

Abstract
The prototypic cannabinoid type 1 (CB1) receptor antagonist/inverse agonist, rimonabant, is comprised of a pyrazole core surrounded by a carboxyamide with terminal piperidine group (3-substituent), a 2,4-dichlorophenyl group (1-substituent), a 4-chlorophenyl group (5-substituent), and a methyl group (4-substituent). Previous structure-activity relationship (SAR) analysis has suggested that the 3-position may be involved in receptor recognition and agonist activity. The goal of the present study was to develop CB1-selective compounds and explore further the SAR of 3-substitution on the rimonabant template. 3-Substituted analogs with benzyl and alkyl amino, dihydrooxazole, and oxazole moieties were synthesized and evaluated in vitro and in vivo. Several notable patterns emerged. First, most of the analogs exhibited CB1 selectivity, with many lacking affinity for the CB2 receptor. Affinity tended to be better when [3H]5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide (SR141716), rather than [3H](−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol (CP55,940), was used as the binding radioligand. Second, many of the analogs produced an agonist-like profile of effects in mice (i.e., suppression of activity, antinociception, hypothermia, and immobility); however, their potencies were not well correlated with their CB1 binding affinities. Further assessment of selected analogs showed that none were effective antagonists of the effects of Δ9-tetrahydrocannabinol in mice, their agonist-like effects were not blocked by rimonabant, they were active in vivo in CB1(−/−) mice, and they failed to stimulate guanosine-5′-O-(3-[35S]thio)-triphosphate binding. Several analogs were inverse agonists in the latter assay. Together, these results suggest that this series of 3-substituted pyrazole analogs represent a novel class of CB1-selective cannabinoids that produce agonist-like effects in mice through a non-CB1, non-CB2 mechanism.
Introduction
Rimonabant, formerly known as SR141716 [(−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol], is the prototypic antagonist of cannabinoid type 1 (CB1) receptors (Rinaldi-Carmona et al., 1994). Its discovery in 1994 intensified interest in scientific research on cannabinoids by providing a valuable pharmacological tool for investigating the structure of the CB1 receptor and determining the role of this receptor within the (then) newly discovered endocannabinoid system (Devane et al., 1992). Later research suggested that rimonabant may not be a neutral CB1 antagonist, but rather may have inverse agonist effects (Landsman et al., 1997; Pan et al., 1998). In vivo, rimonabant has been reported to antagonize various effects of cannabinoid agonists from several classes, including the tetrahydrocannabinols [e.g., Δ9-tetrahydrocannabinol (THC); Compton et al., 1996], bicyclic cannabinoids [e.g., (−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol (CP55,940); Wiley et al., 1995a], aminoalklyindoles [e.g., (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN55,212-2); Fattore et al., 2001)], and anandamide-like cannabinoids (Murillo-Rodríguez et al., 2001). When administered alone, rimonabant decreases feeding behavior (Wiley et al., 2005), has discriminative stimulus effects (Järbe et al., 2004), and stimulates locomotor activity (Compton et al., 1996), the last of which is not related to rimonabant interaction with the CB1 receptor (Bass et al., 2002). In humans, rimonabant was originally marketed as an antiobesity agent and an aid to smoking cessation until its adverse psychiatric effects were revealed during advanced clinical trials (Christensen et al., 2007). Nevertheless, rimonabant remains an excellent template for the investigation of structural requirements for the recognition and activation of CB1 receptors.
Previous structure-activity relationship (SAR) studies have examined rimonabant analogs that retain a central pyrazole structure with manipulation of one of four other areas of the molecule: 1) substitution for carboxyamide and/or piperidine substituent (3-substituent); 2) substitution for the 2,4-dichlorophenyl group (1-substituent); 3 substitution for chlorophenyl group (5-substituent); or 4) substitution for the methyl (4-substituent) (Table 1). Of these various substituents, the 1-substituent is the most unique and is hypothesized to be related to the antagonist properties of rimonabant (Thomas et al., 1998), whereas the 3-substituent has been suggested to be involved in receptor recognition (Wiley et al., 2001) and its inverse agonist effects (Hurst et al., 2006). In an earlier study (Wiley et al., 2001), we reported that some 3-substituted rimonabant analogs possessed in vivo effects in mice that are characteristic of cannabinoid agonists and partial agonists, including suppression of locomotor activity, antinociception, hypothermia, and catalepsy (Martin et al., 1991). Here, we evaluated further the structure-activity relationship of novel 3-substituent rimonabant analogs in vitro and in vivo, with the dual purpose of 1) development of better understanding of the influence of this part of the pyrazole template on CB1 receptor affinity and functioning and 2) discovery of a CB1 receptor-selective agonist. Whereas many traditional cannabinoid agonists such as THC, CP55,940, and WIN55,212-2 bind to both CB1 and CB2 receptors with good affinity, rimonabant shows good selectivity for the CB1 (versus CB2) receptor (Showalter et al., 1996); hence, our goal was to optimize CB1 agonist activity by manipulating the 3-substituent while retaining the pyrazole core in an effort to maintain or improve selectivity. In this study, we have evaluated the in vitro and in vivo effects of structural modification at the 3-position of rimonabant analogs, with the purpose of understanding the role of this position in both agonism and receptor activation.
TABLE 1.
Cannabinoid receptor binding and in vivo effects of 1-(2,4-dichlorophenyl)-4-methyl-5-(4-chlorophenyl)-1H-pyrazoles with alkyl amide 3-substituents

| Compound | R | Receptor Affinitiesa |
Effects in Miceb |
|||||
|---|---|---|---|---|---|---|---|---|
| [3H]CP55,940 | [3H]SR144528 | CB2 | SA | %MPE | RT | RI | ||
| Rimonabant | 6 | 2 | 702c | Stimulated | Inactive | Inactive | Inactive | |
| (0.2) | (0.1) | (62) | (65) | (65) | (65) | (65) | ||
| Δ9-THC | 67 | 764 | 36c | N.T. | ||||
| (3) | (76) | (10) | 0.9d | 2.7d | 2.5d | |||
| CP55,940 | 1 | 31 | 0.7c | N.T. | N.T. | N.T. | N.T. | |
| (0.02) | (8) | (0.02) | ||||||
| O-4334 | 19 | 6 | 1035 | Stimulated | 50% | Inactive | Inactive | |
| (2) | (0.4) | (138) | (61) | (61) | (61) | (61) | ||
| O-4333 | 34 | 11 | 862 | Inactive | Inactive | −3 | Inactive | |
| (2) | (0.8) | (54) | (55) | (55) | (55) | (55) | ||
| O-4332 | 484 | 299 | 2263 | ∼6–20 | ∼6–20 | ∼6–20 | 6.9 | |
| (15) | (23) | (214) | (4–14) | |||||
| O-4331 | 249 | 145 | 742 | 12 | 10 | 12 | 8.2 | |
| (18) | (26) | (94) | (10–14) | (6–18) | (8–18) | (6–12) | ||
| O-4335 | 172 | 56 | 2341 | 18 | 21 | 16 | 40 | |
| (16) | (6) | (362) | (12–27) | (16–25) | (12–21) | (21) | ||
| O-2154 |  |
38 | 10 | 731 | 70 | 50 | −4.2 | 35 |
| (9) | (0.5) | (106) | (6) | (6) | (6) | (6) | ||
| O-4336 | 61 | 15 | 669 | 8 | 6 | −4.3 | 29 | |
| (3) | (0.7) | (106) | (6–10) | (4–7) | (19) | (19) | ||
| O-4371 | 39 | 9 | 555 | 13 | 13 | 17 | 19 | |
| (0.3) | (0.9) | (13) | (9–19) | (8–23) | (11–23) | (13–30) | ||
| O-4373 | 147 | 44 | 1703 | 5 | 7 | 7 | 10 | |
| (11) | (2) | (124) | (2–12) | (5–12) | (5–10) | (5–17) | ||
| O-4337 | 420 | 153 | 1593 | <7 | 12 | 9 | ∼6 | |
| (66) | (11) | (158) | (7–16) | (7–14) | ||||
| O-2155 | 222 | 47 | 1848 | 86 | 89 | −4.6 | 47 | |
| (78) | (7) | (569) | (22) | (22) | (22) | (22) | ||
| O-4339 | 162 | 50 | 947 | 11 | 24 | 11 | 28 | |
| (24) | (4) | (61) | (4–26) | (13–41) | (4–19) | (17–45) | ||
| O-4372 | 77 | 27 | 298 | 97 | 79 | −5.7 | 74 | |
| (6) | (2) | (98) | (63) | (63) | (63) | (63) | ||
| O-4423 |  |
318 | 73 | 3658 | 90 | 77 | −4.5 | 55 |
| (29) | (5) | (1263) | (62) | (62) | (62) | (62) | ||
N.T., not tested.
Ki values are nanomolar S.E.M. values for receptor affinities are shown in parentheses.
Values shown are ED50 (95% confidence intervals in parentheses). Single-dose tests are indicated by magnitude of effect, with dose that was tested in parentheses. All doses are expressed as micromoles per kilogram.
Data from Showalter et al. (1996).
Data from Wiley et al. (1998).
Materials and Methods
Subjects
Male ICR mice (25–32 g), obtained from Harlan (Dublin, VA) and housed in groups of five, were used for assessment of locomotor suppression, antinociception, hypothermia, and catalepsy. Separate mice (n = 5–6 per dose/dose combination, unless otherwise indicated) were used for testing each dose of each compound in this battery of procedures. A subset of pyrazole analogs were also tested in vivo in male and female CB1 knockout [CB1(−/−)] and wild-type [CB1(+/+)] mice, bred on a C57BL/6 background, as described previously (Zimmer et al., 1999). These mice were derived from breeding pairs of heterozygotes (obtained from A. Zimmer, National Institute of Mental Health, Bethesda, MD) and born at Virginia Commonwealth University. Because of limited supply, the transgenic mice were tested with more than one compound or dose of compound. All mice had free access to food in their home cages and were kept in a temperature-controlled (20–22°C) environment with a 12-h light/dark cycle (lights on at 7:00 AM). The in vivo studies reported here were carried out in accordance with guidelines published in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Apparatus
Measurement of spontaneous activity in mice occurred in standard activity chambers interfaced with a Digiscan Animal Activity Monitor (Omnitech Electronics, Inc., Columbus, OH). A standard tail-flick apparatus and a digital thermometer (Thermo Fisher Scientific, Waltham, MA) were used to measure antinociception and rectal temperature, respectively.
Drugs
Pyrazole analogs (synthesized in the laboratory at Organix, Inc.), THC (National Institute on Drug Abuse, Bethesda, MD), and rimonabant (National Institute on Drug Abuse) were mixed in a vehicle of ethanol, Emulphor (Rhone-Poulenc, Inc., Princeton, NJ), and saline in a 1:1:18 ratio. All injections were administered intravenously at a volume of 0.1 ml/10 kg.
Procedures
Membrane Preparations.
Chinese hamster ovary cells stably expressing the human CB1 or CB2 receptor were cultured in a 50:50 mixture of Dulbecco's modified Eagle's medium and Ham F-12 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 mg/ml G418, and 5% fetal calf serum. Cells were harvested by replacement of the media with cold phosphate-buffered saline containing 0.4% EDTA followed by agitation. Membranes were prepared by homogenization of cells in 50 mM Tris-HCl, 3 mM MgCl2, and 1 mM EGTA, pH 7.4, centrifugation at 50,000g for 10 min at 4°C, and resuspension in the same buffer at 1.5 mg/ml. Membranes were stored at −80°C until use.
Radioligand Binding.
Membranes were diluted with assay buffer B (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, and 0.2 mM EGTA). Reactions containing membrane (10 μg of protein) were incubated with 0.5 nM [3H]SR141716 (CB1) or 1 nM [3H]CP55,940 (CB1 or CB2) and varying concentrations of test compounds in assay buffer B containing 0.5% BSA. Nonspecific binding was measured in the presence and absence of 5 μM unlabeled SR141716 (CB1) or 10 μM unlabeled WIN 55,212-2 (CB2). The assay was incubated for 60 min at 30°C and terminated by rapid filtration under vacuum through Whatman (Clifton, NJ) GF/B glass fiber filters that were presoaked in Tris buffer containing 5 g/liter BSA (Tris-BSA), followed by five washes with cold Tris-BSA. Bound radioactivity was determined by liquid scintillation spectrophotometry at 45% efficiency for 3H.
[35S]GTPγS Binding.
Before assays, samples were thawed on ice, centrifuged at 50,000g for 10 min at 4°C, and resuspended in assay buffer A (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl). Reactions containing 10 μg of membrane protein were incubated for 90 min at 30°C in assay buffer A containing 10 μM GDP, 0.1 nM [35S]GTPγS, 0.1% bovine serum albumin, and various concentrations of test compounds. Nonspecific binding was determined in the presence of 20 μM unlabeled GTPγS. Reactions were terminated by rapid vacuum filtration through GF/B glass fiber filters, and radioactivity was measured by liquid scintillation spectrophotometry at 95% efficiency for 35S.
Tetrad Tests in Mice.
Each mouse was tested in a battery of four tests, in which cannabinoid agonists produced a characteristic profile of in vivo effects (Martin et al., 1991): suppression of locomotor activity, antinociception in the tail-flick assay, decreased rectal temperature, and ring immobility. Before injection, rectal temperature and baseline latency in the tail-flick test were measured in the mice. The latter procedure involved exposing the mouse's tail to an ambient heat source (i.e., bright light) and recording latency (in seconds) for tail removal. Typical control latencies were 2 to 4 s. A 10-s maximal latency was used to avoid damage to the mouse's tail. After measurement of temperature and baseline tail-flick latency, mice were injected intravenously with vehicle or drug. Five minutes later they were placed into individual activity chambers for 10 min. Spontaneous activity was measured as the total number of beam interruptions during the entire session, which was expressed as the percentage of inhibition of the control (vehicle) group's activity. Tail-flick latency was measured at 20 min after injection. Antinociception was expressed as the percentage of maximum possible effect (MPE) by using a 10-s maximum test latency. Rectal temperature was measured at 30 min after injection and expressed as the difference between preinjection and postinjection rectal temperatures. At 40 min after injection, the mice were placed on a 5.5-cm ring attached at a height of 16 cm to a ring stand, and the amount of time the animals remained motionless during a 5-min period was recorded. The time that each animal remained motionless on the ring was divided by 300 s and multiplied by 100 to obtain a percentage immobility rating. Whenever quantity of compound allowed, a full dose-effect curve determination in the tetrad tests was conducted; however, insufficient quantities of some of the compounds resulted in probe tests with a single dose.
Evaluation of antagonism of THC's effects in the tetrad was accomplished by intravenous injection of the 3-substituted pyrazole analog followed 10 min later by an intravenous injection of 3 mg/kg THC. Given that many of the compounds produced in vivo effects that were agonist-like and did not function as antagonists (against THC), rimonabant reversal of the in vivo effects of selected compounds was also assessed. For these tests, vehicle or rimonabant was injected intravenously 10 min before intravenous injection of the test compound. In both types of antagonist evaluations, in vivo tests were then conducted by using the same time course and procedure described above.
Selected pyrazole analogs were also evaluated in CB1(−/−) and CB1(+/+) mice in three in vivo assays: spontaneous activity, rectal temperature, and ring immobility. Because these mice were tested more than once, tail-flick assays were not performed to avoid repeated exposure of the tail to a painful stimulus. All other experimental parameters were identical as those described for the ICR mice.
Data Analysis.
Rectal temperature values were expressed as the difference between control temperature (before injection) and temperatures after drug administration (Δ°C). Spontaneous activity was measured as total number of photocell beam interruptions during the 5-min session and expressed as the percentage of inhibition of activity of the vehicle group. During assessment for catalepsy, the total amount of time (in seconds) that the mouse remained immobile on the ring apparatus was measured and used as an indication of catalepsy-like behavior. This value was divided by 300 s and multiplied by 100 to obtain a percentage of immobility. Data analysis was based on a scheme we have used in numerous previous studies with cannabinoids, with maximal cannabinoid effects in each procedure estimated as follows: 90% inhibition of spontaneous activity, 100% MPE in the tail-flick procedure, −6°C change in rectal temperature, and 60% ring immobility. ED50 was defined as the dose at which half-maximal effect occurred. For compounds that produced one or more cannabinoid effects, ED50 was calculated separately by using least-squares linear regression on the linear part of the dose-effect curve for each measure in the mouse tetrad, plotted against log10 transformation of the dose. Rimonabant reversibility of the pharmacological effects of 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-morpholinopropyl)-1H-pyrazole-3-carboxamide (O-4332) and four other selected analogs in mice was analyzed with separate factorial (rimonabant treatment condition × O-4332 dose or pyrazole analog, respectively) ANOVAs for each dependent measure. Separate factorial (genotype × treatment compound) ANOVAs were also used to analyze the effects of selected compounds in CB1(−/−) and CB1(+/+) mice. Significant main effects and interactions were further analyzed with Tukey post hoc tests (α = 0.05) as necessary.
For the CB1 and CB2 receptor binding experiments, displacement IC50 values were originally determined by Hill plots and then converted to Ki values by using the method of Cheng and Prusoff (1973). All experiments were performed in triplicate, with data reported as mean values ± S.E.M. Nonlinear regression analysis was conducted to obtain EC50 and Emax values of agonist-stimulated [35S]GTPγS binding by iterative curve fitting with JMP (SAS for Macintosh; SAS Institute, Cary, NC). Values were reported as the means ± S.E.M. of at least four experiments, each performed in triplicate. Percentage of maximum stimulation was calculated as percentage of the stimulation produced by a maximally effective concentration (3 μM) of CP55,950, normalized to 100%.
Pearson product-moment correlation coefficients (with associated significance tests) were calculated between CB1 binding affinity with [3H]CP55,940 (expressed as log Ki) and in vivo potency for each measure (expressed as log ED50 in μmol/kg) for all active cannabinoid compounds that bound to the CB1 receptor and for which an ED50 value was available for the measure. The Pearson product-moment correlation provided a measure of the strength and direction of relationship between each pair of quantitative variables. In addition, a correlation between CB1 binding affinities using the two different radioligands, [3H]CP55,940 and [3H]SR141716, was calculated for all compounds.
Results
To further explore the structure-activity relationship at the 3-position of rimonabant, several alkylamide analogs were synthesized with varying chain length and varying functional groups at the terminal end of the chain. Table 1 shows CB1 and CB2 receptor binding affinities and in vivo potencies of these rimonabant analogs. Assessment of control compounds showed that rimonabant had good affinity for the CB1 receptor, regardless of which of the two radioligands was used for the displacement assay, albeit it displaced [3H]SR141716 at 3-fold lower concentrations than it displaced [3H]CP55,940. In contrast, THC and CP55,940 exhibited better affinity for the CB1 receptor when tritiated agonist was used as a radioligand than when tritiated antagonist was used. Whereas THC also had good binding affinity at CB2 receptors, rimonabant was relatively selective for CB1 receptors, having poor affinity at CB2 receptors. In vivo, rimonabant stimulated locomotor activity and was inactive in the other three tetrad tests. In contrast, THC produced cannabimimetic effects in all in vivo tests.
All of the 3-substituted analogs of rimonabant in Table 1 shared the property of CB1 receptor selectivity. The best CB2 receptor affinity (Ki) of 298 nM, which was observed with O-4372, was still only moderate, with CB2 receptor affinities of the other compounds ranging from 555 to 3658 nM for O-4371 and O-4423, respectively. Furthermore, the analogs shared with rimonabant the property of better displacement of [3H]SR141716 versus [3H]CP55,940, ranging from 1.6- to 4.7-fold when affinity was assessed with the former radioligand. Although none of the analogs had better CB1 receptor affinity than rimonabant, the benzyl amide analogs (O-4333 and O-4334) resulted in reasonable CB1 affinities, but were not active in vivo. In contrast, the alkylamide analogs (O-4331 and O-4332) showed substantially decreased CB1 receptor affinities. We were surprised to find, however, that these compounds possessed cannabimimetic activity in the tetrad tests, with reasonable potencies (2.5–20 μg/kg) that belied their relatively poor CB1 receptor affinities.
For the bromo and cyano series (Table 1), CB1 receptor affinities improved as the carbon chain was lengthened, with best affinities (as measured by [3H]SR141716 displacement) exhibited by compounds with a pentylbromo (O-4371) or pentylcyano (O-4372) substitution. At comparable carbon chain lengths from ethyl to pentyl, a terminal bromo group resulted in better CB1 receptor affinity compared with a terminal cyano group. Addition of a terminal double bond (O-4373) or branching of the carbon chain (O-4423) did not notably improve CB1 receptor affinity. Regardless of magnitude of CB1 receptor affinity, however, all of these 3-substituent bromo and cyano side chain analogs were active in the in vivo tetrad tests. Potencies ranged from 5 to 28 μg/kg, but did not necessarily correspond with CB1 receptor affinities. For example, O-4337 had poor CB1 receptor affinity (Ki = 420 nM; CP55,940 displacement); yet, it was active in all four tests at similar or lower potencies than O-4371, a compound with one of the best CB1 receptor affinities (Ki = 39 nM; CP55,940 displacement).
Table 2 shows CB1 and CB2 receptor binding data and in vivo potencies for 3-substituted pyrazole analogs in which the 3-amido moiety of rimonabant was replaced with dihydrooxazole moiety (O-4338), oxazole moiety (O-6668), or amino substituents (O-6729, O-6730, O-6731, and O-6740). These compounds maintained the CB1 receptor selectivity that was observed with previous compounds and had good to fair affinity for this receptor (range 45–290 nM; SR141716 displacement). For this series, CB1 receptor binding affinities were more similar between the two radioligands used for the displacement assays, with ratios of [3H]CP55,940 to [3H]SR141716 binding affinities ranging from 0.5 to 2.3. Half of the compounds showed approximately equivalent CB1 receptor binding affinities regardless of the radioligand used in the displacement assay. Compounds O-4338 and O-6668 retained potent in vivo activity, as was exhibited by many of the compounds in Table 1. In contrast, compounds that lacked an amide moiety or dihydrooxazole moiety (O-6740, O-6729, O-6730, and O-6731) were inactive in all in vivo assays at doses up to 30 mg/kg.
TABLE 2.
Cannabinoid receptor binding and in vivo effects of 1-(2,4-dichlorophenyl)-4-methyl-5-(4-chlorophenyl)-1H-pyrazoles with various 3-substituents
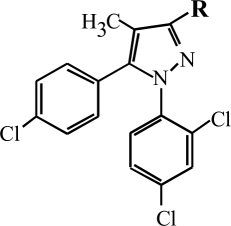
| Compound | R | Receptor Affinitiesa |
Effects in Miceb |
|||||
|---|---|---|---|---|---|---|---|---|
| [3H]CP55,940 | [3H]SR144528 | CB2 | SA | %MPE | RT | RI | ||
| O-4338 |  |
680 | 290 | >10,000 | 2.5 | 6.1 | 9.3 | 16 |
| (78) | (23) | (2–5) | (1.7–23) | (5.7–15) | (5–47) | |||
| O-6668 | 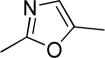 |
68 | 45 | >10,000 | <2 | 6 | ∼2 | 12 |
| (7) | (4) | (5–10) | (7–21) | |||||
| O-6730 |  |
83 | 89 | 5415 | Inactive | Inactive | −2.6 | Inactive |
| (25) | (14) | (543) | (71) | (71) | (71) | (71) | ||
| O-6729 | 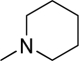 |
185 | 120 | 4573 | Inactive | Inactive | Inactive | Inactive |
| (42) | (11) | (456) | (71) | (71) | (71) | (71) | ||
| O-6731 | 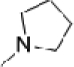 |
117 | 247 | 5342 | 89 | 52 | −4.4 | 16 |
| (12) | (25) | (516) | (72) | (72) | (72) | (72) | ||
| O-6740 | 159 | 265 | >10,000 | Inactive at 8.5 and 28 μg/kg | ||||
| (45) | (26) | Lethal at 85 μg/kg | ||||||
Ki values are nanomolar S.E.M. values for receptor affinities are shown in parentheses.
Values shown are ED50 (95% confidence intervals in parentheses). Single dose tests are indicated by magnitude of effect, with dose that was tested in parentheses. All doses are expressed as micromoles per kilogram.
Table 3 presents binding and in vivo results with 3-substituted pyrazole analogs in which the amide group of rimonabant was replaced with an isosteric dihydrooxazole moiety. The unsubstituted dihydrooxazole analog O-4338 (Table 2) showed the best selectivity for CB1 receptors, having no measureable affinity for CB2 receptors. Although selective, O-4338 nonetheless exhibited poor CB1 receptor affinity (Ki = 680 nM; CP55,940 displacement). In an effort to retain selectivity while improving affinity, a number of alkyl ether-substituted dihydrooxazole analogs were synthesized (Table 3). Like O-4338, none of these ether analogs had measurable binding affinity at CB2 receptors. Furthermore, CB1 receptor affinity was enhanced in all of the compounds, 2- to 8-fold compared with O-4338. Length of the alkyl ether group also affected CB1 affinity, with optimal length of pentyl to heptyl, although substitution of an ethylbromo (O-6629) or ethylazide (O-6658) group produced superior improvement in CB1 affinity. The ratio of CB1 receptor binding measured with the two radioligands was similar to that of the compounds in Table 1 and ranged from 1.2- to 3.8-fold differences. With one exception (O-6659), CB1 receptor binding affinities were consistently better when [3H]SR141716 was used as the displacement ligand than when [3H]CP55,940 was used. It is noteworthy that, with the exception of O-4424, all of these compounds were also active in the in vivo tetrad of tests. O-4424 could not be assessed because of insufficient quantities. When sufficient quantities allowed, a dose-effect curve was determined in vivo, with potencies ranging from 2 to 36 μg/kg.
TABLE 3.
Cannabinoid receptor binding and in vivo effects of 1-(2,4-dichlorophenyl)-4-methyl-5-(4-chlorophenyl)-1H-pyrazoles with dihydrooxazole 3-substituents
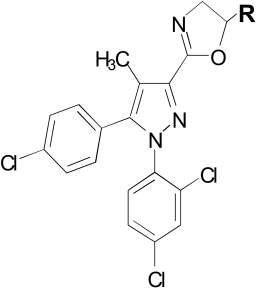
| Compound | R | Receptor Affinitiesa |
Effects in Miceb |
|||||
|---|---|---|---|---|---|---|---|---|
| [3H]CP55,940 | [3H]SR144528 | CB2 | SA | %MPE | RT | RI | ||
| O-4424 | 169 | 45 | >10,000 | N.T. | N.T. | N.T. | N.T. | |
| (0.4) | (3) | |||||||
| O-6211 | 138 | 52 | >10,000 | 2 | 6 | 4 | 12 | |
| (3) | (3) | (1–4) | (4–10) | (2–4) | (8–16) | |||
| O-6212 | 86 | 41 | >10,000 | 81 | 100 | −3.7 | Ataxia | |
| (6) | (1) | (59) | (59) | (59) | (59) | |||
| O-6617 | 85 | 43 | >10,000 | 12 | 8 | 19 | Ataxia | |
| (3) | (1) | (6–17) | (6–12) | (13–25) | (58) | |||
| O-6618 | 89 | 44 | >10,000 | 9 | 13 | 13 | 36 | |
| (10) | (2) | (4–15) | (9–17) | (9–15) | (22–52) | |||
| O-6213 | 182 | 91 | >10,000 | <17 | 9 | 10 | 10 | |
| (18) | (6) | (5–14) | (7–14) | (7–15) | ||||
| O-6215 | 332 | 221 | >10,000 | 9 | 13 | 7 | Ataxia | |
| (52) | (12) | (5–13) | (9–18) | (5–11) | (54) | |||
| O-6214 | 88 | 56 | >10,000 | 42 | 100 | −2.7 | Ataxia | |
| (4) | (2) | (59) | (59) | (59) | (59) | |||
| O-6629 | 24 | 20 | >10,000 | <2 | 1.5 | 1.3 | 4 | |
| (5) | (1) | (0.6–4) | (0.6–2) | (2–6) | ||||
| O-6658 | 57 | 46 | >10,000 | 1 | 2 | 2 | 12 | |
| (11) | (2) | (0.4–2) | (0.9–4) | (1–4) | (6–24) | |||
| O-6659 | 109 | 128 | >10,000 | 6 | 16 | 12 | ∼59 | |
| (7) | (6) | (4–12) | (10–26) | (6–18) | ||||
N.T., not tested.
Ki values are nanomolar S.E.M. values for receptor affinities are shown in parentheses.
Values shown are ED50 (95% confidence intervals in parentheses). Single dose tests are indicated by magnitude of effect, with dose that was tested in parentheses. All doses are expressed as micromoles per kilogram.
Figure 1, top and middle shows the relationship between CB1 receptor binding affinities and potencies for each of the tetrad tests. Correlations are notably low (r = −0.4–0.4; p > 0.05 for all values) for all in vivo tests. In contrast, the correlation between CB1 receptor binding affinities measured with two different radioligands was strong in magnitude and statistically significant (r = 0.89; p < 0.05) (Fig. 1, bottom).
Fig. 1.

Top and middle, scatterplots and regression lines of CB1 affinities (log Ki), measured by [3H]SR141716 displacement, plotted against log ED50 for each of the four in vivo tests (SA, spontaneous activity; RT, change in rectal temperature; RI, ring immobility). Bottom, scatterplot and regression line of CB1 affinities (log Ki) as measured with two radioligands, the cannabinoid receptor agonist [3H]CP55,940 and the cannabinoid receptor antagonist [3H]SR141716. Pearson product-moment correlations are shown for the two measures graphed in each panel. * indicates significant correlation (p < 0.05).
Because the compounds presented in Tables 1 to 3 are analogs of the CB1 receptor antagonist rimonabant, selected compounds provided in sufficient quantity were evaluated to determine reversal of the cannabimimetic effects of 3 mg/kg THC in the tetrad (Table 4). Of the 14 compounds tested, only four (O-4333, O-4334, O-2155, and O-4336) showed any reversal of THC's effects, with none of the compounds producing full blockade of all of the effects of THC. Whereas O-4333 and O-4334 were inactive in the tetrad when tested alone, O-2155 and O-4336 produced agonist-like effects in the tetrad (Table 1). It is noteworthy that O-4333 and O-4334 were among the compounds with the best CB1 receptor binding affinities.
TABLE 4.
Evaluation of antagonism of the in vivo effects of 3 mg/kg THC
Effects of the indicated compound on the agonist effects of 3 mg/kg THC in assays of SA, tail flick, RT, and RI are shown. Structures of all compounds are shown in Table 1, except for O-4338 (Table 2). Degree of reversal of THC's effects is indicated with compound dose at which antagonism was evaluated shown in parentheses.
| Compound | Reversal of THC |
|---|---|
| O-4331 | None (30 mg/kg) |
| O-4332 | None (30 mg/kg) |
| O-4333 | Partial-RI (3 mg/kg) |
| O-4334 | Partial-SA, RI (3 mg/kg) |
| O-2154 | None (3 mg/kg) |
| O-2155 | Partial-SA (10 mg/kg) |
| O-4335 | None (30 mg/kg) |
| O-4336 | Partial-SA, RT (3 mg/kg) |
| O-4337 | None (30 mg/kg) |
| O-4339 | None (30 mg/kg) |
| O-4371 | None (3 mg/kg) |
| O-4372 | None (30 mg/kg) |
| O-4373 | None (30 mg/kg) |
| O-4338 | None (30 mg/kg) |
None of the compounds activated CB1 receptors, as demonstrated by their lack of stimulation of [35S]GTPγS binding (data not shown). However, several compounds exhibited inverse agonist effects, with Emax values ranging from −46 to −69% relative to maximum CP55,940-induced stimulation (Table 5).
TABLE 5.
Evaluation of inverse agonism in [35S]GTPγS assay
Effects of indicated compounds on [35S]GTPγS binding are shown. Percentage of maximum in comparison with CP55,940 is shown. S.E.M.s are shown in parentheses. Structures of compounds O-2154 and O-4332 are shown in Table 1, and structures of O-6729, O-6730, O-6740, O-6659, and O-6668 are shown in Table 3.
| Compound | [35S]GTPγS |
|
|---|---|---|
| EC50 | Emax | |
| μM | % max | |
| O-2154 | 51 (48) | −46 (3) |
| O-4332 | 187 (36) | −65 (7) |
| O-6729 | 18 (3) | −69 (2) |
| O-6730 | 76 (51) | −66 (10) |
| O-6740 | 116 (17) | −65 (0.3) |
| O-6659 | 47 (19) | −64 (8) |
| O-6668 | 16 (2) | −64 (5) |
Figure 2 shows the effects of O-4332 in combination with vehicle or 10 mg/kg rimonabant on locomotor activity (top left), antinociception (top right), rectal temperature (bottom left), and ring immobility (bottom right). Factorial ANOVA revealed a significant main effect of O-4332 dose for locomotor activity (F3,40 = 28.6; p < 0.05), with significant decreases compared with vehicle produced by 1, 3, and 10 mg/kg O-4332 as shown by Tukey post hoc analysis. Likewise, a significant main effect of O-4332 dose was observed for antinociception (F3,40 = 25.9; p < 0.05), with 3 and 10 mg/kg (but not 1 mg/kg) doses of O-4332 producing antinociception compared with vehicle. A significant interaction was shown for the rectal temperature measure (F3,40 = 6.7; p < 0.05). Tukey post hoc analysis revealed that 3 and 10 mg/kg, but not 1 mg/kg, doses of O-4332 produced hypothermia compared with the vehicle/vehicle condition. Rimonabant (10 mg/kg) also slightly, but significantly, reduced rectal temperature. In addition, this dose of rimonabant attenuated the hypothermic effect of 3 mg/kg, but not 10 mg/kg, O-4332, suggesting that rimonabant reversibility of O-4332's hypothermic effect could be overcome by raising the dose of O-4332. A significant interaction was also seen with the ring immobility measure (F3,40 = 3.6; p < 0.05). Tukey post hoc analysis revealed that all doses of O-4332 increased percentage ring immobility compared with vehicle, with rimonabant (10 mg/kg) attenuation of this effect observed at 1 and 3 mg/kg doses of O-4332, but not at the 10 mg/kg dose.
Fig. 2.

Effects of O-4332 in combination with vehicle (open bars) and 10 mg/kg rimonabant (filled bars) on locomotor activity expressed as number of photocell beam breaks (top left), antinociception expressed as percentage of maximum possible effect (top right), change in rectal temperature (bottom left), and percentage ring immobility (bottom right). Each bar represents the mean (± S.E.M.) of six mice. # indicates significant (p < 0.05) main effect of O-4332 dose compared with vehicle. * indicates significant interaction, with post hoc difference in effect of O-4332 dose compared with vehicle/vehicle condition. ** indicates significant interaction, with post hoc difference in effect of O-4332 and rimonabant combination compared with the same dose of O-4332 with vehicle.
Figure 3 shows the effects of 10 mg/kg doses of four other selected analogs (O-6211, O-6629, O-6658, and O-6668) in combination with vehicle or 10 mg/kg rimonabant on locomotor activity (top left), antinociception (top right), rectal temperature (bottom left), and ring immobility (bottom right). Factorial ANOVA revealed a significant main effect of the compounds for locomotor activity (F4,82 = 89.0; p < 0.05), with significant decreases compared with vehicle produced by each of the four compounds revealed by Tukey post hoc analysis. The pretreatment × compound interaction was also significant (F4,82 = 3.1; p < 0.05), with post hoc analysis showing that the 10 mg/kg dose of rimonabant increased locomotion compared with vehicle treatment. For antinociception, significant main effects of pretreatment condition and compound were observed (F1,86 = 4.7; p < 0.05 and F4,86 = 263.6; p < 0.05, respectively). Post hoc analysis revealed that each of the four compounds produced antinociception compared with the groups that received vehicle or rimonabant, and rimonabant produced a significant, but small in magnitude, overall increase in antinociception. Similar significant main effects of pretreatment condition and compound were obtained for the rectal temperature measure (F1,86 = 7.6; p < 0.05 and F4,86 = 125.3; p < 0.05, respectively), with each compound producing a significant decrease in temperature (regardless of whether vehicle or rimonabant pretreatment occurred). Overall, rimonabant slightly, but significantly, increased the magnitude of this temperature decrease. For the ring immobility measure, a significant main effect of compound and a significant interaction were observed (F1,72 = 14.4; p < 0.05 and F3,72 = 10.6; p < 0.05, respectively). Three of the four compounds (O-6211, O-6629, and O-6668) significantly increased time spent immobile on the ring apparatus. For O-6211 and O-6629, immobility was increased further by pretreatment with rimonabant, whereas O-6668-induced immobility was decreased by rimonabant, although it was still significantly enhanced compared with vehicle. Data for O-6658 were unavailable for this measure, because the mice that received this compound could not maintain balance on the ring and fell off repeatedly.
Fig. 3.

Effects of 10 mg/kg doses of selected pyrazole analogs (O-6211, O-6629, O-6658, and O-6668) in combination with vehicle (Veh) (open bars) and 10 mg/kg rimonabant (filled bars) on locomotor activity expressed as number of photocell beam breaks (top left), antinociception expressed as percentage of maximum possible effect (top right), change in rectal temperature (bottom left), and percentage of ring immobility (bottom right). Each bar represents the mean (± S.E.M.) of six mice, with the exception that n = 24 for vehicle/vehicle and rimonabant/vehicle groups and n = 4–5 for ring immobility measure for O-6211 and O-6668. # indicates significant (p < 0.05) main effect of the indicated compound compared with vehicle. * indicates significant interaction, with post hoc difference in effect of compound and rimonabant condition compared with vehicle and compound condition.
Figure 4 shows the effects of 30 mg/kg O-4332 and 10 mg/kg O-6629, O-6658, and O-6668 in CB1(−/−) and CB1(+/+) mice on locomotor activity (top), ring immobility (middle), and rectal temperature (bottom). All of the selected compounds suppressed locomotor activity, increased ring immobility, and decreased rectal temperature in both genotypes, as indicated by significant main effects with follow-up post hoc analysis for each measure (F4,57 = 75.3, p < 0.05; F4,59 = 77.5, p < 0.05; and F4,54 = 158.1, p < 0.05, respectively), with significant differences compared with vehicle produced by each of the four compounds that were revealed by Tukey post hoc analyses.
Fig. 4.

Effects of 30 mg/kg O-4332 and 10 mg/kg O-6629, O-6658, and O-6668 in CB1(−/−) and CB1(+/+) mice (open and filled bars, respectively) on locomotor activity expressed as number of photocell beam breaks (top), percentage of ring immobility (middle), and change in rectal temperature (bottom). Each bar represents the mean (± S.E.M.) of three to six mice, with the exception that n = 15 to 19 for each genotype in the vehicle condition and n = 2 for ring immobility measure for O-6658 in CB1(+/+) mice. # indicates significant (p < 0.05) main effect of compound in both genotypes compared with vehicle (Veh). * indicates significant interaction, with post hoc difference in effect of compound between genotypes.
Discussion
Previous research has shown that manipulation of the 3-substituent of rimonabant results in orderly changes in CB1 receptor recognition (Lan et al., 1999; Wiley et al., 2001; Francisco et al., 2002), with some of the manipulations resulting in pharmacological activity in mice that was cannabinoid agonist-like (Wiley et al., 2001). The results here seem to reinforce the hypothesis that the 3-substituent is involved in cannabimimetic activity, because many of these compounds showed good potency in decreasing locomotor activity and producing antinociception and hypothermia. Furthermore, they exhibited good CB1 receptor selectivity, with some of the compounds having no measurable affinity for the CB2 receptor. In particular, substitution of a dihydrooxazole moiety at the 3-position of the pyrazole core, with or without addition of a terminal ether alkyl group, eliminated CB2 affinity, whereas most analogs in the series still retained good to moderate CB1 affinity. Previous research has suggested that a structural feature of pyrazole analogs that enhances CB1 binding affinity and is crucial to their inverse agonism is the carboxamide oxygen (Hurst et al., 2006). The present results suggest that moderate CB1 affinity and inverse agonism were retained by compounds with 3-substituted dihydrooxazole and oxazole moieties (see compounds in Tables 2 and 3), but lacking a carboxamide oxygen. These compounds also exhibited the greatest CB1 receptor selectivity, suggesting that the carboxamide oxygen may play a role in residual CB2 receptor affinity of rimonabant and its analogs that contain it (see Table 1). Nevertheless, compared with compounds possessing only nitrogen-containing substituents (e.g., O-6729, O-6731, and O-6740), better CB1 affinity was observed for compounds with a carboxamide substituent (e.g., O-4333 and O-4334) or oxazole or dihydrooxazole substitutions (e.g., O-6668 and O-6629).
A consistent observation across most of the compounds was the difference in CB1 receptor affinity dependent on binding radioligand, [3H]CP55,940 and [3H]SR141716, with most compounds showing equivalent or better (up to 4.7-fold) affinity when displacing [3H]SR141716. Previous results have demonstrated that cannabinoid agonists of several classes displace [3H]CP55,940 with greater affinity, whereas CB1 receptor antagonists (pyrazole analogs) exhibit greater potency at displacing [3H]SR141716 (Thomas et al., 1998, 2005; Gullapalli et al., 2010). The enhanced affinity shown here by the pyrazole analogs possessing agonist-like effects suggests that the basis for the differences in affinities resides in the structural (versus functional) properties of the compound. The compounds did not stimulate [35S]GTPγS binding; however, antagonism in this assay was not evaluated.
Several aspects of the cannabinoid agonist-like activity of the 3-substituted pyrazole analogs in mice remain puzzling. First, potencies for producing cannabinoid agonist-like effects were not highly correlated with CB1 receptor binding affinities. In contrast, investigations of the SARs of cannabinoid agonists based on the structural templates of THC and CP55,940 (Compton et al., 1993) or WIN55,212-2 (Wiley et al., 1998) typically report strong correlations between CB1 receptor binding affinities and potencies for these measures in mice. A second inconsistency is the lack of stimulation of CB1 receptors in [35S]GTPγS binding, a functional assay of activation of G protein-coupled receptors. All other classes of cannabinoid agonists produce this effect (Selley et al., 1996; Breivogel and Childers, 2000), whereas rimonabant has been shown to be an inverse agonist in this assay (Landsman et al., 1997). Indeed, several of the analogs presented here also exhibited inverse agonism, suggesting greater similarity to CB1 receptor antagonists than agonists in vitro. In addition, structural variants of O-4333 and O-4334 with identical 3-substituents, but that also contained 4-cyanomethyl substitution on the pyrazole core, potently antagonized CP55,940-induced stimulation of [35S]GTPγS (Cooper et al., 2010). O-4333 and O-4334 also partially blocked the effects of THC in vivo without having agonist activity when administered alone. Yet, with the exception of these two compounds and a couple of others (O-2155 and O-4336) that possessed remnants of antagonist activity in vivo, the selected compounds that were tested as antagonists did not block the in vivo agonist effects of THC, as previous reports have shown that rimonabant does (Rinaldi-Carmona et al., 1995; Compton et al., 1996). The active 3-substituted pyrazoles presented here also are not likely to be producing their effects through activation of CB2 receptors. Although research provides evidence of the possibility of CB2 receptors in the brain (Van Sickle et al., 2005; Onaivi et al., 2006), the localization and function of these receptors (e.g., neuronal versus glial) remain uncertain (Cabral et al., 2008). In addition, challenge tests with the CB2 receptor antagonist 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528) have shown that this compound did not reverse the cannabimimetic effects of THC analogs in this battery of tests (Wiley et al., 2002), suggesting that CB2 receptors do not mediate the effects. Finally, the compounds with the most potency in vivo were CB1 receptor selective, having very low or absent affinity for CB2 receptors.
In an effort to determine whether the cannabinoid agonist-like action of these compounds might be produced by interaction with CB1 receptors despite the contradictory results of the SAR analysis, the effects of rimonabant were evaluated in combination with O-4332, one of the compounds that was active in all tests. Although the binding affinity for CB1 receptors was poor, it was comparable with that of JWH-104 (deoxy-Δ9(11)-THC-dimethylheptyl), a compound that was previously shown to have cannabimimetic effects in these tests through low efficacy activation of the CB1 receptor (Wiley et al., 2002). In the present study, the agonist effects of O-4332 were replicated; however, rimonabant failed to block O-4332-induced hypomobility and antinociception, suggesting that these effects were not CB1 receptor-mediated. Rimonabant also did not reverse the in vivo effects of O-6211, O-6629, O-6658, and O-6668, each of which had much better CB1 receptor selectivity and binding affinities than O-4332. A similar pattern of activity in all tests without rimonabant reversibility has been noted with antipsychotics (Wiley and Martin, 2003), suggesting that activity in the test battery is selective, but not specific, for cannabinoids. Conceivably, then, the pyrazole analogs could be working via a noncannabinoid mechanism. Conversely, rimonabant readily blocks the effects of cannabinoid agonists. For example, it reversed the effects of the low-efficacy cannabinoid agonist JWH-104 (Wiley et al., 2002) and has been shown to block the in vivo effects of various classes of cannabinoid agonists in several species (Rinaldi-Carmona et al., 1994; Wiley et al., 1995b; Compton et al., 1996; Huestis et al., 2001). Instances in which rimonabant did not reverse the effects of cannabinoids also have been reported, particularly for anandamide (Adams et al., 1998); however, the influence of differences in pharmacokinetic factors between THC- and anandamide-like cannabinoids cannot be ruled out. Likewise, pharmacokinetic factors may have also influenced the in vivo results obtained here and is one direction for future research.
In addition to the assessment of rimonabant reversal of their in vivo effects, selected pyrazoles were evaluated in CB1(−/−) and CB1(+/+) mice. The finding that these compounds produced cannabinoid agonist-like effects in both genotypes adds further support to the hypothesis that they are not producing their in vivo effects via activation of CB1 receptors, despite the fact that some of them bind quite well to this receptor. Given these seemingly contradictory results, a crucial question is the mechanism through which these novel pyrazoles are producing their cannabinoid agonist-like in vivo effects. Although this mechanism may be noncannabinoid (as discussed in the preceding paragraph), the fact that the structure of the compounds closely resembles that of rimonabant, a known cannabinoid antagonist, combined with the finding that many of these compounds actually do bind to CB1 receptors suggests that their effects reflect cannabinoid activity. Furthermore, it is conceivable that this activity may be mediated through a CB3 receptor that has been hypothesized (for a review, see Wiley and Martin, 2002), but not yet identified definitively. Candidates for this putative receptor include deorphanized G protein-coupled receptors such as GPR55, transient receptor potential vanilloid 1 ion channels, or peroxisome proliferator-activated receptors (e.g., peroxisome proliferator-activated receptor γ) (for a comprehensive review, see Pertwee et al., 2010).
In summary, this study reports the discovery of a novel class of 3-substituted pyrazole analogs with good to moderate affinity for CB1 receptors and without measurable affinity for CB2 receptors. Although many of these compounds produced in vivo effects in mice that were similar to those observed with CB1 agonists such as THC, the poor correlation between CB1 binding affinity and potency in the tetrad tests suggests that these effects were not mediated by action of the compounds at CB1 receptors. Further support for this hypothesis is derived from the finding that selected “agonist-like” compounds did not activate the CB1 receptor in the [35S]GTPγS assay nor were the in vivo effects of the compounds antagonized by rimonabant. In addition, these compounds produced similar profiles of in vivo effects in CB1(−/−) and CB1(+/+) mice. Together, these results strongly suggest that 3-substituted analogs of rimonabant represent a novel class of cannabinoids that structurally resemble CB1 receptor antagonists, but produce a profile of activity in mice similar to that of cannabinoid agonists through a non-CB1, non-CB2 mechanism. To date, this novel mechanism has not been identified, but may be the putative CB3 receptor.
Acknowledgments
We thank Ramona Winckler and Irina Beletskaya for technical assistance in performing experiments. The late Dr. Billy Martin initiated the research described herein and this article is dedicated to his memory.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA-03672, DA-005488].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- CB
- cannabinoid
- SR141716
- 5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide
- CP 55,940
- (−)-cis-3-[2-hydroxy-4(1,1-dimethyl-heptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexanol
- GTPγS
- guanosine-5′-O-(3-thio)-triphosphate
- MPE
- maximum possible effect
- SA
- spontaneous activity
- RT
- change in rectal temperature
- RI
- ring immobility
- SAR
- structure-activity relationship
- THC
- Δ9-tetrahydrocannabinol
- WIN 55,212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone
- BSA
- bovine serum albumin
- ANOVA
- analysis of variance
- SR144528
- 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide
- JWH-104
- deoxy-Δ9(11)-THC-dimethylheptyl
- O-4332
- 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-morpholinopropyl)-1H-pyrazole-3-carboxamide.
Authorship Contributions
Participated in research design: Wiley and Selley.
Conducted experiments: Selley.
Contributed new reagents or analytic tools: Wang, Kottani, Gadthula, and Mahadevan.
Performed data analysis: Wiley and Selley.
Wrote or contributed to the writing of the manuscript: Wiley, Selley, and Mahadevan.
References
- Adams IB, Compton DR, Martin BR. (1998) Assessment of anandamide interaction with the cannabinoid brain receptor: SR 141716A antagonism studies in mice and autoradiographic analysis of receptor binding in rat brain. J Pharmacol Exp Ther 284:1209–1217 [PubMed] [Google Scholar]
- Bass CE, Griffin G, Grier M, Mahadevan A, Razdan RK, Martin BR. (2002) SR-141716A-induced stimulation of locomotor activity. A structure-activity relationship study. Pharmacol Biochem Behav 74:31–40 [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR. (2000) Cannabinoid agonist signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation, and adenylyl cyclase inhibition. J Pharmacol Exp Ther 295:328–336 [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. (2008) CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153:240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) on an enzymatic reaction. Biochem Pharmacol 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713 [DOI] [PubMed] [Google Scholar]
- Compton DR, Aceto MD, Lowe J, Martin BR. (1996) In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): inhibition of Δ9-tetrahydrocannabinol-induced responses and apparent agonist activity. J Pharmacol Exp Ther 277:586–594 [PubMed] [Google Scholar]
- Compton DR, Rice KC, De Costa BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. (1993) Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther 265:218–226 [PubMed] [Google Scholar]
- Cooper M, Receveur JM, Bjurling E, Nørregaard PK, Nielsen PA, Sköld N, Högberg T. (2010) Exploring SAR features in diverse library of 4-cyanomethyl-pyrazole-3-carboxamides suitable for further elaborations as CB1 antagonists. Bioorg Med Chem Lett 20:26–30 [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949 [DOI] [PubMed] [Google Scholar]
- Fattore L, Cossu G, Martellotta CM, Fratta W. (2001) Intravenous self-administration of the cannabinoid CB1 receptor agonist WIN 55,212-2 in rats. Psychopharmacology (Berl) 156:410–416 [DOI] [PubMed] [Google Scholar]
- Francisco ME, Seltzman HH, Gilliam AF, Mitchell RA, Rider SL, Pertwee RG, Stevenson LA, Thomas BF. (2002) Synthesis and structure-activity relationships of amide and hydrazide analogues of the cannabinoid CB1 receptor antagonist N- (piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716). J Med Chem 45:2708–2719 [DOI] [PubMed] [Google Scholar]
- Gullapalli S, Amrutkar D, Gupta S, Kandadi MR, Kumar H, Gandhi M, Karande V, Narayanan S. (2010) Characterization of active and inactive states of CB1 receptor and the differential binding state modulation by cannabinoid agonists, antagonists and inverse agonists. Neuropharmacology 58:1215–1219 [DOI] [PubMed] [Google Scholar]
- Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, Frank RA. (2001) Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry 58:322–328 [DOI] [PubMed] [Google Scholar]
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, et al. (2006) Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem 49:5969–5987 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington DC [Google Scholar]
- Järbe TU, Harris MY, Li C, Liu Q, Makriyannis A. (2004) Discriminative stimulus effects in rats of SR-141716 (rimonabant), a cannabinoid CB1 receptor antagonist. Psychopharmacology (Berl) 177:35–45 [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A. (1999) Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem 42:769–776 [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. (1997) SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur J Pharmacol 334:R1–R2 [DOI] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. (1991) Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav 40:471–478 [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Cabeza R, Méndez-Díaz M, Navarro L, Prospéro-García O. (2001) Anandamide-induced sleep is blocked by SR141716A, a CB1 receptor antagonist and by U73122, a phospholipase C inhibitor. Neuroreport 12:2131–2136 [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, Myers L, Mora Z, Tagliaferro P, Gardner E, et al. (2006) Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann NY Acad Sci 1074:514–536 [DOI] [PubMed] [Google Scholar]
- Pan X, Ikeda SR, Lewis DL. (1998) SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2+ currents by reversal of tonic CB1 cannabinoid receptor activity. Mol Pharmacol 54:1064–1072 [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, et al. (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Alonso R, Shire D, Congy C, Soubrié P, Brelière JC, Le Fur G. (1995) Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci 56:1941–1947 [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D. (1994) SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350:240–244 [DOI] [PubMed] [Google Scholar]
- Selley DE, Stark S, Sim LJ, Childers SR. (1996) Cannabinoid receptor stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding in rat brain membranes. Life Sci 59:659–668 [DOI] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME. (1996) Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther 278:989–999 [PubMed] [Google Scholar]
- Thomas BF, Francisco ME, Seltzman HH, Thomas JB, Fix SE, Schulz AK, Gilliam AF, Pertwee RG, Stevenson LA. (2005) Synthesis of long-chain amide analogs of the cannabinoid CB1 receptor antagonist N- (piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3- carboxamide (SR141716) with unique binding selectivities and pharmacological activities. Bioorg Med Chem 13:5463–5474 [DOI] [PubMed] [Google Scholar]
- Thomas BF, Gilliam AF, Burch DF, Roche MJ, Seltzman HH. (1998) Comparative receptor binding analyses of cannabinoid agonists and antagonists. J Pharmacol Exp Ther 285:285–292 [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, et al. (2005) Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 310:329–332 [DOI] [PubMed] [Google Scholar]
- Wiley JL, Barrett RL, Lowe J, Balster RL, Martin BR. (1995a) Discriminative stimulus effects of CP 55,940 and structurally dissimilar cannabinoids in rats. Neuropharmacology 34:669–676 [DOI] [PubMed] [Google Scholar]
- Wiley JL, Burston JJ, Leggett DC, Alekseeva OO, Razdan RK, Mahadevan A, Martin BR. (2005) CB1 cannabinoid receptor-mediated modulation of food intake in mice. Br J Pharmacol 145:293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JL, Compton DR, Dai D, Lainton JA, Phillips M, Huffman JW, Martin BR. (1998) Structure-activity relationships of indole- and pyrrole-derived cannabinoids. J Pharmacol Exp Ther 285:995–1004 [PubMed] [Google Scholar]
- Wiley JL, Jefferson RG, Grier MC, Mahadevan A, Razdan RK, Martin BR. (2001) Novel pyrazole cannabinoids: insights into CB(1) receptor recognition and activation. J Pharmacol Exp Ther 296:1013–1022 [PubMed] [Google Scholar]
- Wiley JL, Jefferson RG, Griffin G, Liddle J, Yu S, Huffman JW, Martin BR. (2002) Paradoxical pharmacological effects of deoxy-tetrahydrocannabinol analogs lacking high CB1 receptor affinity. Pharmacology 66:89–99 [DOI] [PubMed] [Google Scholar]
- Wiley JL, Lowe JA, Balster RL, Martin BR. (1995b) Antagonism of the discriminative stimulus effects of Δ9-tetrahydrocannabinol in rats and rhesus monkeys. J Pharmacol Exp Ther 275:1–6 [PubMed] [Google Scholar]
- Wiley JL, Martin BR. (2002) Cannabinoid pharmacology: implications for additional cannabinoid receptor subtypes. Chem Phys Lipids 121:57–63 [DOI] [PubMed] [Google Scholar]
- Wiley JL, Martin BR. (2003) Cannabinoid pharmacological properties common to other centrally acting drugs. Eur J Pharmacol 471:185–193 [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. (1999) Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A 96:5780–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]