

Abstract
Heptosyltransferase I (HepI) is responsible for the transfer of L-glycero-D-manno-heptose to a 3-deoxy-α-D-oct-2-ulopyranosonic acid (Kdo) of the growing core region of lipopolysaccharide (LPS). The catalytic efficiency of HepI with the fully deacylated analogue of Escherichia coli HepI LipidA is 12-fold greater than with the fully acylated substrate, with a kcat/Km of 2.7 × 106 M−1 s−1, compared to a value of 2.2 × 105 M−1 s−1 for the Kdo2-LipidA substrate. Not only is this is the first demonstration that an LPS biosynthetic enzyme is catalytically enhanced by the absence of lipids, this result has significant implications for downstream enzymes that are now thought to utilize deacylated substrates.
The effectiveness of antibiotics for the treatment of Gram-negative infections is hampered by their uptake into cells, because of the presence of an outer membrane containing a high level of lipopolysaccharide (LPS).1 LPS on cell surfaces is important for cell motility, intestinal colonization and bacterial biofilm formation, and contributes substantively to antibiotic resistance.1,2 These characteristics have spurred research toward the development of inhibitors for the LPS biosynthetic enzymes. The structure of the LPS is comprised of three parts: LipidA, core oligosaccharide (Core-OS), and repeating O-Antigen. Heptosyltransferase I (HepI) catalyzes the first step in the LPS synthesis pathway following lipid functionalization of LipidA.3,4 Blocking LPS biosynthesis prior to the addition of an L-glycero-D-manno-heptose (Hep) residue results in increased bacterial sensitivity to hydrophobic antibiotics and phagocytosis by macrophages; thus, HepI is considered an excellent target for inhibitor development.5 In our efforts to determine the enzyme mechanism for drug design, we have found that HepI can efficiently utilize multiple LipidA analogues as substrates, including a completely lipid free LipidA molecule. This is the first enzyme in the LPS biosynthetic pathway that does not have strict selectivity for acylated substrates.3 Furthermore, this finding has important implications for the substrate selectivity of downstream enzymes, which are now thought also to utilize deacylated substrates.
HepI is essential for the transfer of the first Hep moiety in the core oligosaccharide of LPS. It catalyzes formation of an α(1→5) linkage between L-glycero-D-manno-heptose and the first 3-deoxy-α-D-oct-2-ulopyranosonic acid (Kdo) covalently attached to LipidA (Scheme 1).5 A crystal structure of Escherichia coli HepI bound to a donor substrate analogue, ADP-2-deoxy-2-fluoroheptose, reveals binding interactions between ADP-Hep and the sugar donor domain; however, these structures revealed little about interactions of HepI with the acceptor substrate Kdo2-LipidA.6 Our goal was to identify a substrate analogue that would be amenable to crystallography, thereby allowing the residues that confer sugar acceptor specificity to be revealed, while also attempting to identify tighter binding molecules that lead toward the development of HepI inhibitors.
Scheme 1.
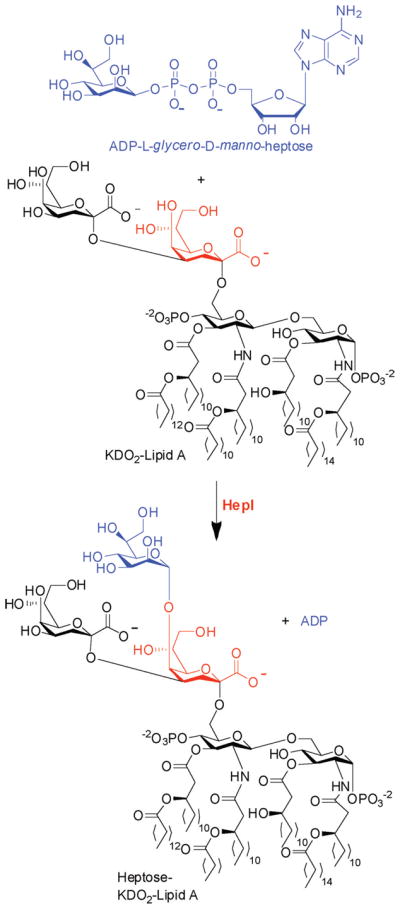
Reaction Catalyzed by E. coli HepI
Deacylation of the LipidA molecules was investigated to improve the solubility of the acceptor substrate, thereby potentially removing the need for detergent, while also removing potential entropic penalties associated with binding. Additionally, because the Kdo2-LipidA molecule is believed to be bound in the membrane during catalysis, we hypothesized that these residues would be relatively unimportant for conferring specificity. Here we report the effect of the acylation state, as well as the number of Kdo molecules, has on Km for the acceptor substrate. Kdo2-LipidA (Figure 1A) was isolated from E. coli HepI knockout strain WBB06 according to published protocols,7 and Kdo-LipidA (Figure 1B, also known as Re-LipidA) was obtained from Sigma-Aldrich. Kdo2-LipidA was O-deacylated by reaction with anhydrous hydrazine. The fully deacylated acceptor molecule (Figure 1D), which was previously synthesized by Brabetz and coworkers,4 was synthesized using a modified method analogous to that used by Bystrova and coworkers.8 First, the O-deacylated Kdo2-LipidA (Figure 1C) was made via reaction with anhydrous hydrazine and then the fully deacylated acceptor resulted from subsequent refluxing with 4 M NaOH.
Figure 1.

Structures of LipidA acceptor substrates: (A) E. coli Kdo2-LipidA, (B) Salmonella minnesota Kdo-LipidA, (C) O-deacylated E. coli Kdo2-LipidA, and (D) fully deacylated E. coli Kdo2-LipidA. The Kdo that acts as a nucleophile for the glysyltransferase reaction is colored red.
Using a coupled assay containing pyruvate kinase and L-lactate dehydrogenase, HepI activity was determined through UV–vis monitoring of production of nicotinamide adenine dinucleotide at 340 nm.6,9 All four LipidA analogues (Figure 1A–D) were found to be competent substrates when the assay was conducted with or without Triton X-100 (Table 1). Formation of the pentasaccharide (Scheme 1) derived from use of the fully deacylated LipidA analogue (Figure 1D) was confirmed by ESI-MS (see the Supporting Information). A roughly 5-fold decrease in the rate of turnover was observed between the fully acylated acceptor substrates and the deacylated forms of Kdo2-LipidA, with similar rates observed for both the O-deacylated and fully deacylated LipidA analogues. This decrease in kcat is more than offset by an increased binding affinity, causing the overall catalytic efficiency to be 4-fold greater for the fully deacylated Kdo2-LipidA than for the fully acylated LipidA (kcat/Km values of 2.1 × 106 and 5.2 × 105 M−1 s−1, respectively) in the presence of Triton X-100 and 12-fold greater (kcat/Km values of 2.7 × 106 and 2.2 × 105 M−1 s−1, respectively) in the absence of detergent.
Table 1.
Kinetic Parameters for the Reaction of HepI with LipidA Acceptor Molecules in the Presence and Absence of Detergent
| acceptor | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| A | 6.6 ± 0.8 | 29 ± 7 | 2.2 × 105 |
| A with Triton X-100 | 4.7 ± 0.3 | 9 ± 1 | 5.2 × 105 |
| B | 8 ± 1 | 46 ± 13 | 1.7 × 105 |
| B with Triton X-100 | 5.5 ± 0.4 | 16 ± 3 | 3.4 × 105 |
| C | 1.06 ± 0.05 | 0.9 ± 0.2 | 1.1 × 106 |
| C with Triton X-100 | 1.47 ± 0.08 | 0.4 ± 0.1 | 3.7 × 106 |
| D | 0.76 ± 0.03 | 0.28 ± 0.08 | 2.7 × 106 |
| D with Triton X-100 | 1.05 ± 0.07 | 0.5 ± 0.1 | 2.1 × 106 |
Furthermore, the <2-fold binding affinity discrimination between Kdo-LipidA and Kdo2-LipidA can be rationalized because in vivo it has been demonstrated that the E. coli Kdo transferase (EcKdtA) is a bifunctional enzyme that catalyzes incorporation of both Kdo units. Because a single incorporation of Kdo is only observed using isolated EcKdtA with limiting quantities of its sugar donor, cytosine monophosphate-Kdo,10 HepI would only encounter Kdo2-LipidA in vivo, thereby eliminating any driving force for selectivity between Kdo-LipidA and Kdo2-LipidA.
Tighter binding of the fully deacylated acceptor analogue, when compared to the acylated acceptors, is enhanced further in the absence of detergent. We hypothesize that when the acyl chains are contained within a detergent micelle, they are isolated from the enzyme, but when free in solution (in the absence of detergent), they make destabilizing interactions with the protein surface or aggregate together. This hypothesis is supported by the observation that HepI aggregates in the presence of Triton X-100, the fully acylated Kdo2-LipidA, and the combination, whereas apo HepI and HepI bound to the fully deacylated LipidA do not show significant aggregation when analyzed by size exclusion chromatography (data not shown). Furthermore, the observation that the fully deacylated acceptor has a higher affinity in the absence of detergent, and the opposite is true for the O-deacylated LipidA analogue, provides further support for our hypothesis. Because the acyl portion of the LipidA acceptor resides in the inner leaflet of the inner membrane when the reaction occurs in vivo, we speculate that the enzyme only recognizes the sugar region of these acceptor molecules for catalysis. This is the first report of enzymatic activity for HepI in the absence of detergent, suggesting that perhaps the sensitivity of the coupled assay for determining initial rates is also an improvement over traditional thin layer chromatography analysis of radioisotope incorporation for LPS elongation.11
The small differences in kinetic parameters among these four substrates suggest that the sugar binding interactions of the acceptor substrate predominate for HepI. Our findings are consistent with a prior report that Kdo2-LipidIVA (a tetraactylated LipidA analogue that is biosynthetically upstream of HepI) can be adventitiously turned over by HepI and has an apparent Km of 4.5 μM.11 The ability of HepI not only to utilize an underacylated LipidA analogue but also to have improved catalytic efficiency in the absence of any lipids has broad implications for enzymes that utilize glycolipid substrates, and especially other enzymes of the LPS biosynthetic pathway.
Furthermore, our finding that fully deacylated Kdo2-LipidA is a better substrate for the enzyme than the native substrate, and that it binds without inducing aggregation, should open up new strategies for structural analysis of the protein–substrate interactions of this and related glycolipid-modifying enzymes. The ability of this enzyme to utilize multiple LipidA analogues suggests that examination of the glycosyltransferases involved in LPS biosynthesis, including HepI, could yield answers that are broadly applicable to questions concerning substrate selectivity in glycosyltransferases in general. We anticipate that these results will ultimately enhance lead discovery in the area of inhibition of LPS biosynthesis for treatment of Gram-negative bacterial infections.
Supplementary Material
Acknowledgments
Funding
D.J.C. thanks the National Institutes of Health Doctoral Studies in Molecular Biophysics Training Grant 5 T32GM008271 for support.
We thank Christian Raetz (Duke University, Durham, NC) for generously providing E. coli HepI knockout strain WBB06.
Footnotes
LPS deacylation procedure, HepI purification, detailed assay conditions, and ESI-MS data from reaction of the fully deacylated Kdo2-LipidA substrate with HepI. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schnaitman CA, Klena JD. Microbiol Rev. 1993;57:655–682. doi: 10.1128/mr.57.3.655-682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams V, Fletcher M. Appl Environ Biotechnol. 1996;62:100–104. doi: 10.1128/aem.62.1.100-104.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brabetz W, Mueller-Loennies S, Holst O, Brade H. Eur J Biochem. 1997;247:716–724. doi: 10.1111/j.1432-1033.1997.00716.x. [DOI] [PubMed] [Google Scholar]
- 5.Coleman WG, Leive L. J Bacteriol. 1979;139:899–910. doi: 10.1128/jb.139.3.899-910.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grizot S, Salem M, Vonsouthi V, Durand L, Moreau F, Dohi H, Vincent S, Escaich S, Ducruix A. J Mol Biol. 2006;363:383–394. doi: 10.1016/j.jmb.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 7.Galanos C, Luederitz O, Westphal O. Eur J Biochem. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 8.Bystrova OV, Shashkov AS, Kocharova NA, Knirel YA, Zahringer U, Pier GB. Biochemistry (Moscow, Russ Fed) 2003;68:918–925. doi: 10.1023/a:1025759217501. [DOI] [PubMed] [Google Scholar]
- 9.Anderson KW, Murphy AJ. J Biol Chem. 1983;258:14276–14278. [PubMed] [Google Scholar]
- 10.Sirisena DM, Brozek KA, MacLachlan PR, Sanderson KE, Raetz CRH. J Biol Chem. 1992;267:18874–18884. [PubMed] [Google Scholar]
- 11.Kadrmas JL, Raetz CRH. J Biol Chem. 1998;273:2799–2807. doi: 10.1074/jbc.273.5.2799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.