

Abstract
Parkinson's disease (PD) is a debilitating neurodegenerative disorder associated with severe motor impairments caused by the loss of dopaminergic innervation of the striatum. Previous studies have demonstrated that positive allosteric modulators (PAMs) of metabotropic glutamate receptor 4 (mGlu4), including N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide, can produce antiparkinsonian-like effects in preclinical models of PD. However, these early mGlu4 PAMs exhibited unsuitable physiochemical properties for systemic dosing, requiring intracerebroventricular administration and limiting their broader utility as in vivo tools to further understand the role of mGlu4 in the modulation of basal ganglia function relevant to PD. In the present study, we describe the pharmacologic characterization of a systemically active mGlu4 PAM, N-(3-chlorophenyl)picolinamide (VU0364770), in several rodent PD models. VU0364770 showed efficacy alone or when administered in combination with l-DOPA or an adenosine 2A (A2A) receptor antagonist currently in clinical development (preladenant). When administered alone, VU0364770 exhibited efficacy in reversing haloperidol-induced catalepsy, forelimb asymmetry-induced by unilateral 6-hydroxydopamine (6-OHDA) lesions of the median forebrain bundle, and attentional deficits induced by bilateral 6-OHDA nigrostriatal lesions in rats. In addition, VU0364770 enhanced the efficacy of preladenant to reverse haloperidol-induced catalepsy when given in combination. The effects of VU0364770 to reverse forelimb asymmetry were also potentiated when the compound was coadministered with an inactive dose of l-DOPA, suggesting that mGlu4 PAMs may provide l-DOPA-sparing activity. The present findings provide exciting support for the potential role of selective mGlu4 PAMs as a novel approach for the symptomatic treatment of PD and a possible augmentation strategy with either l-DOPA or A2A antagonists.
Introduction
Parkinson's disease (PD) is a chronic neurodegenerative disorder that affects approximately 1% of the population worldwide over the age of 55 and is associated with motor symptoms including tremor, muscle rigidity, bradykinesia, and gait/postural imbalances (Jankovic, 2008). The primary neuropathology of PD involves the progressive degeneration of dopamine neurons in the substantia nigra pars compacta (SNc), which provide the major dopaminergic innervation to the striatum and other basal ganglia (BG) nuclei (Hassler, 1938). Although dopamine replacement therapies, such as l-DOPA, remain the gold standard for the symptomatic treatment of PD, these treatments ultimately fail to provide reliable efficacy with disease progression and are associated with numerous dose-limiting side effects, including on/off oscillations, dyskinesias, and cognitive impairments (Stacy and Galbreath, 2008).
Over the last decade, an enhanced understanding of BG circuitry has allowed the investigation of mechanisms that alter BG function in ways that do not rely on dopamine replacement. Within the BG, normal motor movement is regulated by two primary output nuclei, the substantia nigra pars reticulata (SNr) and the internal globus pallidus (GPi; entopeduncular nucleus in nonprimates) (Conn et al., 2005; Niswender and Conn, 2010). Output from the SNr/GPi is maintained by a delicate balance between two distinct striatal GABAergic projection pathways: the direct pathway, which projects directly to the SNr/GPi, and the indirect pathway, which projects from the striatum to the external globus pallidus (GPe). The GPe then sends additional GABAergic projections to the subthalamic nucleus (STN), which in turn provides excitatory input to the SNr/GPi (Conn et al., 2005). Loss of striatal dopaminergic innervation from the SNc results in overactivation of the indirect pathway (excessive inhibitory tone at the level of the GPe and subsequent disinhibition of the STN), leading to increased excitation of the SNr/GPi nuclei and the motor impairments observed in PD (Conn et al., 2005). Recent surgical strategies have focused on correcting this overactivity within the indirect pathway; deep brain stimulation of the STN, for example, has shown efficacy in correcting PD motor symptoms, suggesting that pharmacological manipulation of synaptic transmission in the indirect pathway may provide an important approach for nondopaminergic PD treatment.
Two important targets for potential pharmacologic modulation of the indirect pathway are the adenosine A2A receptor and metabotropic glutamate receptor 4 (mGlu4) (Johnson et al., 2009; Morelli et al., 2009). Antagonism of adenosine A2A receptors, expressed on medium spiny GABAergic neurons that project to the GPe, results in decreased inhibition of the GPe and antiparkinsonian-like effects in animal PD models (Hettinger et al., 2001; Hodgson et al., 2010). In a recent phase II, double-blind clinical trial, positive effects were reported on the motor impairments in patients with Parkinson's disease by using the A2A receptor antagonist preladenant (Hauser et al., 2011). With regard to mGlu4, activation of this receptor, expressed presynaptically on GABAergic neurons within the striato-pallidal synapse, also reduces excessive inhibitory tone within the GPe and reverses motor deficits in rodent PD models (Bradley et al., 1999; Matsui and Kita, 2003; Valenti et al., 2003; Lopez et al., 2007; Sibille et al., 2007; Niswender et al., 2008; Beurrier et al., 2009). Unfortunately, the development of selective orthosteric mGlu4 agonists has been hindered by the high conservation of the glutamate binding site across the mGlu subtypes.
Using an alternative strategy, our group and others have identified highly selective mGlu4 ligands that potentiate receptor activity via action at sites that are less highly conserved and topographically distinct relative to the glutamate binding site, termed positive allosteric modulators (PAMs). For example, (±)-cis-2-(3,5-dicholorphenylcarbamoyl)cyclohexanecarboxylic (VU0155041) and N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide represent two chemically and structurally distinct classes of mGlu4 PAMs that bind to allosteric sites on mGlu4 and potentiate the effects of glutamate (Maj et al., 2003; Marino et al., 2003; Niswender et al., 2008). These early mGlu4 PAMs provided important proof-of-concept data suggesting antiparkinsonian-like efficacy in preclinical PD models, but had limited utility as in vivo tools because of unsuitable physiochemical properties (e.g., limited solubility and central penetration) for systemic dosing studies. We have reported the discovery of a novel series of biaryl amides with mGlu4 PAM activity that exhibited submicromolar potency at rat and human mGlu4 as well as improved physiochemical properties for systemic dosing (Engers et al., 2009). Here, we describe the pharmacologic characterization of a systemically active mGlu4 PAM, N-(3-chlorophenyl)picolinamide (VU0364770), derived from this biaryl amide chemical series. Using multiple rodent PD models, we show that VU0364770 exhibits efficacy when administered alone as well as in combination with l-DOPA or the adenosine A2A receptor antagonist preladenant.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (Harlan, Indianapolis, IN), weighing 250 to 300 g, used for the haloperidol and forelimb asymmetry (FLA) studies were group-housed with food and water available ad libitum. Rats for the haloperidol studies were kept under a 12-h light/dark cycle with lights on from 6:00 AM to 6:00 PM and tested during the light phase unless stated otherwise. Rats for the forelimb asymmetry studies were housed under a reversed 12-h light/dark cycle with lights on between 4:00 PM and 4:00 AM and tested during the first half of the dark cycle. For the reaction time (RT) task studies, male Wistar rats (Charles River Laboratories, l'Arbresle, France), weighing 175 to 185 g at the beginning of the experiment, were used and fed 15 to 17 g/day of laboratory chow delivered 3 h after the testing period, so as to maintain 85% of their free feeding body weight with water provided ad libitum. All experimental procedures were approved by the Vanderbilt University Animal Care and Use Committee and followed the guidelines set forth by the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and the requirements of the European Communities Council (directive 86/609/EEC, November 24, 1986).
Drugs
Apomorphine hydrochloride, benserazide hydrochloride, clorgyline, R-(−)-deprenyl hydrochloride, haloperidol, l-DOPA methyl ester, and 6-hydroxydopamine hydrobromide were obtained from Sigma (St. Louis, MO). l-Glutamate was obtained from Tocris Bioscience (Ellisville, MO). Preladenant, VU0364770, N-(6-(trifluoromethyl)pyridin-2-yl)picolinamide (VU0364772), and the A2A antagonist (Neurocrine Biosciences, San Diego, CA) (Slee et al., 2008) were synthesized in-house in the laboratory of Dr. C. R. Hopkins. Clorgyline, R-(−)-deprenyl hydrochloride, VU0364770, VU0364772, and preladenant were suspended in an aqueous solution of 10% Tween 80. Benserazide and l-DOPA methyl ester were dissolved in 0.9% saline, whereas haloperidol and the Neurocrine adenosine 2A receptor antagonist were dissolved in 0.85% lactic acid (Sigma) and 0.5% Cremophor EL (Sigma), respectively. Except for apomorphine and l-DOPA, which are unstable at neutral pH, all drug formulations were adjusted to a pH of approximately 7 by using 1 N sodium hydroxide. Drugs were administered in a volume of 1 to 2 ml/kg i.p./s.c.) or 10 ml/kg p.o.
The chemical synthesis for the biaryl amides VU0364770 and VU0364772 was completed by an in-house synthesis using the following methods.
N-(3-Chlorophenyl)picolinamide.
To a solution of 3-chloroaniline (29.5 ml, 0.28 mol) in dimethylformamide (300 ml) at 0°C was added 4-dimethylaminopyridine (50 mg) and N,N-diisopropylethylamine (118 ml, 0.84 mol) (Scheme 1). Picolinoyl chloride hydrochloride (50 g, 0.28 mol) was added portionwise over 30 min, and after an additional 15 min, the ice bath was removed. After 16 h at room temperature, the reaction was added to EtOAc/NaHCO3 (saturated) (1:1; 1000 ml). The organic layer was separated and washed with water (5 × 200 ml) and brine (200 ml), dried (MgSO4), filtered, and concentrated under reduced pressure. After recrystallization of the crude solid from EtOH, the desired product was obtained as an off-white solid (63.5g, 84% yield): Rf = 0.47 (33% EtOAc/hexanes); Mp = 81 to 83°C; analytical LCMS: single peak (214 nm), 3.417 min; 1H NMR (400 MHz, CDCl3): δ 10.06 (br s, 1H), 8.62 (d, J = 4.4 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.95 to 7.91 (m, 2H), 7.63 (dd, J = 8.0, 1.2 Hz, 1H), 7.51 (dd, J = 7.6, 4.8 Hz, 1H), 7.31 (dd, 8.4, 8.0 Hz, 1H), 7.13 (dd, J = 8.0, 1.2 Hz, 1H); high-resolution MS, calculated for C12H10N2OCl (M+H+), 233.0482; found 233.0483.
Scheme 1.

N-(3-chlorophenyl)picolinamide.
N-(6-(Trifluoromethyl)pyridin-2-yl)picolinamide.
To a solution of 6-(trifluoromethyl)pyridin-2-amine (5.0 g, 30.8 mmol) in dimethylformamide (60 ml) at 0°C was added 4-dimethylaminopyridine (10 mg) and N,N-diisopropylethylamine (13.0 ml, 92.4 mmol) (Scheme 2). Picolinoyl chloride hydrochloride (5.8 g, 32.4 mmol) was added portionwise over 10 min, and after an additional 15 min, the ice bath was removed. After 12 h at room temperature, the reaction was added to EtOAc/NaHCO3 (saturated) (1:1; 100 ml). The organic layer was separated and washed with water (3 × 20 ml) and brine (20 ml), dried (MgSO4), filtered, and concentrated under reduced pressure. After recrystallization, the desired product was obtained as a white solid (4.7 g, 57%): Rf = 0.46 (33% EtOAc/hexanes); Mp = 141 to 142°C; analytical LCMS: single peak (214 nm), 3.546 min; 1H NMR (400 MHz, CDCl3): δ 10.68 (br s, 1H), 8.68 to 8.64 (m, 2H), 8.30 (d, J = 7.6 Hz, 1H), 7.94 (dd, J = 7.6, 7.6 Hz, 1H), 7.93 (dd, J = 8.0, 7.6 Hz, 1H), 7.53 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H); high-resolution MS, calculated for C12H9N3OF3 (M+H+), 268.0698; found 268.0695.
Scheme 2.

N-(6-(trifluoromethyl)pyridin-2-yl)picolinamide.
HCl Salt Formation for In Vivo Studies.
To a solution of the amide in dichloromethane (0.2M) at 0°C was added 4 M HCl in 1,4-dioxane (5 eq.) dropwise. After 15 min, the ice bath was removed. The solvent was removed after additional 30 min at room temperature to provide a pure HCl salt of the appropriate amide.
In Vitro Studies
Cell Culture.
Human mGlu4/Gqi5/Chinese hamster ovary cells were grown in 90% Dulbecco's modified Eagle's medium (DMEM), 10% dialyzed fetal bovine serum (FBS), 100 units/ml penicillin/streptomycin, 20 mM HEPES, pH 7.3, 1 mM sodium pyruvate, 20 μg/ml proline, 2 mM glutamine, 400 μg/ml G418 sulfate (Mediatech, Herndon, VA), and 5 nM methotrexate (EMD Chemicals, Gibbstown, NJ). Rat mGlu4/HEK/G protein-regulated inwardly rectifying K (potassium) channel (GIRK) cells, as well as HEK/GIRK lines expressing rat mGlu2, mGlu3, mGlu7, mGlu8, and human mGlu6, were cultured in 45% DMEM, 45% F-12, 10% FBS, 20 mM HEPES, 2 mM [scap]l-glutamine, 100 units/ml penicillin/streptomycin, 1× nonessential amino acids, 1 mM sodium pyruvate, 700 μg/ml G418, and 0.6 μg/ml puromycin. Rat mGlu1 and mGlu5 HEK cell lines were grown in DMEM, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, 1× nonessential amino acids, 1 mM sodium pyruvate, and 500 μg/ml G418. All cell culture reagents were from Invitrogen (Carlsbad, CA) unless otherwise noted.
Calcium Mobilization Assays.
Human mGlu4/Gqi5/Chinese hamster ovary cells (30,000 cells/20 μl/well) were plated in black-walled, clear-bottomed, tissue culture-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, 100 units/ml penicillin/streptomycin, and 1 mM sodium pyruvate (plating medium). The cells were grown overnight at 37°C in the presence of 5% CO2. The next day, the medium was removed and replaced with 20 μl of 1 μM Fluo-4 AM (Invitrogen) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 and diluted in assay buffer (Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid; Sigma) for 45 min at 37°C. Dye was removed and replaced with 20 μl of assay buffer. For concentration-response curve experiments, compounds were serially diluted 1:3 into 10 point concentration response curves in DMSO, transferred to daughter plates by using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA), and diluted in assay buffer to a 2× final concentration. Ca2+ flux was measured by using the Functional Drug Screening System 6000 (FDSS6000; Hamamatsu Photonics Systems, Hamamatsu, Japan). After establishment of a fluorescence baseline for 4 s (four images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), 20 μl of test compounds were added to the cells, and the response was measured. At 142 s later, 10 μl (5×) of an EC20 concentration of glutamate was added to the cells, and the response of the cells was measured. After an additional 120 s, 12 μl (5×) of an EC80 concentration of agonist was added, and readings taken for an additional 40 s. Calcium fluorescence was recorded as fold over basal fluorescence, and raw data were normalized to the maximal response to glutamate. Potency (EC50) and maximum response (percentage of Glu maximum) for compounds were determined by using a four-parameter logistical equation in Prism (GraphPad Software, Inc., San Diego, CA). For efficacy experiments, a constant amount of compound was applied before the addition of a full glutamate concentration-response curve, and the left shift of the EC50 of the curves was calculated as “fold shift.”
GIRK-Mediated Thallium Flux Assay.
Cells were plated into 384-well, black-walled, clear-bottom, poly-d-lysine-coated plates (Greiner Bio-One) at a density of 15,000 cells/20 μl/well in DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 100 units/ml penicillin/streptomycin (assay media). Plated cells were incubated overnight at 37°C in the presence of 5% CO2. The next day, the medium was removed from the cells, and 20 μl/well of 330 nM Fluo Zn2 (Invitrogen; prepared as a stock in DMSO and mixed in a 1:1 ratio with pluronic acid F-127) in assay buffer [Hanks' balanced salt (Invitrogen) containing 20 mM HEPES, pH 7.3] was added to the plated cells. Cells were incubated for 1 h at room temperature, and the dye was replaced with the 20 μl of assay buffer.
Test Compound Preparation.
Glutamate was diluted in thallium buffer [125 mM sodium bicarbonate (added fresh the morning of the experiment), 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES, pH 7.3] at 5× the final concentration to be assayed. For concentration-response curve experiments, compounds were serially diluted 1:3 into 10 point concentration- response curves in DMSO, transferred to daughter plates using the Echo, and diluted in assay buffer to a 2× final concentration. Cell plates and compound plates were loaded onto a Hamamatsu FDSS6000 kinetic imaging plate reader. Baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and test compounds were added in a 20-μl volume and incubated for 2.5 min before the addition of 10 μl of thallium buffer ± agonist. After the addition of agonist, data were collected for an additional 2 min.
Data were analyzed by using Excel (Microsoft, Redmond, WA). Raw data were opened in Excel, and each data point in a given trace was divided by the first data point from that trace (static ratio). For experiments in which PAMs were added, data were again normalized by dividing each point by the fluorescence value immediately before the agonist addition to correct for any subtle differences in the baseline traces after the compound incubation period. The slope of the fluorescence increase beginning 5 s after thallium/agonist addition and ending 15 s after thallium/agonist addition was calculated. Curves were fitted by using a four-point logistical equation using GraphPad Prism. For efficacy experiments, a constant amount of compound was applied before the addition of a full glutamate concentration-response curve, and the left shift of the EC50 of the curves was calculated as fold shift.
Selectivity Studies
Rat mGlu1 and mGlu5.
HEK cells expressing rat mGlu1. The effects of VU0364770 on rat mGlu1 and mGlu5 were assessed by using calcium mobilization and measuring the glutamate concentration-response relationship in the presence and absence of 10 μM VU0364770. Using a double-addition protocol, VU0364770 was added to the cells, followed 2.5 min later by a full concentration-response of glutamate. Shifts of the concentration-response relationship were used to assess potential potentiator (left shift of more than 2-fold) or antagonist (right shift of more than 2-fold or depression of the maximum response by at least 75%) activity of VU0364770. Compounds were further assessed for mGlu5 antagonist activity by performing a full concentration-response curve, starting at 30 μM and serially diluted it by using 1:3 dilutions, in the presence of an EC80 concentration of glutamate.
Rat mGlu2, mGlu3, mGlu7, and mGlu8 and Human mGlu6.
Compound activity at the rat group II and III mGlus was assessed by using thallium flux through GIRKs as described above for mGlu4 fold-shift experiments. Cells were plated at a density of 15,000 cells/20 μl/well in assay media. The effects of VU0364770 were assessed by measuring the glutamate concentration-response relationship in the presence and absence of 10 μM VU0364770. Using a double-addition protocol, VU0364770 was added to the cells, followed 2.5 min later by a full concentration-response of glutamate or, in the case of mGlu7, (2S)-2-amino-4-phosphonobutanoic acid. As above, shifts of the concentration-response relationship were used to assess potential potentiator (left shift of more than 2-fold) or antagonist (right shift of more than 2-fold or depression of the maximum response by at least 75%) activity of VU0364770. Compounds were further assessed for mGlu6 PAM activity by performing a full concentration-response curve, starting at 30 μM and serially diluted it using 1:3 dilutions, in the presence of an EC20 concentration of glutamate.
GPCRProfiler Assay.
GPCRProfiler (Millipore Corporation, Billerica, MA) assay buffer was HBSS-supplemented with 20 mM HEPES and 2.5 mM probenecid (Sigma P8761). Probenecid was prepared by placing 710 mg into a 15-ml conical vial; 5 ml of 1 N NaOH was then added, and the mixture was vortexed until dissolved. An additional 5 ml of HBSS/20 mM HEPES was then added, and 10 ml of probenecid solution was then mixed into 1 liter of HBSS/20 mM HEPES, pH 7.4 with NaOH. Cells were seeded (from cultures that were less than 90% confluent) at 12,500 cells/well of a 384-well plate. Plates were then incubated at room temperature for time lengths that varied depending on the specific cell line and then incubated at 37°C for 24 h before assay. Assays was performed using Fluo-8 No Wash Ca2+ dye (ABD Biosciences, Sunnyvale, CA). A 2× dye concentration was prepared in GPCRProfiler assay buffer. Cells were washed with GPCRProfiler assay buffer, and 20 μl of buffer was retained in the plate; 20 μl/well of 2× dye was added, and the plates were incubated at 30°C, 5% CO2 for 90 min. Compound plates were prepared to add 20 μl/well during the first addition, and compounds were prepared at 3× the final concentration (in this case, 10 μl with a final volume after compound addition of 60 μl). To assess allosteric modulation, a second plate was prepared by using a 4× stock of reference agonist, which was diluted in 4-fold dilutions into an eight-point concentration series. Twenty microliters was then added to the plate for a final volume of 80 μl. Plates were read on a fluorometric imaging plate reader. Sixty reads at 1 s/read were performed, followed by 60 reads at 2 s/read. Data were analyzed as follows: the response of the compound (VU364770) alone was normalized to the maximal response of the relevant agonist to identify potential agonist activity of VU0364770. In these studies, VU0364770 did not induce responses alone on any of the receptors examined. To assess the modulator effects of VU0364770, complete agonist concentration responses were performed in the presence and absence of 10 μM VU0364770. The potency observed in the presence of VU0364770 was divided by the potency in the absence of compound. For values in which this number was below 1, indicating a left shift of agonist potency, the following calculation was performed: 1/[potency of agonist + VU0364770/potency of agonist − VU0364770], and the inverse of this value was plotted for ease of identifying antagonists (positive numbers indicating right shift of the agonist curve in the presence of compound) or potentiators/PAMs (negative numbers indicating left shift of the agonist curve in the presence of compound).
Microsomal Stability.
The metabolic stability of each compound was investigated in human and rat hepatic microsomes (BD Biosciences, San Jose, CA) by using substrate depletion methodology (percentage of parent compound remaining). In separate 96-well plates for each time point, a mixture of 0.1 M potassium phosphate-buffered, pH 7.4, 1 μM test compound, 0.5 mg/ml microsomes, and 1 mM NADPH (t = 3, 7, 15, 25, or 45 min) or buffer (t = 0) were continually incubated at 37°C under ambient oxygenation. At the respective time, each plate's reaction was precipitated by the addition of 2 volumes of ice-cold acetonitrile containing glyburide as an internal standard (50 ng/ml). The plates were centrifuged at 3000 rpm (4°C) for 10 min. The resulting supernatants were transferred and diluted 1:1 (supernatant/water) into new 96-well plates in preparation for LC/MS/MS analysis. Each compound was assayed in triplicate within the same 96-well plate. The in vitro half-life (t1/2, min; Eq. 1), intrinsic clearance (CLint, ml/min/kg; Eq. 2), and subsequent predicted hepatic clearance (CLHEP, ml/min/kg; Eq. 3) was determined by using the following equations:
where k represents the slope from linear regression analysis (percentage of test compound remaining).
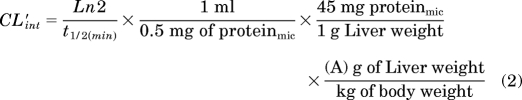 |
Plasma Protein Binding.
The protein binding of each compound was determined in human and rat plasma via equilibrium dialysis using Single-Use RED Plates with inserts (Thermo Fisher Scientific, Waltham, MA). Plasma (220 μl) was added to the 96-well plate containing test compound (5 μl) and mixed thoroughly. Subsequently, 200 μl of the plasma-compound mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μl of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated for 4 h at 37°C with shaking. At completion, 50-μl aliquots from each chamber were diluted 1:1 (50 μl) with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min) and supernatants were transferred and diluted 1:1 (supernatant/water) into a new 96-well plate, which was then sealed in preparation for LC/MS/MS analysis. Each compound was assayed in triplicate within the same 96-well plate.
CYP450 Inhibition.
A four-in-one, 96-well plate assay for determining IC50 values against human CYP450 1A2, 2C9, 2D6, and 3A4 was developed based on previous reports (Youdim et al., 2008). Human liver microsomes (final concentration of 0.1 mg/ml) and a substrate mixture containing the CYP450 probe substrates phenacetin (10 μM), diclofenac (5 μM), dextromethorphan (5 μM), and midazolam (2 μM) were added to a potassium phosphate-buffered solution (0.1 M, pH 7.4) and warmed to 37°C. The reaction mixture was divided evenly into the 96-well plate, and various dilutions of each inhibitor/compound of interest (in duplicate) were then added to this reaction mixture such that the final concentration of each compound ranged from 100 nM to 30 μM. This mixture was allowed to preincubate for 15 min while shaken at 37°C. Buffer or NADPH (1 mM) was added, and the reaction mixture was incubated for an additional 8 min at 37°C before quenching with 2 volumes of ice-cold acetonitrile containing 50 ng/ml of carbamazepine as internal standard. The plates were centrifuged at 4000 rpm (4°C) for 10 min, and the supernatant was removed and diluted with water (1:4, v/v) in preparation for LC/MS/MS analysis. The IC50 values for each compound were obtained for the individual CYP450 enzymes by quantitating the inhibition of metabolite formation for each probe: acetaminophen (1A2), 4-hydroxydiclofenac (2C9), dextrorphan tartrate (2D6), and 1-hydroxymidazolam (3A4). Miconazole was included as a positive control broad-spectrum CYP450 inhibitor. For discrete 2C19 inhibition experiments, a similar assay design was used with the following exceptions: the probe substrate was S-mephenytoin (40 μM), the NADPH incubation with the reaction mixture went for 30 min, the supernatant was reconstituted 1:1 with water for analysis, and the metabolite used for quantitation was 4-hydroxymephenytoin.
Monoamine Oxidase Inhibition.
The potential inhibition of monoamine oxidase A (MAO-A) and monoamine oxidase-B (MAO-B) by VU0364770 was investigated according to the protocol. In brief, male Sprague-Dawley rat brain mitochondria (0.5 ng/ml; Celsis, Baltimore, MD) in 100 mM potassium phosphate buffer were preincubated with either 50 nM clorgyline or 500 nM R-(−)-deprenyl hydrochloride for 15 min at 37°C to irreversibly inactivate MAO-A or MAO-B, respectively. The inactivated MAO-A- or MAO-B-containing mitochondria were added to an equal volume of VU0364770 (0.1–30 μM in potassium phosphate buffer, final concentration) and incubated at 37°C for 5 min before the nonselective MAO substrate kynuramine dihydrobromide (30 μM final concentration) was added. The assay was stopped by the addition of acetonitrile containing 50 nM carbamazepine as internal standard followed by centrifugation (4000g) for 10 min at 4°C. The concentration of the kynuramine metabolite 4-hydroxyquinoline in the supernatant was determined by LC/MS/MS using a CTC Analytics (Zwingen, Switzerland) HTS PAL/AB Sciex (Foster City, CA) API 4000 system. Mean 4-hydroxyquinoline concentrations were used to fit concentration-response curves by variable slope nonlinear regression for inhibition of MAO-A or MAO-B activity by VU0364770 using GraphPad Prism (version 4.03) to obtain IC50 values.
In Vivo Studies
In Vivo Pharmacokinetic Studies.
Male Sprague-Dawley rats (n = 2), weighing approximately 250 to 300 g, were purchased from Harlan and implanted with catheters in the carotid artery and jugular vein. The cannulated animals were acclimated to their surroundings for approximately 1 week before dosing and provided food and water ad libitum. Parenteral administration of compounds to rats was achieved via a jugular vein catheter at a dose of 1 mg/kg (20% DMSO/80% saline) and a dose volume of 1 ml/kg. Blood collections via the carotid artery were performed at predose, and 2, 7, 15, and 30 min, and 1, 2, 4, 7 and 24 h postdose. Catheters were flushed with 0.2 ml of saline containing 10% heparin every 2 days after testing procedures to maintain the patency of each catheter. Samples were collected into chilled EDTA-fortified tubes and centrifuged for 10 min at 3000 rpm (4°C), and the resulting plasma was aliquoted into 96-well plates for LC/MS/MS analysis. Pharmacokinetic parameters were obtained from noncompartmental analysis (WinNonLin, V5.3; Pharsight, Mountain View, CA) of individual concentration-time profiles after the parenteral administration of a test article. For systemic exposure studies, measuring both systemic plasma and central nervous system tissue exposure, VU0364770 was administered subcutaneously in 10% Tween 80, and 0.5 to 4 h later blood and whole brain samples were collected. Whole blood was collected into chilled EDTA-fortified tubes, centrifuged for 10 min at 3000 rpm (4°C), and stored at −80°C until LC/MS/MS analysis. The brain samples were rinsed in phosphate-buffered saline, snap-frozen, and stored at −80°C. Before LC/MS/MS analysis, brain samples were thawed to room temperature and subjected to mechanical homogenation by using a Mini-Beadbeater and 1.0-mm zirconia/silica beads (BioSpec Products, Bartlesville, OK).
Monoamine Oxidase Inhibition In Vivo.
To determine whether VU0364770 inhibits MAO in vivo, we compared the effects of VU0364770 with selective MAO-A or MAO-B inhibitors on the brain levels of dopamine and its metabolites when administered alone or in combination with l-DOPA/benserazide. In brief, rats were pretreated with vehicle, VU0364770 (100 mg/kg s.c.), clorgyline (4 mg/kg i.p.), or deprenyl (2 mg/kg i.p.) followed 60 min later by administration of vehicle or a combination of l-DOPA (4.5 mg/kg i.p.) and benserazide (15 mg/kg i.p.). Two hours after the initial drug treatment dopamine-rich brain regions were dissected for analysis of monoamines and their acidic metabolites by HPLC with electrochemical detection. Trunk blood and the remaining brain tissue were collected for the determination of plasma and brain concentrations of l-DOPA by HPLC/MS. In brief, frozen rat brains were mechanically homogenized in isopropanol/water (70:30) by using a Mini-Beadbeater and 1.0-mm zirconia/silica beads (BioSpec Products) and extracted by using 3 volumes of acetonitrile containing an internal standard (50 ng/ml carbamazepine). After centrifugation at 4000g for 5 min the supernatants were diluted 1:1 with water and analyzed by LC/MS using a Shimadzu (Columbia, MD) LC-10AD pump connected to a LEAP Technologies (Carrboro, NC) CTC PAL auto-sampler and an AB Sciex API-4000 triple-quadrupole instrument.
Haloperidol-Induced Catalepsy.
Catalepsy was assessed by using a horizontal bar placed 6 cm from the testing surface. The forepaws of each rat were placed gently on the bar with the body positioned at an angle of ∼45° to the testing surface. The latency in seconds required for the rat to remove one or both forepaws from the bar was manually measured. Rats randomly assigned to treatment groups were injected with haloperidol (0.75 or 1.5 mg/kg i.p.) and returned to their home cages for 60 min. Rat were examined for catalepsy 30 min after the administration of either VU0364770 (1–56.6 mg/kg s.c.), VU0364772 (1–56.6 mg/kg s.c.), A2A antagonist (56.6 mg/kg p.o.), preladenant (0.03–30 mg/kg p.o.), or vehicle. In the interaction studies rats were administered VU0364770 (10 or 30 mg/kg) + vehicle, VU0364770 (10 or 30 mg/kg) + preladenant (0.1–1 mg/kg), or vehicle + preladenant (0.1–1 mg/kg) 30 min before testing.
Rotarod Test.
The effects of VU0364770 on motor performance were evaluated by using a rotarod (MED Associates, St. Albans, VT). All rats were given an initial training trial of 120 s, followed by two additional training trials of 85 s, approximately 10 min apart, using a rotarod (7.5 cm in diameter) rotating at a constant speed of 20 revolutions/min. After initial training trials, a baseline trial of 120 s was conducted, and any rats that did not reach the 120-s criteria were excluded from the study. Rats were then treated with vehicle or VU0364770 (10, 30, 56.6, or 100 mg/kg s.c.), and tested 30 min later. The time each animal remained on the rotarod was recorded, and animals that did not fall off of the rotarod were given a maximal score of 120 s.
Locomotor Activity.
Locomotor activity studies were conducted by using a SmartFrame Open Field System (Kinder Scientific, San Diego, CA) equipped with 16 × 16 infrared photobeams located 1 in above the floor of the chamber. Rats were pretreated with vehicle or VU364770 (10–56.6 mg/kg s.c.) for 30 min and then placed in the open-field chambers for a 30-min test session. Ambulation was measured as the total number of photobeam breaks per 5-min interval and recorded with a Pentium I computer equipped with Motor Monitor System software (Kinder Scientific). Data were analyzed by one-way ANOVA followed by Dunnett's test using JMP version 5.1.2 statistical software (SAS Institute, Cary, NC).
6-OHDA Lesions-Induced Forelimb Asymmetry and Reaction Time Deficits
Unilateral 6-OHDA Lesions.
Animals were injected with buprenorphine (0.03 mg/kg s.c.) and anesthetized with isoflurane before being placed in a stereotaxic frame. Unilateral lesions of the nigrostriatal dopamine projections were made by injecting 6-hydroxydopamine (6-OHDA) into the left median forebrain bundle (AP, −3.6 mm; mediolateral, 1.8 mm; dorsoventral, −8.6 mm) (Paxinos and Watson, 2007). In brief, a 5-μl microsyringe with an outer diameter of 0.47 mm (Thermo Fisher Scientific) was lowered to the injection site where 4 μl of 6-OHDA hydrobromide (3.6 μg/μl in ice-cold saline containing 0.015% ascorbic acid) were injected over a period of 4 min. The sham lesion procedure involved lowering the injection cannula 2 mm dorsal to the lesion site without delivering an intracerebral injection. After suturing the scalp rats were injected with 5% dextrose in Ringer's solution (3 ml s.c.) to prevent postsurgical dehydration. To minimize weight loss encountered in some 6-OHDA-lesioned rats, the regular chow was supplemented with Bacon Softies (Bio-Serv, Frenchtown, NJ).
Apomorphine-Induced Rotation.
Three weeks after surgery rats were tested for apomorphine-induced rotation (Marshall and Ungerstedt, 1977). In brief, rats were injected with apomorphine (0.05 mg/kg s.c.), connected to a tether, and immediately placed into a cylinder-style rotometer (AccuScan Instruments, Inc., Columbus, OH). The number and direction of full 360° turns were automatically recorded over a period of 30 min. The total number of turns (contralateral + ipsilateral) and the net number of contralateral turns (contralateral − ipsilateral) were calculated. Lesioned rats with less than 45 net contralateral turns/30 min were excluded from subsequent studies.
Forelimb Asymmetry Assay.
6-OHDA lesion-induced forelimb asymmetry was assessed by using the cylinder test (Schallert et al., 2000). This assay measures the rats' forelimb use during spontaneous exploration of a Plexiglas cone. In brief, rats were gently placed into a clear Plexiglas cone (bottom diameter 21 cm; top diameter 37 cm; CleverSys, Reston, VA), and their behavior was recorded over a period of 10 min by a video camera placed underneath the Plexiglas cone by using Pinnacle Studio software (Pinnacle Systems, Mountain View, CA). Video recordings were analyzed offline by an observer blinded to both lesion status and drug treatment. The VLC media player (http://www.videolan.org/vlc/download-windows.html) was used to replay videos at various playback speeds (0.125–1.0 × real-time speed) to facilitate detailed analysis of forepaw placement during rearing and landing.
Forelimb usage was analyzed as outlined by Lundblad et al. (2004). To avoid scoring incidental contact with the cylinder wall only weight-bearing contacts as evidenced by splayed digits were scored. Subsequent wall contacts were counted only after a rat completely withdrew its paw from the wall, resulting in its weight being exclusively supported by its hind limbs. All floor landings from a rearing or grooming position onto either an individual forelimb or onto both forelimbs simultaneously were scored. The following parameters were used to calculate a composite measure of forelimb usage, termed FLA Index Score, according to the formula below:
 |
where Wunaffected is the number of wall contacts of the unaffected (ipsilateral) forelimb; Wimpaired is the number of wall contacts of the impaired (contralateral) forelimb; Lunaffected is the number of landings on the unaffected (ipsilateral) forelimb; and Limpaired is the number of landings on the impaired (contralateral) forelimb.
The FLA Index Score provides continuous data ranging between −100 (100% preference of the unaffected limb) and +100 (100% preference of the impaired limb), with a score of zero indicating a lack of forepaw preference.
Bilateral 6-OHDA Lesions.
Animals were anesthetized by an intramuscular injection of xylazine (15 mg/kg) and ketamine (100 mg/kg i.p.) and placed in a stereotaxic instrument (David Kopf Instruments, Tujunga, CA) with the incisor bar positioned −3.0 mm under the interaural line for surgical procedures based on coordinates of the stereotaxic atlas of Paxinos and Watson (2007). Lesioned animals received a bilateral injection of 6-OHDA hydrochloride (Sigma) (4 μg/μl, 3 μl per side) in the striatum at the following coordinates: AP +0.2 mm; mediolateral ±3.5 mm; dorsoventral −4.8 mm according to bregma. The sham control group received the vehicle alone (ascorbate solution, 0.1 mg/ml) in the dorsal striatum. The flow rate (0.5 μl/min) and volume of infusion were controlled by a micropump (CMA/400; CMA/Microdialysis, Stockholm, Sweden) using a 10-μl Hamilton (Reno, NV) microsyringe, connected by a Tygon tubing fitting to the 30-gauge stainless-steel injector needles. Three additional min were allowed for diffusion of the toxin. After surgery, rats were allowed a 7-day recovery period before behavioral testing.
Lesion Verification.
Rats were decapitated under deep isoflurane anesthesia and the brains were extracted. Tissue punches of the dorsolateral striatum (ipsilateral and contralateral to the lesion) were frozen on dry ice, and tissue concentrations of dopamine, 3,4-dihypdroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were analyzed by HPLC with electrochemical detection. For immunohistochemical verification rats were deeply anesthetized with isoflurane and transcardially perfused with 0.1 M phosphate buffer containing 0.9% sodium chloride, pH 7.4, followed by 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Brains were postfixed overnight and cryoprotected in 0.1 M phosphate buffer containing 30% sucrose. Using a freezing microtome, 42-μm-thick coronal sections through the striatum and substantia nigra were cut and processed by immunohistochemistry to reveal tyrosine hydroxylase (TH) immunoreactivity. In brief, sections were treated with methanolic peroxide [10% methanol, 0.6% hydrogen peroxide in 50 mM Tris-buffered saline (TBS)] to block endogenous peroxidase activity and washed in TBS, and nonspecific binding was blocked using TBS+ (4% normal horse serum, 0.2% Triton X-100 in TBS). Sections were incubated for 2 days in a mouse monoclonal antibody against TH (ImmunoStar, Hudson, WI) that was diluted 1:3000 in TBS+. After incubations in biotinylated donkey anti-mouse antibody (1:1000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA) and streptavidin-conjugated horseradish peroxidase (1:1600; Jackson ImmunoResearch Laboratories Inc.) TH immunoreactivity was revealed by reacting sections in TBS containing 0.025% 3,3″-diaminobenzidine, 0.6% nickel ammonium sulfate, 0.06% cobalt chloride, and 0.001% hydrogen peroxide to reveal a blue-black precipitate. Sections were mounted from 0.15% gelatin, dried, and coverslipped by using DPX mounting media (Electron Microscopy Sciences, Hatfield, PA). Immunostained sections were analyzed by using a Nikon (Tokyo, Japan) AZ 100M microscope.
Reaction Time Performance.
As described in detail elsewhere (Lopez et al., 2007, 2008), rats were trained in operant boxes (Campden Instruments Ltd., Cambridge, UK) to quickly release a lever at the cue light onset occurring after one of four randomly generated foreperiods. During a 3-month training period, rats were required to release the lever within a period of 600 ms to be rewarded with a 45-mg sucrose pellet in daily sessions of 100 trials. RTs were measured as the time elapsing from the visual trigger signal onset to the lever release. The number of correct and incorrect (nonrewarded) responses with RTs above the 600-ms time limit (“delayed responses”) was measured in 100-trial daily sessions. Delayed responses represent an index of akinetic symptoms because the subjects could not react rapidly enough after the trigger cue. Premature responses occurring before the cue onset were recorded independently and were not rewarded. The premature responses during the foreperiod preceding the visual cue represents a loss of attentional control and impulsive behavior, leading to a disruption of movement preparation. Performance of the rats was measured for 7 consecutive days (preoperative baseline) before surgery. After a 1-week recovery period, all the animals were tested in the RT task. Sham (n = 12) and 6-OHDA (n = 25) animals were divided into subgroups receiving three doses of VU 0364770 (10, 20, and 56.6 mg/kg s.c.) in a pseudo-random order following a Latin-square design at postsurgical days 16, 20, 24, and 28. Another group of 6-OHDA lesioned rats (n = 4) and sham (n = 3) received injections of the negative control VU0364772 at a high dose of 30 mg/kg.
Histological Control of DA Lesion: [3H]Mazindol Binding to Dopamine Uptake Sites.
Animals were anesthetized, and their brains were quickly removed after decapitation, frozen in dry ice, and kept at −80°C until cryostat sectioning. Coronal (10 μm thick) tissue sections were cut at −20°C at the level of the striatum (between coordinates AP 1.56 and −0.6 mm; related to bregma) (Paxinos and Watson, 2007). The loss of DA terminals in the striatum was indexed through the extent of DA denervation by analysis of [3H]mazindol binding to DA uptake sites as described previously (Amalric et al., 1995). The binding of [3H]mazindol to dopamine uptake sites in the striatum was measured according to the procedure described by Javitch et al. (1985). In brief, sections were air-dried and rinsed for 5 min at 4°C in 50 mM Tris buffer with 120 mM NaCl and 5 mM KCl. They were then incubated for 40 min with 15 nM [3H]mazindol (PerkinElmer, Zaventem, Belgium; specific activity 17 Ci/mM) in 50 mM Tris buffer containing 300 mM NaCl and 5 mM KCl added with 0.3 mM desipramine to block the noradrenalin transporter. Nonspecific binding was determined by incubating some sections in the same solution plus 30 mM benztropine. Sections were rinsed twice for 3 min in the incubation medium without mazindol and for 10 s in distilled water and air-dried. Autoradiographs were generated by apposing the sections to 3H-sensitive screens (Raytest, Courbevoie, France) for 21 days.
Liquid Chromatography/Mass Spectrometry Analysis.
In vivo samples were analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 triple-quadrupole instrument that was coupled with Shimadzu LC-10AD pumps and a LEAP Technologies CTC PAL auto-sampler. Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3-μm column (Fortis Technologies Ltd, Cheshire, UK) thermostated at 40°C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), and mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 30% B after a 0.2-min hold, linearly increased to 90% B over 0.8 min, held at 90% B for 0.5 min, and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 ml/min. The source temperature was set at 500°C, and mass spectral analyses were performed using multiple reaction monitoring, with transitions specific for each compound using a Turbo-Ionspray source in positive ionization mode (5.0-kV spray voltage). All data were analyzed by using AB Sciex Analyst 1.4.2 software.
CYP450 inhibition samples were analyzed via ESI mass spectrometry using an AB Sciex API-4000 triple-quadrupole instrument that was coupled to a Shimadzu LC-10AD pumps and a LEAP Technologies CTC PAL auto-sampler. Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3-μm column thermostated at 40°C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), and mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 10% B after a 0.2 min hold, was linearly increased to 90% B over 1.3 min, and returned to 10% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 ml/min. The source temperature was set at 500°C, and mass spectral analyses were performed by using multiple reaction monitoring, with transitions specific for each compound by using a Turbo-Ionspray source in positive ionization mode (5.0-kV spray voltage). All data were analyzed by using AB Sciex Analyst 1.4.2 software.
Microsomal stability and plasma protein binding samples were analyzed on a Thermo Fisher Scientific TSQ Quantum Ultra triple quad detector via ESI mass spectrometry with two Thermo Fisher Scientific Accella pumps and a LEAP Technologies CTC PAL autosampler. Analytes were separated by gradient elution on a dual column system with two Waters (Milford, MA) Acquity BEH C18, 2.1 × 50 mm, 1.7-μm columns heated at 50°C. HPLC mobile phase A was 95:5:0.1 water/acetonitrile/formic acid, and mobile phase B was 95:5:0.1 acetonitrile/water/formic acid. Pump 1 ran the gradient: 95:5 (A/B) at 800 μl/min hold 0 to 0.5 min, linear ramp to 5:95 (A/B) 0.5 to 1.0 min, 5:95 (A/B) hold 1.0 to 1.9 min, return to 95:5 (A/B) at 1.9 min. While pump 1 ran the gradient method, pump 2 equilibrated the second column isocratically with 95:5 (A/B). The total run time was 2.0 min per injection. All compounds are optimized by using Thermo Fisher Scientific's QuickQuan software.
Statistics
Data are presented as means ± S.E.M. Statistical comparisons were made by Student's t tests or one-way ANOVA followed by Dunnett's test as indicated using JMP version 5.1.2 statistical software (SAS Institute) or GraphPad Prism version 4.03.
Results
VU0364770 Is a Selective Positive Allosteric Modulator of mGlu4 in Recombinant Systems.
The functional activity of VU0364770 at mGlu4 was measured in a calcium mobilization assay using cells coexpressing human mGlu4 with the chimeric G protein, Gqi5, which links the Gi/o-coupled mGlu4 receptor to the phospholipase Cβ/Ca2+−pathway. VU0364770 produced a concentration-dependent potentiation of the response to an EC20 concentration of glutamate with EC50 value of 1.1 ± 0.2 μM and increased the maximal response to glutamate from 100 to 227 ± 17% (Fig. 1A). As a test of compound efficacy, the ability of a 30 μM concentration of VU0346770 to left-shift the glutamate concentration-response curve (CRC) was examined. In these experiments, VU0364770 shifted the glutamate CRC 31.4 ± 4.0-fold to the left (Fig. 1B). As described previously by our group, we also evaluated the pharmacological properties of VU0364770 by using an alternative approach that takes advantage of the ability of endogenous Gβγ subunits of Gi/o-coupled GPCRs to alter the kinetics of GIRKs to conduct the ion thallium (Niswender et al., 2008). In these studies, HEK293 cells stably coexpressing heteromeric GIRK1/2s and the rat mGlu4 receptor were preincubated with VU0364770 and then stimulated with glutamate in the presence of thallium. In these experiments, VU0364770 exhibited a potency of 290 ± 80 nM (Fig. 1C) and shifted the glutamate concentration-response curve 18.1 ± 1.7-fold to the left (Fig. 1D). VU0364770 did not exhibit intrinsic allosteric agonist activity in the in vitro assays performed. Although in both assays a left shift of the glutamate curve was evident, as observed with other mGlu4 PAMs, we routinely observed a large increase in the maximal response to glutamate in the calcium assay but not in the GIRK/thallium flux assay. We interpret this result to indicate a potential preferential coupling of mGlu4 to native Gi/o G proteins, where glutamate alone can induce the maximal response possible within the system, versus the chimeric Gqi5 G protein, which may be a nonideal G protein-coupling event. In addition, we would note that the Hill slopes between the potency assays differed, with a shallow slope (0.67) observed in the calcium assay and a Hill slope of 1.2 for the thallium flux assay. Although in our experience this is not consistently observed with mGlu4 PAMs, this may again reflect differences in coupling profiles and cooperativity between assays. Collectively, these in vitro studies suggest that VU0364770 is a potent PAM of multiple signaling pathways that enhances the response of the rat and human mGlu4 receptors to the endogenous agonist glutamate.
Fig. 1.

VU0364770 is a potent and effective PAM of mGlu4. A and B, VU0346770 exhibits a potency of 1.1 ± 0.2 μM at human mGlu4 in the presence of an EC20 concentration of glutamate (A) and shifts the glutamate concentration-response curve 31.4 ± 4.0-fold to the left (B). C and D, VU0364770 exhibits a potency of 290 ± 80 nM at rat mGlu4 (C) and induces an 18.1 ± 1.7-fold left shift of the glutamate concentration-response curve (D). Data were previously presented in tabular form (Engers et al., 2009).
We next evaluated the in vitro functional selectivity of VU0364770. VU0364770 was tested by using Ricerca's (Painesville, OH; formerly MDS Pharma's) Lead Profiling Screen (binding assay panel of 68 GPCRs, ion channels, and transporters screened at 10 μM). VU0364770 did not significantly bind to any of the 68 targets (no inhibition of radioligand binding >50% at 10 μM), with the exception of an 80% displacement of radioligand at the human norepinephrine transporter. Because of concerns that this chemical scaffold might possess activity at MAO, we performed full IC50 determinations for VU0364770 at the MAO-A and MAO-B isoforms; these studies resulted in Ki values of 8.5 and 0.72 μM for human MAO-A and human MAO-B, respectively.
Because VU0364770 is an allosteric modulator of mGlu4, there is the potential that it might exhibit activity at allosteric sites on other GPCRs. If so, this might not be detected in radioligand binding assays because these rely on orthosteric radioligands. To further assess the selectivity of VU0364770 for mGlu4 relative to other GPCRs, as well as determine its selectivity profile among the eight mGlu receptors, we measured the effects of this compound on the functional responses of a panel of 176 GPCRs. The studies for 168 family A, B, and C GPCRs were performed by using a Millipore functional screen, and the experiments for the mGlus were performed at Vanderbilt by using a similar experimental format. In these experiments, a 10 μM concentration of VU0364770 was incubated with cells for several minutes before the application of increasing concentrations of an agonist appropriate for each receptor. This method allows the detection of potential agonist (when compound is added before agonist), potentiator (observed as a left shift of the agonist CRC), or antagonist (observed as a right shift or decrease in the maximal response) activity at another receptor. VU0364770 exhibited a remarkably clean ancillary pharmacology profile, with little to no agonist, potentiator, or antagonist activity at receptors tested in the Millipore panel (Fig. 2). When tested at a 10 μM concentration at each mGlu receptor, VU0364770 exhibited weak PAM activity (4.3-fold left shift of the glutamate CRC) at mGlu6 and antagonist activity (3.3-fold right shift of the glutamate CRC) at mGlu5 (compare to the 16.5-fold left shift of the glutamate concentration-response for mGlu4at 10 μM; Fig. 2). When further evaluated in a full concentration-response curve format, VU0364770 exhibited antagonist activity at mGlu5 with a potency of 17.9 ± 5.5 μM and PAM activity at mGlu6 with a potency of 6.8 ± 1.7 μM (compare with the potency of VU0364770 on the rat mGlu4 receptor of 290 ± 80 nM; Fig. 1C). Taken together, these findings indicate that VU0364770 possesses a relatively clean ancillary pharmacology profile that allows a clear interpretation of the findings from the behavioral studies of mGlu4 activation in vivo.
Fig. 2.

Selectivity profile of VU0364770 among family A, B, and C GPCRs. A 10 μM concentration of VU0364770 was applied to cells expressing various GPCRs; VU0364770 induced no response when applied alone (data not shown). A subsequent addition of a full agonist concentration-response curve for each receptor allowed measurement of potential PAM or antagonist activity. The potency observed in the presence of VU0364770 was divided by the potency in the absence of compound. For values in which this number was below 1, indicating a left shift of agonist potency, the following calculation was performed: (1/[potency of agonist + VU0364770/potency of agonist − VU0364770]), and the inverse of this value was plotted for ease of identifying antagonists (positive numbers indicating right shift of the agonist curve in the presence of compound) or potentiators/PAMs (negative numbers indicating left shift of the agonist curve in the presence of compound). As an example, the dose ratio calculated by the presence of compound/absence of compound for mGlu4 was 0.06. This value was divided into 1 to give 16.7, and the inverse of this number, −16.7, was plotted onto the graph. Representative examples of family A GPCRs tested are shown (A) as well as all tested family B and C GPCRs (B). 5-HT, serotonin; M, muscarinic; A, adenosine; Alpha, α adrenergic; Beta, β adrenergic; CB, cannabinoid; D, dopamine; H, histamine; Mu/Delta/Kappa, opioid; P2Y, purinergic; AT, angiotensin; B, bradykinin; CRF, corticotrophin-releasing factor; GAL, galanin; Y, neuropeptide Y; OX, orexin; SSt, somatostatin; CXCR, chemokine; CCR, chemokine; EP, prostanoid; FFA, free fatty acid; S1P, lysophospholipid; CGRP, calcitonin gene-related peptide; CCK, cholecystokinin; GIP, glucagon; GLP, glucagon; PTH, parathyroid hormone; VPAC, vasoactive intestinal peptide; CaS, calcium sensing; GABAB, GABA.
VU0364770 Exhibits Suitable In Vitro and In Vivo Pharmacokinetic Properties for Systemic Dosing in Animal Models.
Before conducting in vivo studies with VU0364770, we characterized both the in vitro and in vivo pharmacokinetic properties of this ligand (Tables 1 and 2). As shown in Table 1, after intravenous administration, VU0364770 was rapidly cleared from the systemic circulation (165 ml/min/kg) and exhibited a volume of distribution of 2.92 l/kg. VU0364770 is a highly protein-bound ligand displaying free fractions of 2.7 and 1.8% in human and rat plasma, respectively. With the exception of CYP1A2, VU0364770 did not inhibit CYP450 enzyme activity to any appreciable level (Table 1). VU0364770 also showed an improved in vivo pharmacokinetic profile relative to previously reported mGlu4 PAMs with enhanced central penetration and a total brain-to-plasma ratio of more than 1 after systemic administration of a 10 mg/kg dose (Table 2).
TABLE 1.
In vitro pharmacokinetic and CYP450 profile of VU0364770
| Intravenous Clearance (Cassette) | |
|---|---|
| T1/2, min | 22.1 |
| Cl, ml/min/kg | 165 |
| Vss, l/kg | 2.92 |
| CYP450 1A2, μM | 0.55 |
| CYP450 2C9 | >30 |
| CYP450 2D6 | >30 |
| CYP450 3A4 | >30 |
TABLE 2.
In vivo pharmacokinetic profiles of VU0364770 and VU0364772
Area under the curve (AUC) data were derived from two to three rats per treatment group for the following time points: 15 and 30 min and 1, 3 and 6 h post-treatment administration.
| VU0364770 (10 mg/kg s.c.) | VU0364772 (10 mg/kg s.c.) | |
|---|---|---|
| AUC(0–6 h), nM · h | 2020 | 715 |
| Cmax, nM | 666 | 137 |
| tmax, h | 1 | 1 |
| Brain/plasma | 1.4 | 0.8 |
| Fu,B | 0.006 | 0.004 |
| Fu,P | 0.018 | 0.009 |
VU0364770 Produces a Dose-Dependent Reversal of Haloperidol-Induced Catalepsy.
As shown in Fig. 3, VU0364770 dose-dependently reversed haloperidol (0.75 mg/kg)-induced catalepsy in rats, significant at doses of 10 to 56.6 mg/kg, after subcutaneous dosing (F6,69 = 8.04; p < 0.001; Fig. 3A). The reversal of haloperidol-induced catalepsy produced by VU0364770 was more potent, but of a comparable magnitude to the effects observed with a previously reported A2A antagonist developed at Neurocrine Biosciences (Slee et al., 2008). The duration of action for the anticataleptic effects of VU0364770 was approximately 2 h (Fig. 4), which corresponded with a significant decrease in brain exposure (Table 2). The anticataleptic effects of VU0364770 were both time-dependent (F3,65 = 9.74; p < 0.01) and dose-dependent (F1,65 = 39.84; p < 0.001), with a significant interaction between these factors (F3,65 = 9.81; p < 0.01). In contrast, VU0364772, a close structural analog of VU0364770 with no in vitro mGlu4 PAM activity, had no effect on reversing haloperidol-induced catalepsy at doses up to 56.6 mg/kg s.c., whereas the positive control A2A antagonist showed efficacy in the same experiment (F6,45 = 30.3; p < 0.001; see Fig. 3B). The findings with VU0364772 were consistent with the interpretation that VU0364770 mediates its effects through activation of mGlu4. Finally, VU0364770 produced a dose-related reversal of haloperidol-induced catalepsy across a dose range that had no effect on spontaneous locomotor activity or performance on the rotarod test after a pretreatment time of 30 min and at doses up to 100 mg/kg s.c. (Fig. 5).
Fig. 3.

The mGlu4 PAM VU0364770 produces a dose-dependent reversal of haloperidol (0.75 mg/kg i.p.)-induced catalepsy in rats. A, for comparison, the effects of VU0364770 were compared with a top dose of a previously published A2A antagonist from Neurocrine. B, in contrast, the structural analog VU0364772 (inactive at mGlu4in vitro) had no effect across the dose range tested on haloperidol-induced catalepsy. Catalepsy was measured as the latency to withdraw the forepaws from a horizontal bar with a cutoff of 30 s. Bar graphs represent the means ± S.E.M. of 10 to 12 rats/treatment group. **, p < 0.05 versus the vehicle control group by Dunnett's test. VEH, vehicle.
Fig. 4.

VU0364770 exhibits anticataleptic efficacy up to 2 h after administration. VU0364770 exhibited a reversal of haloperidol-induced catalepsy for up to 2 h after pretreatment. Catalepsy was measured as the latency to withdraw the forepaws from a horizontal bar with a cutoff of 30 s. Bar graphs represent the means ± S.E.M. of 10 to 12 rats/treatment group. ***, P < 0.001 versus the vehicle control group by Dunnett's test.
Fig. 5.

VU0364770 has no effect on spontaneous locomotor activity in the open field (A) or performance on the rotorod test (B). Changes in locomotor activity were represented as ambulations (total photobeam breaks/5-min intervals). Performance on the rotorod was measured as the latency to fall off the rotorod in seconds. Each data point or bar graph represents the means ± S.E.M. of six to eight rats/treatment group. VEH, vehicle.
VU0364770 Enhances the Efficacy of the A2A Antagonist Preladenant to Reverse Haloperidol-Induced Catalepsy.
Previous studies have shown that selective A2A antagonists produce robust antiparkinsonian-like effects in animal models of dopamine depletion, and more recently in clinical trials with patients with PD, when given alone or in combination with l-DOPA (Varty et al., 2008; Hodgson et al., 2010; Pinna et al., 2010; Hauser et al., 2011). In another report, coadministration of a group III mGlu agonist with an A2A antagonist produced an enhanced reduction of haloperidol-induced catalepsy that was greater in magnitude than that observed with either compound administered alone (Lopez et al., 2008). Using the highly selective and potent A2A antagonist preladenant, currently under clinical trials for the symptomatic treatment of patients with PD, we evaluated the potential for a functional interaction between the mechanisms of our mGlu4 PAM VU0364770 and preladenant in reversing haloperidol-induced catalepsy in rats. We first determined the dose-related effect of both compounds in this behavioral model after administration alone. As shown in Fig. 6, A and B, both VU0364770, at 30 and 56.6 mg/kg s.c., and preladenant, at 1 to 30 mg/kg p.o., reversed cataleptic behavior in rats induced by a 1.5 mg/kg dose of haloperidol (F5,59 = 13.46, p < 0.001 and F7,78 = 21.43, p < 0.001, respectively). When given in combination with a 10 mg/kg (F7,76 = 10.20; p < 0.001) or 30 mg/kg (F7,83 = 12.25; p < 0.001) dose of VU0364770 subcutaneously, which produced no or little effect when administered alone, there was a marked leftward shift of the preladenant dose-response curve, with efficacy observed at the previously ineffective doses of 0.1 and 0.3 mg/kg p.o. preladenant (Fig. 6, C-E). At the end of these behavioral studies, brain and plasma exposure levels for VU0364770 and preladenant in each dose combination were assessed to rule out the possibility that the observed behavioral interactions between these two compounds were caused by a pharmacokinetic drug-drug interaction, resulting in altered metabolism of one or both compounds. As shown in Table 3, the total brain and plasma concentrations of VU0364770 and preladenant increased in a dose-proportional manner across the dose range with no alterations in brain/plasma ratios. Neither compound exhibited a substantial dose- or compound interaction-related change in brain-to-plasma ratio. In addition, brain levels of haloperidol remained constant across all treatment groups. These exciting preliminary findings raise the possibility that the mechanisms of selective potentiation of mGlu4 and A2A antagonism may provide a greater than additive or synergistic antiparkinsonian effect in preclinical PD models and potentially in individuals with PD.
Fig. 6.

VU0364770 and the A2A antagonist preladenant produce an enhanced anticataleptic effect in rats when administered in combination. A and B, when injected alone, both VU0364770 (A) and preladenant (B) produce a dose-dependent reversal of haloperidol (1.5 mg/kg, i.p.)-induced catalepsy in rats. C and D, the anticataleptic effects of preladenant were significantly enhanced when coadministered with either an ineffective (10 mg/kg) or moderately effective (30 mg/kg) dose of VU0364770. E, the robust leftward shift of the percentage inhibition of the preladenant dose-response curves when given in combination with VU0364770 (10 or 30 mg/kg s.c.) in comparison with the dose-response effects of either compound alone. Catalepsy was measured as the latency to withdraw the forepaws from a horizontal bar with a cutoff of 30 s. Each data point or bar graph represents the means ± S.E.M. of 8 to 10 rats/treatment group. *, p < 0.05; **, <0.001 versus the vehicle control group by Dunnett's test.
TABLE 3.
Brain and plasma concentrations of VU0364770 alone or in combination with the A2A antagonist preladenant
Data represent the mean ± S.E.M. of six animals.
| Dose |
VU0364770 |
Preladenant |
Haloperidol, Total Brain | |||||
|---|---|---|---|---|---|---|---|---|
| VU0364770 | Preladenant | Total Plasma | Total Brain | Brain/Plasma Ratio | Total Plasma | Total Brain | Brain/Plasma Ratio | |
| ng/ml | ng/g | ng/ml | ng/g | ng/g | ||||
| 10 mg/kg | 0 mg/kg | 259 ± 31 | 607 ± 59 | 2.60 ± 0.49 | N.D. | N.D. | N.D. | 312 ± 77 |
| 10 mg/kg | 0.1 mg/kg | 320 ± 41 | 521 ± 59 | 1.85 ± 0.41 | 1.4 ± 0.1 | N.D. | N.D. | 251 ± 35 |
| 10 mg/kg | 0.3 mg/kg | 395 ± 26 | 739 ± 57 | 1.91 ± 0.19 | 6.0 ± 0.8 | 23.1 ± 5.0 | 4.6 ± 1.5 | 308 ± 79 |
| 10 mg/kg | 1.0 mg/kg | 539 ± 47 | 951 ± 74 | 1.79 ± 0.09 | 23.2 ± 2.0 | 108.7 ± 9.8 | 4.8 ± 0.5 | 275 ± 52 |
| 30 mg/kg | 0 mg/kg | 271 ± 38 | 502 ± 56 | 1.90 ± 0.11 | N.D. | N.D. | 302 ± 69 | |
| 30 mg/kg | 0.1 mg/kg | 237 ± 20 | 457 ± 39 | 1.94 ± 0.09 | N.D. (3/6)* | N.D. (5/6)** | 301 ± 52 | |
| 30 mg/kg | 0.3 mg/kg | 367 ± 34 | 501 ± 27 | 1.40 ± 0.10 | 6.9 ± 1.3 | 19.6 ± 1.9 | 3.1 ± 0.3 | 318 ± 69 |
| 30 mg/kg | 1.0 mg/kg | 362 ± 29 | 669 ± 49 | 1.89 ± 0.14 | 19.7 ± 2.2 | 107.9 ± 6.2 | 5.8 ± 0.6 | 385 ± 109 |
N.D., not detectable in six of six animals.
Not detectable in three of six animals.
Not detectable in five of six animals.
VU0364770 Reverses Forelimb Asymmetry Alone and in Combination with l-DOPA.
We next evaluated the ability of VU0364770 to reverse forelimb asymmetry in rats that received unilateral 6-OHDA lesions of dopamine neurons. Dopamine neurons were selectively lesioned, and rats were assessed behaviorally to ensure unilateral loss of motor function. Three weeks after surgery, rats with unilateral 6-OHDA lesion exhibited marked rotation behavior with increases in the number of total rotations (t60 = 15.95; p < 0.001) as well as the number of net contralateral rotations (t60 = 14.81; p < 0.001); sham-lesioned controls did not exhibit any rotation behavior in response to apomorphine (Table 4).
TABLE 4.
Apomorphine (0.05 mg/kg s.c.)-induced rotation in sham- and unilaterally 6-OHDA-lesioned rats
Data represent mean ± S.E.M.
| Sham (n = 30) | 6-OHDA (n = 32) | |
|---|---|---|
| Total rotationsa | 13.20 ± 1.13 | 211.19 ± 12.77*** |
| Net rotationsb | −0.67 ± 1.23 | 205.63 ± 13.43*** |
Contralateral + ipsilateral rotations.
Contralateral − ipsilateral rotations.
P < 0.001 (t test).
Animals with verified lesions, as assessed by induction of contralateral rotations, as well as sham control rats, were used to measure the antiparkinsonian effects of VU0364770 and other agents by determining the effects on forelimb asymmetry. As shown in Fig. 7A, l-DOPA produced a robust dose-dependent reversal of the FLA index score in rats, significant at doses of 2.5 and 4.5 mg/kg p.o. (F3,39 = 18.01; p < 0.001). In comparison with the vehicle treatment group with a FLA index score of −88, VU0364770 (100 mg/kg s.c.) produced a modest improvement in forelimb asymmetry of approximately 32% as indicated by a change in FLA index score to −60 (F3,47 = 73.6; p < 0.001; see Fig. 7B). However, when coadministered with an ineffective dose of l-DOPA (1.5 mg/kg p.o.), VU0364770 (100 mg/kg s.c.) produced a robust reduction in forelimb asymmetry of more than 75% as measured by a change in FLA index score to −30 (Fig. 7B). Brain and plasma exposure levels of l-DOPA and VU0364770 were not altered when administered in combination in comparison with dosing alone (Table 5). The observed efficacy of VU0364770 in this behavioral model suggests that selective activation of mGlu4 may provide an important novel approach for the treatment of motor symptoms in patients with PD as a potential monotherapy and/or in combination with l-DOPA treatment. The present findings also suggest that selective potentiation of mGlu4 may provide a potential l-DOPA sparing mechanism of action that will be important to elucidate further in future studies.
Fig. 7.

VU0364770 produces a reversal of forelimb asymmetry induced by unilateral 6-OHDA lesion of the median forebrain bundle in rats alone or in combination with an inactive dose of l-DOPA. A, the dose-dependent reversal of forelimb asymmetry (as measured by the forelimb asymmetry index score) by l-DOPA. B, the moderate improvement of forelimb asymmetry by VU0364770 (100 mg/kg s.c.) or robust reversal in combination with l-DOPA (1.5 mg/kg p.o.). Each bar graph represents the means ± S.E.M. of 5 to 15 (A) and 9 to 15 (B) rats/treatment group. *, p < 0.05; **, P < 0.01 versus the vehicle control group by Dunnett's test.
TABLE 5.
Brain and plasma concentrations of L-DOPA and VU0364770 after systemic administration of VU0364770 (100 mg/kg s.c.) alone or in combination with l-DOPA (1.5 mg/kg i.p.) and benserazide (15 mg/kg i.p.)
Data are means ± S.E.M. of five to seven animals; no significant treatment effects (P > 0.23, t-test).
| Pretreatment (t = 0 min) | Treatment (t = 60 min) | L-DOPA (t = 120 min) |
VU0364770 (t = 120 min) |
||
|---|---|---|---|---|---|
| Plasma | Brain | Plasma | Brain | ||
| ng/ml | ng/g | ng/ml | ng/g | ||
| Vehicle | Vehicle | N.D. | N.D. | ||
| Vehicle | l-DOPA/benserazide | 2350 ± 49 | 433 ± 46 | ||
| VU0364770 | Vehicle | N.D. | N.D. | 316 ± 34 | 1476 ± 14 |
| VU0364770 | l-DOPA/benserazide | 2330 ± 12 | 403 ± 17 | 308 ± 47 | 1823 ± 24 |
N.D., not detected.
After completion of studies of forelimb asymmetry, 6-OHDA lesions were verified by either TH immunohistochemistry or tissue neurochemistry. Unilateral infusion of 6-OHDA into the median forebrain bundle caused a massive loss of TH immunoreactivity, a marker of midbrain dopamine neurons, in the ipsilateral substantia nigra and striatum (Fig. 8). Two-way ANOVA of the striatal tissue dopamine concentrations revealed significant effects of treatment (lesion versus sham: F1,116 = 34.3, p < 0.001), side (ipsilateral versus contralateral: F1,116 = 30.4, p < 0.001), and a lesion × side interaction (F1,116 = 28.68, p < 0.001). Thus, the median forebrain bundle lesions reduced striatal dopamine concentration by more than 99% in the ipsilateral striatum without altering dopamine in the contralateral, nonlesioned striatum, whereas sham lesions did not alter dopamine levels in the striatum (Table 6).
Fig. 8.

Immunohistochemical verification of the loss of nigrostriatal dopamine neurons after unilateral 6-OHDA injection into the median forebrain bundle. Coronal sections illustrate the loss of TH immunoreactive axons and cell bodies in the caudate-putamen and SNc, respectively in the 6-OHDA-lesioned hemisphere (B and D), but not in sham-lesioned rats (A and C). A modest decrease in TH labeling is also observed in the ventral tegmental area of lesioned animals.
TABLE 6.
Concentrations of dopamine and its metabolites in the dorsolateral striatum of sham- and unilaterally 6-OHDA-lesioned rats
Data represent means ± S.E.M.
| Treatment | Dopamine | DOPAC | HVA |
|---|---|---|---|
| ng/mg protein | |||
| Sham (n = 28) | |||
| Contralateral | 125.39 ± 3.65 | 12.76 ± 0.37 | 7.24 ± 0.24 |
| Ipsilateral | 123.62 ± 2.58 | 11.93 ± 0.26 | 7.10 ± 0.24 |
| Percentage depletion | 1.41 | 6.50 | 1.93 |
| 6-OHDA (n = 31) | |||
| Contralateral | 120.40 ± 2.96 | 12.51 ± 0.56 | 7.05 ± 0.22 |
| Ipsilateral | 0.70 ± 0.07***### | 0.50 ± 0.05***### | 0.48 ± 0.29***### |
| Percentage depletion | 99.24 | 96.00 | 93.19 |
P < 0.001 vs. sham ipsilateral.
P < 0.001 vs. 6-OHDA contralateral (Bonferroni test).
VU0364770 Reverses Attentional Deficits in the Reaction Time Task.
In the present study, we evaluated the potential dose-related effects of VU0364770 on performance deficits in the RT task induced by bilateral 6-OHDA striatal lesions in rats. As previously established, bilateral 6-OHDA striatal infusions recapitulate many aspects of the earlier stages of PD, including akinetic and/or attentional dysfunction and reduction of dopamine levels by 74 and 53% in rostral and caudal areas of the striatum, respectively (Amalric et al., 1995; Turle-Lorenzo et al., 2006). As shown in Fig. 9, bilateral 6-OHDA striatal infusions produced akinetic and/or attentional dysfunction measured by impaired RT performance. Akinesia (e.g., increased delayed response) to the trigger visual cue was found in 56% of the animals (Fig. 9B) showing restricted DA nerve terminal loss in the dorsolateral part of the striatum (sensorimotor area) as assessed by the loss of tyrosine hydroxylase immunoreactivity (Fig. 9C). A predominant increase of premature responding was observed in the remaining 44% of rats (Fig. 9D), which exhibited more widespread loss of dopaminergic terminals extending into ventromedial and rostral (limbic) parts of the striatum (Fig. 9F; Supplemental Fig. 1). As shown in Fig. 9B, a 56.6 mg/kg s.c. dose of VU0364770 tended to reduce the akinetic deficits expressed by the increased number of delayed responses, although this effect did not reach significance. Performance measured postsurgery and after vehicle or VU0364770 56 mg/kg s.c. injections was significantly different from preoperative level of performance (F13,39 = 6.9; p < 0.05). Although VU0364770 had no effect at lower doses (10 and 30 mg/kg; data not shown), the increased number of premature responses was significantly reduced by a 56.6 mg/kg dose of VU0364770 in comparison with vehicle injection (F10,30 = 4.09; p < 0.05). The level of performance of sham-operated animals was not modified by surgery or VU0364770 injections (Fig. 9, A and D). In addition, VU0364772 did not modify performance of sham and 6-OHDA groups in the RT task (data not shown).
Fig. 9.

VU0364770 exhibits efficacy in bilateral 6-OHDA-lesioned animals exhibiting attentional and akinetic deficits in the reaction time task. A, B, D, and E, the mean number ± S.E.M. of delayed responses (A and B) and premature responses (D and E) of sham and 6-OHDA-lesioned rats averaged by blocks of six sessions for the preoperative (PRE) and postoperative (POST) periods and during the sessions after administration of vehicle (VEH) or VU0364770 (56.6 mg/kg s.c.) are shown. VU0364770 slightly, but nonsignificantly, decreased the number of delayed responses induced by bilateral 6-OHDA lesion of the nigrostriatal pathway in rats. However, VU0364770 reversed the increased number of premature responses. *, p < 0.05 versus the preoperative performance; λ, p < 0.05 versus vehicle injection (ANOVA followed by Student's t test). C and F, coronal sections through the striatum stained for tyrosine hydroxylase immunoreactivity to visualize the loss of striatal dopamine terminals in rats exhibiting the “akinetic” and “premature” reaction time deficits, respectively. Detailed illustrations of the lesion extent are shown in Supplemental Fig. 1.
VU0364770 Has No Effect on Monoamine Turnover in the Striatum.
In vitro profiling for off-target activity of VU0364770 revealed significant binding to human MAO-A and MAO-B enzymes. In a follow-up functional assay using a rat mitochondrial preparation, VU0364770 moderately inhibited MAO-A and MAO-B activity with an EC50 of 9.16 and 2.97 μM, respectively. To control for the possibility that the observed efficacy of VU0364770 was confounded by the possible inhibition of MAO-A or MAO-B activity after systemic administration, we evaluate the effects of VU0364770 (100 mg/kg s.c.) in a side-by-side comparison with the MAO-B inhibitor R-(−)-deprenyl (2 mg/kg i.p.) and the MAO-A inhibitor clorgyline (4 mg/kg i.p.) on striatal concentrations of dopamine and its acidic monoamine metabolites DOPAC and HVA. The doses of MAO-A and MAO-B inhibitors were chosen based on the maximum doses that produced inhibition of MAO-A or MAO-B without crossover effects at the other enzyme isoform; the pretreatment time for all three compounds was selected based on the in vivo pharmacokinetic parameters of VU0364770. One-way ANOVAs revealed significant effects of treatment on the tissue concentration of striatal dopamine (F3,27 = 3.62; p < 0.05), DOPAC (F3,27 = 5.38; p < 0.01), and HVA (F3,27 = 4.50; p < 0.05). At the top dose of VU0364770 used in the behavioral studies, there was no effect on striatal concentrations of dopamine or its metabolites DOPAC and HVA (Fig. 10). The MAO-B inhibitor deprenyl also had no effect on dopamine and metabolite concentrations in the striatum. However, the MAO-A inhibitor clorgyline significantly increased striatal dopamine and decreased DOPAC (Fig. 10). Because the predominant MAO isoform in rats is MAO-A (Buu et al., 1987; Paterson et al., 1991), it is not surprising that there was a robust increase in striatal dopamine levels observed with the MAO-A inhibitor clorgyline, but not with the MAO-B inhibitor deprenyl. Furthermore, the lack of effect of VU0364770 on striatal dopamine levels is consistent with the interpretation that the effects of VU0364770 in the behavioral studies are caused by selective potentiation of mGlu4, not enhanced striatal dopamine levels caused by the inhibition of MAO-A or MAO-B activity in vivo.
Fig. 10.

The selective mGlu4 PAM VU0364770 (100 mg/kg s.c.) does not affect striatal concentrations of dopamine and its acidic metabolites. The MAO-A inhibitor clorgyline increases dopamine and decreases DOPAC concentrations in the dorsal striatum. In contrast, VU0364770 and the MAO-B inhibitor deprenyl exhibit no effect on striatal dopamine and metabolite levels. Each bar graph represents the means ± S.E.M. of six rats/treatment group. *, p < 0.05 versus the vehicle control group by Dunnett's test.
Discussion
Limitations with current dopamine replacement therapies for the symptomatic treatment of PD have given rise to alternative strategies focused on normalizing synaptic transmission within the indirect pathway of the BG. Selective mGlu4 PAMs can decrease excessive inhibitory tone at the striatopallidal synapse and reduce motor impairments in animal PD models (Valenti et al., 2003; Lopez et al., 2007; Sibille et al., 2007; Niswender et al., 2008; Engers et al., 2009). Unfortunately, early-generation mGlu4 PAMs possessed unsuitable physiochemical properties for systemic dosing to allow broader investigation of the role of mGlu4 in BG circuitry. The present pharmacologic characterization of VU0364770 shows that this systemically active mGlu4 PAM is an important tool for the modulation of mGlu4 activity in vivo. VU0364770 exhibits nanomolar potency, high selectivity for mGlu4 relative to other mGlus and other targets, enhanced physiochemical properties for in vivo dosing, and robust efficacy in rodent PD models when systemically administered alone or in combination with l-DOPA or A2A antagonists.
The characterization of VU0364770 as a potent and selective mGlu4 PAM with systemic activity provides an important advance in further demonstrating the feasibility of achieving high selectivity for mGlu4 by targeting allosteric sites. Whereas VU0364770 exhibits weak mGlu5 antagonist activity in vitro, the relative selectivity of this ligand for mGlu4 versus mGlu5 is approximately 40-fold. Based on our pharmacokinetic analyses (Table 2), the levels of VU0364770 in the brain are not predicted to be high enough for an mGlu5-mediated effect. Moreover, the lack of efficacy observed using the inactive structural analog VU0364772 provides a critical confirmation that the antiparkinsonian-like effects of VU0364770 are mediated via mGlu4. In the in vitro assays examined, VU0364770 did not exhibit any intrinsic agonist activity at mGlu4. These effects are similar to those observed with the first-generation mGlu4 PAM N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide, but are in contrast to the intrinsic agonist activity observed with another compound, VU0155041 (Marino et al., 2003; Niswender et al., 2008). Although the extent to which intrinsic agonist activity may boost efficacy in vivo remains unclear, the effects observed with VU0364770 after systemic administration suggest that there is sufficient endogenous tone at central mGlu4 to observe efficacy with mGlu4 PAMs in models of more extensive dopamine depletion that may represent later stages of PD. However, the present data suggest that VU0364770 exhibits only modest effects on akinesia induced by partial bilateral lesions of the striatum, which are thought to represent a model of the earlier stages of PD. It is possible that distinctions in VU0364770 efficacy in these two dopamine lesion models may involve a changing role of mGlu4 modulation of BG circuitry as striatal dopamine levels decrease. For example, if STN projections to the GPe provide a primary source of glutamate for the activation of mGlu4 in GABAergic striatopallidal terminals, increased activity of the STN under conditions of more severe lesions would provide enhanced glutamate activation of the receptor and subsequently greater potentiation by mGlu4 PAMs. In addition, previous studies have shown that activation of mGlu4 reduces transmission at glutamatergic synapses projecting from the STN into the SNc (Conn et al., 2005; Valenti et al., 2005). In the early stages of PD, modeled by partial 6-OHDA striatal lesions, activation of mGlu4 expressed at the subthalamonigral synapse could lead to decreased excitatory drive on still intact SNc dopamine neurons. If so, this may further disrupt the functional balance between the direct and indirect pathways. In the later stages of PD, as modeled by the 6-OHDA median forebrain bundle lesions or D2 receptor antagonism, reducing transmission at excitatory synapses onto DA neurons may have less impact and may not counteract actions at the striatopallidal synapse.
When considering the potential efficacy of mGlu4 potentiation in various stages of PD, it is important to note that the measure of akinesia in the partial lesion model is fundamentally different from the measures used in severe depletion models. In addition, it is currently unclear whether the differential reversal of akinetic and attentional deficits by VU0364770 is related to the degree of dopamine depletion in the dorsolateral striatum or the more widespread lesion affecting adjacent striatal territories that are innervated by other cortical areas, including the medial prefrontal cortex. The loss of control during the “hold period” before the visual cue presentation leads to impulsive responsiveness and attentional dysfunction, relating to the cognitive deficits after damage to the frontal lobes and those described in patients with PD (Owen, 2004; Turle-Lorenzo et al., 2006). Although future studies will address these areas, our present findings provide strong support for the continued development of selective mGlu4 PAMs as a potential monotherapy for the motor and cognitive symptoms associated with PD.
The potentiation of the antiparkinsonian-like effects of VU0364770 with an inactive dose of l-DOPA represents the first report of a pharmacologic interaction between selective activation of mGlu4 and l-DOPA-mediated mechanisms in vivo. Although clinically available dopamine replacement therapies provide effective treatment for the motor symptoms observed in the early stages of PD, these treatments fail to deliver consistent efficacy with disease progression and are associated with many motor and cognitive adverse side effects (Jankovic, 2008; Stacy and Galbreath, 2008). Pharmacologic augmentation of l-DOPA treatment through the enhancement of the magnitude and/or duration of a given l-DOPA dose may allow potential l-DOPA-sparing strategies to minimize or delay the development of motor fluctuations and dyskinesias (Rezak, 2007). In the present study, the enhanced antiparkinsonian effects of VU0364770, in combination with an inactive dose of l-DOPA in the forelimb asymmetry model, seem to be the result of a true pharmacodynamic interaction (Table 5). In addition, we established that the enhanced efficacy observed with both compounds was not the result of increased striatal dopamine levels induced by potential off-target inhibition of MAO-A and/or MAO-B activity. It is noteworthy that the magnitude of the observed efficacy with the combination of VU0364770 and l-DOPA was greater than the efficacy observed with either compound alone, suggesting a greater than additive or synergistic interaction and a potentially novel l-DOPA sparing mechanism (Tallarida, 2010).
Another important consideration related to these data is the possibility that a selective mGlu4 PAM might induce dyskinesias or potentiate l-DOPA-induced dsykinesias by decreasing GABAergic neurotransmission in the GPe (Matsui and Kita, 2003; Valenti et al., 2003). However, although dyskinetic activity would be predicted if all GABAergic neurotransmission was shut down in the GPe, mGlu4 PAMs are postulated to have a more subtle modulatory effect on GABAergic signaling. In ongoing studies, we are further investigating these l-DOPA-sparing effects to determine whether an improved therapeutic index for l-DOPA therapy can be achieved when given in combination with an mGlu4 PAM.
The enhanced antiparkinsonian-like effects observed with VU0364770 in combination with the A2A antagonist preladenant provide the first demonstration of a pharmacologic interaction between a selective mGlu4 PAM and an A2A antagonist in an animal model of PD. Previous preclinical and clinical studies have demonstrated that A2A antagonists produce robust antiparkinsonian effects when given alone or in combination with l-DOPA (Varty et al., 2008; Hodgson et al., 2010; Pinna et al., 2010; Hauser et al., 2011). Lopez et al. (2008) reported that coadministration of the group III mGlu agonist (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid with either the A2A antagonists 1,3,7-trimethyl-8-(3-chlorostyryl)caffeine or 3,7-dihydro-8-[(1E)-2-(3-methoxyphenyl)ethenyl]-7-methyl-3-[3-(phosphonooxy)propyl-1-(2-propynyl)-1H-purine-2,6-dione disodium salt hydrate enhanced reductions in haloperidol-induced catalepsy that were greater than the effects observed with either compound alone. Based on overlapping, but nonidentical, expression patterns of adenosine A2A receptor and mGlu4 within the BG circuitry, the observed pharmacologic interaction between these two receptors may involve synergistic effects on the same and/or different neuronal pathways. For example, one possible functional interaction may involve the combined antagonism of adenosine A2A receptors on medium spiny GABAergic neurons that project to the GPe and the activation of mGlu4 expressed presynaptically at the striato-pallidal synapse, because both mechanisms have been reported to reduce excessive inhibitory tone at the level of the GPe and reduce preclinical motor impairments (Bradley et al., 1999; Hettinger et al., 2001; Valenti et al., 2003; Niswender et al., 2008; Hodgson et al., 2010). Another possible site of this functional interaction between A2A and mGlu4 may be at the level of the corticostriatal nerve terminals, because blockade of presynaptically expressed A2A receptors on these striatal synapses inhibits glutamatergic transmission (Tozzi et al., 2007), whereas activation of mGlu4 using the group III agonists (2S)-2-amino-4-phosphonobutanoic acid and (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid reduces glutamatergic activity at corticostriatal synapses (Pisani et al., 1997; Corti et al., 2002). These exciting preliminary findings raise the possibility that the mechanisms of selective potentiation of mGlu4 and A2A antagonism in multiple regions of the BG circuitry may provide a greater than additive or synergistic antiparkinsonian effect in preclinical PD models and potentially in individuals with PD.
In summary, VU0364770 is the first systemically active and selective mGlu4 PAM with robust efficacy in preclinical models of PD when administered alone or in combination with l-DOPA or the A2A antagonist preladenant. The present findings provide important evidence for the utility of selective mGlu4 PAMs as a novel treatment strategy for the symptomatic treatment of PD and as a possible augmentation strategy with either l-DOPA or A2A antagonists.
Supplementary Material
Acknowledgments
Lesion analysis was performed in part through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants NS048334, NS31373]; the National Institutes of Health National Institute of Mental Health [Grant MH084659]; the Michael J. Fox Foundation; and Era-Net NEURON [Grant ANR-08-NEUR-006-01]. Vanderbilt is a Specialized Chemistry Center within the Molecular Libraries Probe Centers Networks.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- PD
- Parkinson's disease
- A2A
- adenosine 2A
- BG
- basal ganglia
- FLA
- forelimb asymmetry
- mGlu
- metabotropic glutamate receptor
- PAM
- positive allosteric modulator
- SNc
- substantia nigra pars compacta
- SNr
- substantia nigra pars reticulata
- GPi
- internal globus pallidus
- GPe
- external globus pallidus
- STN
- subthalamic nucleus
- VU0155041
- (±)-cis-2-(3,5-dicholorphenylcarbamoyl)cyclohexanecarboxylic
- VU0364770
- N-(3-chlorophenyl)picolinamide
- VU0364772
- N-(6-(trifluoromethyl)pyridin-2-yl)picolinamide
- GPCR
- G protein-coupled receptor
- GIRK
- G protein-regulated inwardly rectifying K (potassium) channel
- HEK
- human embryonic kidney
- DMSO
- dimethyl sulfoxide
- 6-OHDA
- 6-hydroxydopamine
- HBSS
- Hanks' balanced salt solution
- DMEM
- Dulbecco's modified Eagle's medium
- DOPAC
- 3,4-dihydroxyphenylacetic acid
- HVA
- homovanillic acid
- ANOVA
- analysis of variance
- MAO
- monoamine oxidase
- FBS
- fetal bovine serum
- LC
- liquid chromatography
- MS
- mass spectrometry
- CL
- clearance
- HPLC
- high-performance liquid chromatography
- AP
- anteroposterior
- TH
- tyrosine hydroxylase
- TBS
- Tris-buffered saline
- RT
- reaction time
- DA
- dopamine
- ESI
- electrospray ionization
- CRC
- concentration-response curve.
Authorship Contributions
Participated in research design: Jones, Bubser, Thompson, Dickerson, Turle-Lorenzo, Amalric, Blobaum, Bridges, Morrison, Jadhav, Daniels, Conn, and Niswender.
Conducted experiments: Jones, Bubser, Thompson, Dickerson, Turle-Lorenzo, Blobaum, Bridges, Morrison, Jadhav, Italiano, Bode, Daniels, and Niswender.
Contributed new reagents or analytic tools: Jones, Blobaum, Bridges, Morrison, Jadhav, Engers, Daniels, Lindsley, Hopkins, and Niswender.
Performed data analysis: Jones, Bubser, Thompson, Dickerson, Turle-Lorenzo, Amalric, Blobaum, Bridges, Morrison, Jadhav, Italiano, Bode, Daniels, and Niswender.
Wrote or contributed to the writing of the manuscript: Jones, Bubser, Thompson, Dickerson, Amalric, Blobaum, Bridges, Morrison, Daniels, Lindsley, Hopkins, Conn, and Niswender.
References
- Amalric M, Moukhles H, Nieoullon A, Daszuta A. (1995) Complex deficits on reaction time performance following bilateral intrastriatal 6-OHDA infusion in the rat. Eur J Neurosci 7:972–980 [DOI] [PubMed] [Google Scholar]
- Beurrier C, Lopez S, Révy D, Selvam C, Goudet C, Lhérondel M, Gubellini P, Kerkerian-LeGoff L, Acher F, Pin JP, et al. (2009) Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J 23:3619–3628 [DOI] [PubMed] [Google Scholar]
- Bradley SR, Standaert DG, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. (1999) Immunohistochemical localization of subtype 4a metabotropic glutamate receptors in the rat and mouse basal ganglia. J Comp Neurol 407:33–46 [PubMed] [Google Scholar]
- Buu NT, Angers M, Duhaime J, Kuchel O. (1987) Modification of dopamine and norepinephrine metabolism in the rat brain by monoamine oxidase inhibitors. J Neural Transm 70:39–50 [DOI] [PubMed] [Google Scholar]
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. (2005) Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci 6:787–798 [DOI] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. (2002) Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience 110:403–420 [DOI] [PubMed] [Google Scholar]
- Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. (2009) Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J Med Chem 52:4115–4118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassler R. (1938) Pathologie der paralysis agitans und des postenzephalitischen Parkinsonismus. J Psychol Neurol 48:387–476 [Google Scholar]
- Hauser RA, Cantillon M, Pourcher E, Micheli F, Mok V, Onofrj M, Huyck S, Wolski K. (2011) Preladenant in patients with Parkinson's disease and motor fluctuations: a phase 2, double-blind, randomised trial. Lancet Neurol 10:221–229 [DOI] [PubMed] [Google Scholar]
- Hettinger BD, Lee A, Linden J, Rosin DL. (2001) Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J Comp Neurol 431:331–346 [DOI] [PubMed] [Google Scholar]
- Hodgson RA, Bedard PJ, Varty GB, Kazdoba TM, Di Paolo T, Grzelak ME, Pond AJ, Hadjtahar A, Belanger N, Gregoire L, et al. (2010) Preladenant, a selective A(2A) receptor antagonist, is active in primate models of movement disorders. Exp Neurol 225:384–390 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington DC [Google Scholar]
- Jankovic J. (2008) Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79:368–376 [DOI] [PubMed] [Google Scholar]
- Javitch JA, Strittmatter SM, Snyder SH. (1985) Differential visualization of dopamine and norepinephrine uptake sites in rat brain using [3H]mazindol autoradiography. J Neurosci 5:1513–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Conn PJ, Niswender CM. (2009) Glutamate receptors as therapeutic targets for Parkinson's disease. CNS Neurol Disord Drug Targets 8:475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Acher F, De Leonibus E, Mele A, Amalric M. (2007) Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson's disease. J Neurosci 27:6701–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Johnston TH, Brotchie JM, Schann S, Neuville P, Amalric M. (2008) Functional interaction between adenosine A2A and group III metabotropic glutamate receptors to reduce parkinsonian symptoms in rats. Neuropharmacology 55:483–490 [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. (2004) A model of l-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis 16:110–123 [DOI] [PubMed] [Google Scholar]
- Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, et al. (2003) (−)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology 45:895–906 [DOI] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, et al. (2003) Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc Natl Acad Sci U S A 100:13668–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JF, Ungerstedt U. (1977) Supersensitivity to apomorphine following destruction of the ascending dopamine neurons: quantification using the rotational model. Eur J Pharmacol 41:361–367 [DOI] [PubMed] [Google Scholar]
- Matsui T, Kita H. (2003) Activation of group III metabotropic glutamate receptors presynaptically reduces both GABAergic and glutamatergic transmission in the rat globus pallidus. Neuroscience 122:727–737 [DOI] [PubMed] [Google Scholar]
- Morelli M, Carta AR, Jenner P. (2009) Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol 193:589–615 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, et al. (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74:1345–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen AM. (2004) Cognitive dysfunction in Parkinson's disease: the role of frontostriatal circuitry. Neuroscientist 10:525–537 [DOI] [PubMed] [Google Scholar]
- Paterson IA, Juorio AV, Berry MD, Zhu MY. (1991) Inhibition of monoamine oxidase-B by (−)-deprenyl potentiates neuronal responses to dopamine agonists but does not inhibit dopamine catabolism in the rat striatum. J Pharmacol Exp Ther 258:1019–1026 [PubMed] [Google Scholar]
- Paxinos G, Watson C. (2007) The Rat Brain in Stereotoxic Coordinates, 6th ed, Elsevier Inc., Amsterdam, The Netherlands [Google Scholar]
- Pinna A, Tronci E, Schintu N, Simola N, Volpini R, Pontis S, Cristalli G, Morelli M. (2010) A new ethyladenine antagonist of adenosine A(2A) receptors: behavioral and biochemical characterization as an antiparkinsonian drug. Neuropharmacology 58:613–623 [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Bernardi G. (1997) Activation of group III metabotropic glutamate receptors depresses glutamatergic transmission at corticostriatal synapse. Neuropharmacology 36:845–851 [DOI] [PubMed] [Google Scholar]
- Rezak M. (2007) Current pharmacotherapeutic treatment options in Parkinson's disease. Dis Mon 53:214–222 [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. (2000) CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39:777–787 [DOI] [PubMed] [Google Scholar]
- Sibille P, Lopez S, Brabet I, Valenti O, Oueslati N, Gaven F, Goudet C, Bertrand HO, Neyton J, Marino MJ, et al. (2007) Synthesis and biological evaluation of 1-amino-2-phosphonomethylcyclopropanecarboxylic acids, new group III metabotropic glutamate receptor agonists. J Med Chem 50:3585–3595 [DOI] [PubMed] [Google Scholar]
- Slee DH, Zhang X, Moorjani M, Lin E, Lanier MC, Chen Y, Rueter JK, Lechner SM, Markison S, Malany S, et al. (2008) Identification of novel, water-soluble, 2-amino-N-pyrimidin-4-yl acetamides as A2A receptor antagonists with in vivo efficacy. J Med Chem 51:400–406 [DOI] [PubMed] [Google Scholar]
- Stacy M, Galbreath A. (2008) Optimizing long-term therapy for Parkinson disease: levodopa, dopamine agonists, and treatment-associated dyskinesia. Clin Neuropharmacol 31:51–56 [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. (2010) Combination analysis. Adv Exp Med Biol 678:133–137 [DOI] [PubMed] [Google Scholar]
- Tozzi A, Tscherter A, Belcastro V, Tantucci M, Costa C, Picconi B, Centonze D, Calabresi P, Borsini F. (2007) Interaction of A2A adenosine and D2 dopamine receptors modulates corticostriatal glutamatergic transmission. Neuropharmacology 53:783–789 [DOI] [PubMed] [Google Scholar]
- Turle-Lorenzo N, Maurin B, Puma C, Chezaubernard C, Morain P, Baunez C, Nieoullon A, Amalric M. (2006) The dopamine agonist piribedil with l-DOPA improves attentional dysfunction: relevance for Parkinson's disease. J Pharmacol Exp Ther 319:914–923 [DOI] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. (2005) Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther 313:1296–1304 [DOI] [PubMed] [Google Scholar]
- Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, Conn PJ. (2003) Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci 23:7218–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varty GB, Hodgson RA, Pond AJ, Grzelak ME, Parker EM, Hunter JC. (2008) The effects of adenosine A2A receptor antagonists on haloperidol-induced movement disorders in primates. Psychopharmacology (Berl) 200:393–401 [DOI] [PubMed] [Google Scholar]
- Youdim KA, Lyons R, Payne L, Jones BC, Saunders K. (2008) An automated, high-throughput, 384-well cytochrome P450 cocktail IC50 assay using a rapid resolution LC-MS/MS end-point. J Pharm Biomed Anal 48:92–99 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.