observed. The numerical results here show that a possible explanation for the differences between the results of Doyle et al. (2004) and those of Lee et al. (1994) and Oliver et al. (1999) is due to an increased adhesion between the cell and the gelatin substrate, especially at the leading edge, and/or decreased leading edge contractility of a cell moving on gelatin. We note that the current model incorporates cell–substrate attachments and local extension–contraction at a simple level, and further numerical investigations, with a more detailed treatment of cell–substrate dynamics, is required to address the issue fully. Nevertheless, the model presented here does shed light on the various, somewhat contradictory, experimental results.
The next step towards the long-term goal of developing a more realistic model of cell motility, one that incorporates the interplay of intracellular biochemical dynamics and cell mechanics, is to model a cell moving on a deformable substrate in which the regions of attachment change dynamically. The formulation of such a model involves contact mechanics (Kikuchi & Oden 1988), and a difficulty here is that one must keep track of the surfaces in contact so that they do not penetrate one another. To preclude this, suitable forces must develop when the surfaces are in contact. Furthermore, models involving contact mechanics can also include frictional forces that result when two surfaces in contact move relative to one another. Incorporating frictional interaction into a model of cell–substrate interaction will be advantageous as it will allow us to model weaker adhesions as frictional forces and stronger adhesions as fixed attachments to the substrate. Thereby, we will be able to investigate the effects of strength and spatial and temporal evolution of adhesions on motility and cell traction patterns.
Because the power that drives cell motility stems from biochemical and biophysical processes, another extension of the model is to couple the existing mechanical model to a model of biochemical reactions that determines direction sensing in chemotaxis, the strength and direction of actin-mediated extensions and actomyosin contractions and locations of adhesion sites. Adhesion sites are of particular interest because they are the direct connection between intracellular biochemistry and cell mechanics (Bershadsky et al. 2003; Lele et al. 2006). Since the system of focal adhesions within a cell is generally made up of discrete patches of cell–substrate connections, a hybrid discrete–continuum model is ideally suited to model their dynamics. Of course, many other questions remain, including how to predict the shape of a cell and how it depends on the substrate properties, and how to predict the smooth versus pulsatile motion of different cell types.
To describe tissue growth, we developed a lattice-free, cell-based model in which individual cells are ellipsoidal. The details of intracellular dynamics and cell–cell interactions in the actively dividing region are easily included without compromising the numerical tractability of the method. For example, it is easy to introduce changes in cell-level parameters such as adhesion and growth rates and investigate their consequences. Here, we attempted to understand the behaviour of individual cells at the centre of a tissue in the presence of a constraint that limits cell growth. We also applied the model to specific migration experiments of labelled cells and microspheres on the surface of tumour spheroids in order to elucidate the role of adhesion between cells. This framework is general enough to include chemotaxis in the model, which in turn will allow us to consider tumour cell shedding, a very important feature in the dynamics of highly invasive tumours such as gliomas (Giese et al. 2003). In addition, it has been shown that the cells can switch modes from collective motility to single-cell amoeboid motility by altering cell–cell adhesion and protease function in tumour cell lines (Wolf & Friedl 2006). To capture this plasticity in the motility phenotype, we are developing a framework in which individual cells can mechanically interact with a network of individual ECM fibres. This approach will allow us to test hypotheses concerning this motility switch.
In the cell-based model described herein, only stress and nutrient levels affect cell growth. However, there are many other factors that are involved in cell growth. Of particular importance is the intracellular network that controls cell proliferation, apoptosis or quiescence. There can be multiple growth factors that determine cell fate in a subtle way, such as the EGF and TGF-β signalling pathways involved in epithelial cell growth in an early breast cancer. One of the advantages of cell-based models compared with continuum models is that one can easily integrate such elements into the model in a very localized manner.
In the hybrid model, we described tumour growth in the presence of a surrounding outer gel by treating the quiescent and necrotic tumour regions as continua and adapting a cell-based model in the proliferating region. This unique approach allows us to address numerous experiments that cannot be explored by discrete-cell or continuum models alone. This can lead us to a better understanding of the role of the microenvironment of tumour growth. It is important to note that the three-dimensional microenvironment can lead to very different features in intracellular signalling, cell–cell adhesion and drug responses (Smalley et al. 2006). To study this, we are developing a three-dimensional version of the hybrid model described herein; a preliminary result is that we are able to predict the differences in growth behaviour of epithelial cell growth on a solid substrate as found in Galle et al. (2005). The interaction between substrate and a three-dimensional cell in the three-dimensional microenvironment is important in many biological systems, including in self-renewal processes in a crypt. In another vein, further work on the effect of stress on tumour growth will involve a comparison of our numerical results with the experiments of Helmlinger et al. (1997) in detail.
Acknowledgements
The research herein was supported by the National Science Foundation (USA), the National Institutes of Health (USA), the Alexander von Humboldt Foundation, the Digital Technology Center, and the Minnesota Supercomputing Institute.
Footnotes
One contribution of 12 to a Theme Issue ‘Mechanics in biology: cells and tissues’.
References
- Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. 2002. Molecular biology of the cell, 4th edn. New York, NY: Garland. [Google Scholar]
- Ambrosi D., Mollica F. 2002. On the mechanics of a growing tumour. Int. J. Eng. Sci. 40, 1297–1316. ( 10.1016/S0020-7225(02)00014-9) [DOI] [Google Scholar]
- Anderson K. I., Cross R. 2000. Contact dynamics during keratocyte motility. Curr. Biol. 10, 253–260. ( 10.1016/S0960-9822(00)00357-2) [DOI] [PubMed] [Google Scholar]
- Bausch A. R., Ziemann F., Boulbitch A. A., Jacobson K., Sackmann E. 1998. Local measurements of viscoelastic parameters of adherent cell surfaces by magnetic bead microrheometry. Biophys. J. 75, 2038–2049. ( 10.1016/S0006-3495(98)77646-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky A. D., Balaban N. Q., Geiger B. 2003. Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell Dev. Biol. 19, 677–695. ( 10.1146/annurev.cellbio.19.111301.153011) [DOI] [PubMed] [Google Scholar]
- Burton K., Park J. H., Taylor D. L. 1999. Keratocytes generate traction forces in two phases. Mol. Biol. Cell 10, 3745–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. Y., Funamoto S., Firtel R. A. 2001. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26, 557–566. ( 10.1016/S0968-0004(01)01934-X) [DOI] [PubMed] [Google Scholar]
- Dallon J. C., Othmer H. G. 2004. How cellular movement determines the collective force generated by the Dictyostelium discoideum slug. J. Theor. Biol. 231, 203–222. ( 10.1016/j.jtbi.2004.06.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorie M. J., Kallman R. F., Rapacchietta D. F., Antwerp D. V., Huang Y. R. 1982. Migration and internalization of cells and polystyrene microsphere in tumour cell spheroids. Exp. Cell Res. 141, 201–209. ( 10.1016/0014-4827(82)90082-9) [DOI] [PubMed] [Google Scholar]
- Doyle A., Marganski W. F., Lee J. 2004. Calcium transients induce spatially coordinated increases in traction force during the movement of fish keratocytes. J. Cell Sci. 117, 2203–2214. ( 10.1242/jcs.01087) [DOI] [PubMed] [Google Scholar]
- Dumontier M., Hocht P., Mintert U., Faix J. 2000. Rac1 GTPases control filopodia formation, cell motility, endocytosis, cytokinesis and development in Dictyostelium. J. Cell Sci. 113, 2253–2265. [DOI] [PubMed] [Google Scholar]
- Evans E., Yeung A. 1989. Apparent viscosity and cortical tension of blood granulocytes determined by micropipet aspiration. Biophys. J. 56, 151–160. ( 10.1016/S0006-3495(89)82660-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneberg W., Westphal M. 2001. Dictyostelium cells' cytoplasm as an active viscoplastic body. Eur. Biophys. J. 30, 284–294. ( 10.1007/s002490100135) [DOI] [PubMed] [Google Scholar]
- Flaherty B., McGarry J. P., McHugh P. E. 2007. Mathematical models of cell motility. Cell Biochem. Biophys. 49, 14–28. ( 10.1007/s12013-007-0045-2) [DOI] [PubMed] [Google Scholar]
- Friedl P., Wolf K. 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374. ( 10.1038/nrc1075) [DOI] [PubMed] [Google Scholar]
- Galle J., Loeffler M., Drasdo D. 2005. Modeling the effect of deregulated proliferation and apoptosis on the growth dynamics of epithelial cell populations in vitro. Biophys. J. 88, 62–75. ( 10.1529/biophysj.104.041459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G., et al. 2007. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128, 561–575. ( 10.1016/j.cell.2006.12.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A., Bjerkvig R., Berens M. E., Westphal M. 2003. Cost of migration: invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 21, 1624–1636. ( 10.1200/JCO..05.063) [DOI] [PubMed] [Google Scholar]
- Groebe K., Mueller-Klieser W. 1996. On the relation between size of necrosis and diameter of tumour spheroids. Int. J. Radiat. Oncol. Biol. Phys. 34, 395–401. ( 10.1016/0360-3016(95)02065-9) [DOI] [PubMed] [Google Scholar]
- Heidemann S. R., Kaech S., Buxbaum R. E., Matus A. 1999. Direct observations of the mechanical behaviours of the cytoskeleton in living fibroblasts. J. Cell Biol. 145, 109–122. ( 10.1083/jcb.145.1.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger G., Netti P. A., Lichtenbeld H. C., Melder R. J., Jain R. K. 1997. Solid stress inhibits the growth of multicellular tumour spheroids. Nat. Biotechnol. 15, 778–783. ( 10.1038/nbt0897-778) [DOI] [PubMed] [Google Scholar]
- Henson J. H., Svitkina T. M., Burns A. R., Hughes H. E., MacPartland K. J., Nazarian R., Borisy G. G. 1999. Two components of actin-based retrograde flow in sea urchin coelomocytes. Mol. Biol. Cell 10, 4075–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel G. A. 2000. Nonlinear solid mechanics: a continuum approach for engineering. Chichester, UK: Wiley. [Google Scholar]
- Janmey P. A. 1991. Mechanical properties of cytoskeletal polymers. Curr. Opin. Cell Biol. 2, 4–11. ( 10.1016/0955-0674(91)90159-V) [DOI] [PubMed] [Google Scholar]
- Jurado C., Haserick J. R., Lee J. 2004. Slipping or gripping? Fluorescent speckle microscopy in fish keratocytes reveals two different mechanisms for generating a retrograde flow of actin. Mol. Biol. Cell 16, 507–518. ( 10.1091/mbc.E04-10-0860) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. 2005. Cell migration during gastrulation. Curr. Opin. Cell Biol. 17, 533–541. ( 10.1016/j.ceb.2005.08.006) [DOI] [PubMed] [Google Scholar]
- Keren K., Pincus Z., Allen G. M., Barnhart E. L., Marriott G., Mogilner A., Theriot J. A. 2008. Mechanism of shape determination in motile cells. Nature 453, 475–481. ( 10.1038/nature06952) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi N., Oden J. T. 1988. Contact problems in elasticity: a study of variational inequalities and finite element methods. Philadelphia, PA: Society for Industrial and Applied Mathematics. [Google Scholar]
- Kim Y., Stolarska M. A., Othmer H. G. 2007. A hybrid model for tumour spheroid growth in vitro I: theoretical development and early results. Math. Models Methods Appl. Sci. 1710S), 1773–1798. ( 10.1142/S0218202507002479) [DOI] [Google Scholar]
- Koehl G., McNally J. G. 2002. Myosin II redistribution during rear retraction and the role of filament assembly and disassembly. Cell Biol. Int. 26, 287–296. ( 10.1006/cbir.2001.0855) [DOI] [PubMed] [Google Scholar]
- Laurent V. M., Kasas S., Yersin A., Schäffer T. E., Catsicas S., Dietler G., Verkhovsky A. B., Meister J.-J. 2005. Gradient of rigidity in the lamellipodia of migrating cells revealed by atomic force microscopy. Biophys. J. 89, 667–675. ( 10.1529/biophysj.104.052316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. H., Liu D. T. 1967. Finite-strain elastic–plastic theory with applications to plane-wave analysis. J. Appl. Phys. 38, 19–27. ( 10.1063/1.1708953) [DOI] [Google Scholar]
- Lee J., Jacobson K. 1997. The composition and dynamics of cell–substratum adhesions in locomoting fish keratocytes. J. Cell Sci. 110, 2833–2844. [DOI] [PubMed] [Google Scholar]
- Lee J., Leonard M., Oliver T., Ishihara A., Jacobson K. 1994. Traction forces generated by locomoting keratocytes. J. Cell Biol. 127, 1957–1964. ( 10.1083/jcb.127.6.1957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lele T. P., Thodeti C. K., Ingber D. E. 2006. Force meets chemistry: analysis of mechanochemical conversion in focal adhesions using fluorescence recovery after photobleaching. J. Cell Biochem. 97, 1175–1183. ( 10.1002/jcb.20761) [DOI] [PubMed] [Google Scholar]
- Lim C. T., Zhou E. H., Quek S. T. 2006. Mechanical models for living cells—a review. J. Biomech. 39, 195–216. ( 10.1016/j.jbiomech.2004.12.008) [DOI] [PubMed] [Google Scholar]
- Lo C. M., Wang H. B., Dembo M., Wang Y. L. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144–152. ( 10.1016/S0006-3495(00)76279-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKintosh F. C. 1998. Theoretical models of viscoelasticity of actin solutions and the actin cortex. Biol. Bull. 194, 351–353. ( 10.2307/1543110) [DOI] [PubMed] [Google Scholar]
- Mitchison T. J., Cramer L. P. 1996. Actin-based cell motility and cell locomotion. Cell 84, 371–379. ( 10.1016/S0092-8674(00)81281-7) [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A., Schlaepfe D. D. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68. ( 10.1038/nrm1549) [DOI] [PubMed] [Google Scholar]
- Mogilner A. 2006. On the edge: modeling protrusion. Curr. Opin. Cell Biol. 18, 32–39. ( 10.1016/j.ceb.2005.11.001) [DOI] [PubMed] [Google Scholar]
- Munevar S., Wang Y.-L., Dembo M. 2001. Traction force microscopy of migrating normal and H-ras transformed 3T3 fibroblasts. Biophys. J. 80, 1744–1757. ( 10.1016/S0006-3495(01)76145-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver T., Dembo M., Jacobson K. 1999. Separation of propulsive and adhesive traction stresses in locomoting keratocytes. J. Cell Biol. 145, 589–604. ( 10.1083/jcb.145.3.589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T. D., Blanchoin L., Mullins R. D. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29, 545–576. ( 10.1146/annurev.biophys.29.1.545) [DOI] [PubMed] [Google Scholar]
- Ponti A., Machacek M., Gupton S. L., Waterman-Storer C. M., Danuser G. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science 305, 1782–1786. ( 10.1126/science.1100533) [DOI] [PubMed] [Google Scholar]
- Ponti A., Matov A., Adams M., Gupton S., Waterman-Storer C. M., Danuser G. 2005. Periodic patterns of actin turnover in lamellipodia and lamellae of migrating epithelial cells analyzed by quantitative fluorescent speckle microscopy. Biophys. J. 89, 3456–3469. ( 10.1529/biophysj.104.058701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabodzey A., Alcaide P., Luscinskas F. W., Ladoux B. 2008. Mechanical forces induced by transendothelial migration of human neutrophils. Biophys. J. 95, 1428–1438. ( 10.1529/biophysj.107.119156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E. K., Hoger A., McCulloch A. D. 1998. Stress-dependent finite growth of soft tissues. J. Biomech. 27, 455–467. ( 10.1016/0021-9290(94)90021-3) [DOI] [PubMed] [Google Scholar]
- Saez A., Ghibaudo M., Buguin A., Silberzan P., Ladoux B. 2007. Rigidity-driven growth and migration of epithelial cells on microstructured anisotropic substrates. Proc. Natl Acad. Sci. USA 104, 8281–8286. ( 10.1073/pnas.0702259104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz M. P., Felsenfeld D., Galbraith C. G., Choquet D. 1999. Cell migration as a five-step cycle. Biochem. Soc. Symp. 65, 233–243. [PubMed] [Google Scholar]
- Skalak R.Carlson D.E., Shield R.T. 1981. Growth as a finite displacement field. In Proc. IUTAM Symp. on Finite Elasticity The Hague, The Netherlands: Martinus Nijhoff. [Google Scholar]
- Small J. V. 1989. Microfilament-based motility in non-muscle cells. Curr. Opin. Cell Biol. 1, 75–79. ( 10.1016/S0955-0674(89)80040-7) [DOI] [PubMed] [Google Scholar]
- Small J. V., Stradal T., Vignal E., Rottner K. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12, 112–120. ( 10.1016/S0962-8924(01)02237-1) [DOI] [PubMed] [Google Scholar]
- Smalley K. S. M., Lioni M., Herlyn M. 2006. Life isn't flat: taking cancer biology to the next dimension. In Vitro Cell. Dev. Biol. Anim. 42, 242–247. ( 10.1290/0604027.1) [DOI] [PubMed] [Google Scholar]
- Svitkina T. M., Verkhovsky A. B., McQuade K. M., Borisy G. G. 1997. Analysis of the actin–myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139, 397–415. ( 10.1083/jcb.139.2.397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber L. A., Perucchio R. 2000. Modeling heart development. J. Elasticity 61, 165–197. ( 10.1023/A:1011082712497) [DOI] [Google Scholar]
- Uchida K. K., Kitanishi-Yumura T., Yumura S. 2003. Myosin II contributes to the posterior contraction and the anterior extension during the retraction phase in migrating Dictyostelium cells. J. Cell Sci. 116, 51–60. ( 10.1242/jcs.00195) [DOI] [PubMed] [Google Scholar]
- Vallotton P., Gupton S. L., Waterman-Storer C. M., Danuser G. 2004. Simultaneous mapping of filamentous actin flow and turnover in migrating cells by quantitative fluorescent speckle microscopy. Proc. Natl Acad. Sci. USA 101, 9660–9665. ( 10.1073/pnas.0300552101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiev B., Weijer C. J. 2003. Modelling of Dictyostelium discoideum slug migration. J. Theor. Biol. 223, 347–359. ( 10.1016/S0022-5193(03)00103-6) [DOI] [PubMed] [Google Scholar]
- Verkhovsky A. B., Svitkina T. M., Borisy G. G. 1999. Self-polarization and directional motility of cytoplasm. Curr. Biol. 9, 11–20. ( 10.1016/S0960-9822(99)80042-6) [DOI] [PubMed] [Google Scholar]
- Wilkes R. P., Athanasiou K. A. 1996. The intrinsic incompressibility of osteoblast-like cells. Tissue Eng. 2, 167–181. ( 10.1089/ten.1996.2.167) [DOI] [PubMed] [Google Scholar]
- Wolf K., Friedl P. 2006. Molecular mechanisms of cancer cell invasion and plasticity. Br. J. Dermatol. 154(Suppl. 1), 11–15. ( 10.1111/j.1365-2133.2006.07231.x) [DOI] [PubMed] [Google Scholar]
- Xu J., Tseng Y., Wirtz D. 2000. Strain hardening of actin filament networks. J. Biol. Chem. 275, 35 886–35 892. ( 10.1074/jbc.M002377200) [DOI] [PubMed] [Google Scholar]
- Yam P. T., Wilson C. A., Ji L., Hebert B., Barnhart E. L., Dye N. A., Wiseman P. W., Danuser G., Theriot J. A. 2007. Actin myosin network reorganization breaks symmetry at the cell rear to spontaneously initiate polarized cell motility. J. Cell Biol. 178, 1207–1221. ( 10.1083/jcb.200706012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai M., Butler J. P., Suzuki T., Kanda A., Kurachi M., Tashiro H., Sasaki H. 1999. Intracellular elasticity and viscosity in the body, leading, and trailing regions of locomoting neutrophils. Am. J. Physiol. 277, C432–C440. [DOI] [PubMed] [Google Scholar]