

Abstract
Current knowledge is insufficient to explain why only a proportion of individuals exposed to environmental carcinogens or carrying a genetic predisposition to cancer develop disease. Clearly, other factors must be important and one such element that has recently received attention is the human microbiome, the residential microbes including Bacteria, Archaea, Eukaryotes, and viruses that colonize humans. Here, we review principles and paradigms of microbiome-related malignancy, as illustrated by three specific microbial-host interactions. We review the effects of the microbiota on local and adjacent-neoplasia, present the estrobolome model of distant effects, and discuss the complex interactions with a latent virus leading to malignancy. These are separate facets of a complex biology interfacing all the microbial species we harbor from birth onward toward early reproductive success and eventual senescence.
Introduction
Cancer, which manifests as the uncontrolled proliferation of host cells, is a leading cause of death in human societies worldwide. Disease and death result from local and distant spread of malignant cells, as well as from their metabolic and systemic effects. Although virtually all human tissues containing cells with replicating potential may be affected by cancer, there is host population-specificity in the timing and tissues involved, in histology, and in natural history. While many genetic predispositions to increased cancer risk in general and to the development of specific malignancies have been identified, environmental effects dominate for virtually all of the major human cancers (Lichtenstein et al., 2000).
Significant exposures to environmental carcinogens, including toxic chemicals, ionizing radiation, and microbial pathogens, are extremely varied (Schottenfeld et al., 2006). However, current knowledge is insufficient to explain why only a proportion of heavy smokers develop lung cancer, or who among those with hepatitis B infection will develop hepatomas. Clearly, other environmental factors must be important (Wynder and Gori, 1977) and one such element factor that has received attention more recently is the human microbiome.
Since their earliest origins (Ley et al., 2008a), animals have been colonized by residential micro-organisms including bacteria, fungi, protozoa, and viruses, that are collectively described as the microbiome (Lederberg and McCray, 2001). With the development of new scientific tools, there has been increasing interest in the composition, function, stability, and host-specificity of the microbiome, and the aggregate of its genes, the metagenome (Benson et al., 2010; Ley et al., 2006; Li et al., 2008). This review focuses on the relationships of the microbiome to human malignancy. We present general principles and explore three models of microbiome constituents affecting cancer risk and pathogenesis. The three paradigms we describe here illustrate different aspects of the emerging pathogenetic processes involving the microbiome.
General Principles
The Microbiome
Humans are colonized by residential microbes including Bacteria, Archaea, Eukaryotes, and viruses (Turnbaugh et al., 2007). The proportion of bacterial cells represented in the human body is estimated to be ~90%, and of all genes, >99%. Initial colonization occurs at the time of birth and we progressively acquire a population of ~1014 bacterial cells at equilibrium, which essentially remain for life and this process is recapitulated in every human lifetime. A human virome consisting of persistent colonizing viruses also exists, but is far less explored. Preliminary examination of the fecal virome has shown numerous bacteriophages, but with no obvious relationship to neoplasia (Reyes et al., 2010).
Each anatomical niche possesses its own mixture of microbial populations. Although generally conserved at higher taxonomic levels and in functional properties (Arumugam et al., 2011) between all humans, interindividual microbiota variation at lower levels (genus, species, and strain) is enormous (Qin et al., 2010). The microbiome composition appears to evolve over the human life span, but the exact magnitude of such changes is unknown. The individual organisms and cells in the microbiome both compete and cooperate with one another (Blaser and Kirschner, 2007), and the metagenome has both functional and genetic plasticity (Arumugam et al., 2011; Muegge et al., 2011).
Interactions with hosts
We do not carry our microbial load passively. There is increasing evidence for a rich, complex, dynamic, and individual-specific microbial interaction with hosts. Interactions involve microbial signaling of host cells that affect metabolic, neurological, inflammatory, immunologic and host-defense functions, among others (Barton et al., 2007; Dethlefsen et al., 2007; Ley et al., 2008b; Muegge et al., 2011). The nature of host responses also shapes microbiome populations and metabolism (Vijay-Kumar et al., 2010). Indeed, a long-term well-choreographed Nash equilibrium may model host-microbial co-evolution (Blaser and Kirschner, 2007). The range of equilibrium arrangements is broad; simplified formulations are shown in Figure 1. Host interactions with the microbiome are both local, e.g. in the gastrointestinal tract lumen, or distant, involving hormonal intermediates, microbial metabolites, and immunologic messengers.
Figure 1. Equilibrium between co-evolved microbes and host cells. Panel A: Single organism equilibrium.
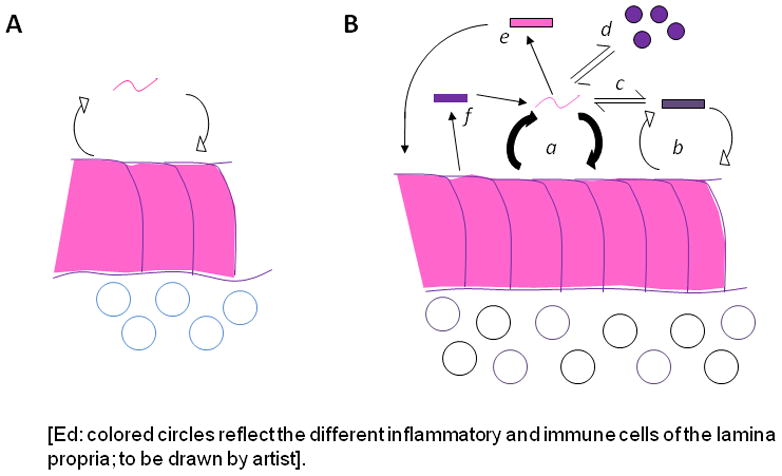
In this model, there is a counter-regulatory (negative feedback) interaction involving metabolic and physical signals between microbe and host. Panel B: Multiple organisms in equilibrium. In this much more complex system, organisms may have individual equilibrium relationships (e.g. a and b) with host cells as in Panel A. However, the interaction between these two microbes (c) will affect their individual interactions. Similarly, another microbe (d) might interact exclusively with an organism (a), but not with the host, with the extent of the interaction affecting the equilibrium relationships. An alternative is that an interactive microbe (a) can interact with a second microbe (e) that directly signals the host, but does not receive direct host signals back. Finally, the host might have a specific interaction with another microbe (shown as f), which can have a unidirectional interaction with microbes (e.g. with organism a) but not directly with the host.
Human cancers must be considered against the background of host-microbiome interactions. In general, cancers are log-linear with host age (Nordling, 1953). Three questions are relevant to this review: How are cancers initiated? Why do some but not all tumors progress? What determines susceptibility to treatment? In summary, there is a theoretical basis for the microbiome to participate in each of these phenomena (Figure 2). For example, microbiome-induced recruitment of lamina propria innate and adaptive immune effectors induces epithelial cell proliferation (Israel et al., 2001) and cell proliferation per se is pro-oncogenic (Ames and Gold, 1990). Microbial adjuvancy or suppression affects immune surveillance (Gaboriau-Routhiau et al., 2009; Ivanov et al., 2009), which limits tumor progression. Differing drug metabolism affects susceptibility to chemotherapy (Wallace et al., 2010).
Figure 2. Mechanisms by which the microbiome can enhance malignant transformation and cancer spread.
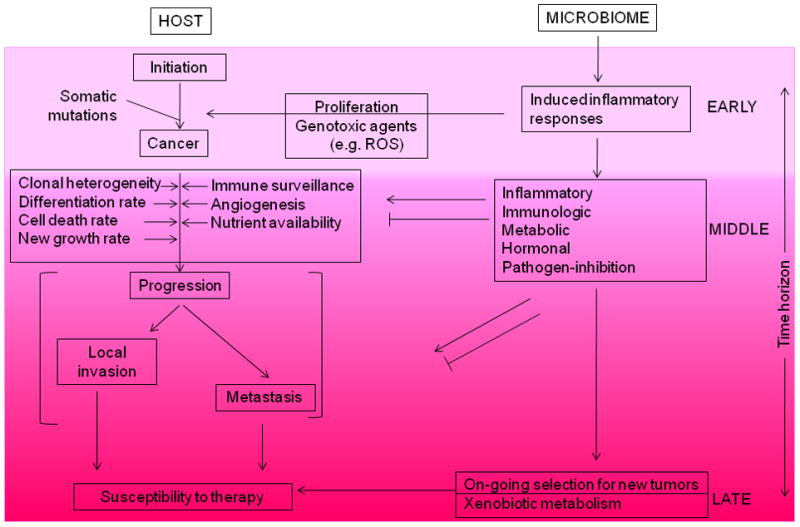
Cancer involves the transformation of physiologically responsive cells into autonomously replicating tumors that have the capacity to invade local tissues or spread widely. Multiple host processes (indicated on the left) govern the success of neoplastic cells to cause cancer. However, the interaction of the microbiome with the host (right) yields effects that can enhance or suppress tumorigenesis. Microbiome-induced inflammation affects the initiation (‘EARLY’) of cancers. Medium-term interactions affect multiple facets that influence whether or not tumors progress, and if so, in which ways. Finally, ‘LATE’ interactions affect the susceptibility of the tumor to therapies.
We are living at a time of great ecological change; global warming at the macro-level and “disappearing microbiota” at the micro-level (Blaser and Falkow, 2009). Thus, long-established human-microbial ecological relationships, and their contribution to cancer risk, are changing. For example, in developed countries, the incidence of gastric cancer is falling, whereas esophageal adenocarcinoma is rising (see below). Nevertheless, several different types of cancer development can be considered (Figure 3) at the crossroads of our ancient and our “post-modern” microbiota. Of the microbe-induced human malignancies, Class A is defined as involving immunologic tissues, Class B requires direct microbial interactions with parenchymal cells, and Class C involves distant effects from local interactions. Several examples (Table 1) may provide a model for not-yet recognized relationships between microbiome and particular cancers (Blaser, 2008). We illustrate general types of relationships between different constituents of the human microbiome and several human cancers through the following paradigms.
Figure 3. Classification of microbiome-associated human malignancies.
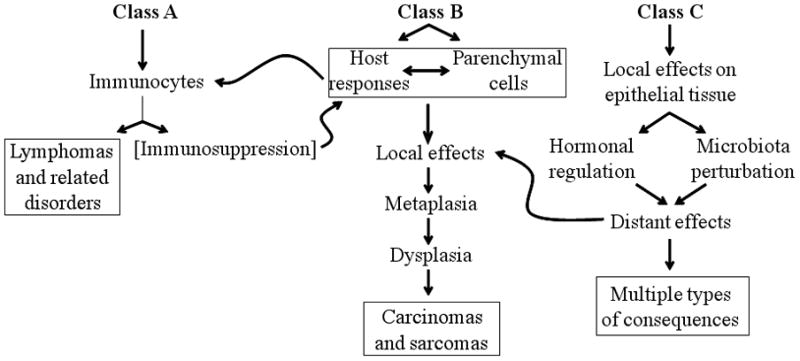
Three types of relationships can be envisaged between the microbiome and mechanisms that give rise of cancers. In Class A, the primary interactions involve immunocytes; in Class B, involve local parenchymal cells, and in Class C, the local interactions produce distant effects. Specific examples of all three classes are indicated in Table 1. Adapted from MJ Blaser (2008), with permission.
Table 1.
Examples of microbe-induced human malignancies, by class
| Microbe(s) | Examples of malignancies by class
|
||||
|---|---|---|---|---|---|
| A | B | C | |||
| EBV* | Lymphomas | Nasopharyngeal carcinoma | |||
| HTLV-1 | ATL | ||||
| HHV-8 | Kaposi’s sarcoma | ||||
| HIV | Lymphomas | Kaposi’s sarcoma | |||
| Hepatitis B | Hepatocellular carcinoma | ||||
| Hepatitis C | Lymphomas | Hepatocellular carcinoma | |||
| H. pylori* | MALT gastric lymphoma | Gastric adenocarcinoma | [Esophageal adenocarcinoma]** | ||
| HPV | Anogenital carcinomas, oropharyngeal carcinoma | ||||
| Schistosomal species | Bladder cancer | ||||
| Liver flukes | Cholangiocarcinoma | ||||
| Hypothesized scenarios: microbiome |
|
[Breast, & endometrial adenocarcinomas] | |||
| ΔMicrobiome† |
|
[Testicular, & prostate adenocarcinomas] | |||
| Microbiome | Colon adenocarcinoma | ||||
Abbreviations: ATL, adult T-cell leukemia/lymphoma; HHV-8, human herpesvirus 8; HTLV-1, human T-cell lymphotropic virus type 1; MALT, mucosa-associated lymphoid tissue.
Represents microbes that are constituents of the ancestral human microbiome.
Brackets indicate that the presence of a specific microbe (e.g. H. pylori), or as yet unidentified member(s) of the microbiome may either inhibit or promote the development of the bracketed malignancy.
In addition to metabolic activities of usual microbial constituents, changes (Δ) in the microbiome also may be involved in inducing some cancers.
Paradigm 1. Active inflammation promoting cancer in a lumenal organ (H. pylori)
H. pylori are microaerophilic, highly motile, curved gram negative bacilli that colonize the human stomach (Atherton and Blaser, 2009). Considerable evidence reveals that H. pylori is ancestral in humans, colonizing our ancestors before the major out-of-Africa migration 58,000 years ago (Linz et al., 2007). Acquired in early childhood and carried for nearly all of the life span, H. pylori was ubiquitous in all human populations in which it was studied (Bardhan, 1997). Also, examination of gastric 16S rRNA genes indicates that H. pylori dominates the gastric microbiota (Andersson et al., 2008; Bik et al., 2006). Thus, H. pylori has been the ancient, dominant, and highly interactive member of the human gastric microbiota. Nevertheless, H. pylori prevalence has steadily decreased over the course of the 20th century (Chen and Blaser, 2008; Roosendaal et al., 1997); this is both a major and surprising shift in human microecology.
As such, we can now measure the consequences of carrying H. pylori or not. As discussed below, the presence or absence of the organism can lead to very different gastric physiologies and each of the 3 classes of microbe-induced malignancies outlined in Figure 3 can be attributed to the presence or absence of H. pylori. Substantial epidemiologic, histologic, and experimental animal studies implicate H. pylori in the causation of gastric adenocarcinoma (Peek and Blaser, 2002), the second leading cause of cancer death worldwide (Class B relationship). In addition, H. pylori is strongly associated with gastric MALT-lymphomas (Parsonnet et al., 1994) (Class A relationship). Conversely, H. pylori has an inverse association with esophageal (gastro-esophageal junction) adenocarcinoma, consistent with a protective effect (Islami and Kamangar, 2008) (Class C relationship).
Gastric cancer
Via secreted substances, physical attachment, as well as the injection, by a type IV secretion system (TFSS), of its own constituents, H. pylori are highly interactive with host cells (Odenbreit et al., 2000), consistent with the Class B model (Figure 3). A major injected constituent is the ~128 KDa CagA protein, which contains several tyrosine phosphorylation (EPIYA) domains that are recognized by host cell kinases, that convert CagA into p-CagA (Backert and Selbach, 2008). As an indication of H. pylori complexity, the EPIYA-rich encoding region contains many DNA repeats, permitting the expansion or diminution of EPIYA site number. Strains with C or D type EPIYAs signal host epithelial cells via the Src/Shp-2/MAPK pathway whereas EPIYA lacking proteins signal through gp130/JAK/STAT3 (Lee et al., 2010). Since these are cross-inhibitory pathways, the population biology of H. pylori within a particular locale determines, with very fine tuning, the nature of host responses, enabling persistence, but also leading to tissue responses and injury. Individuals carrying H. pylori strains that possess CagA are at increased risk for the development of precursor lesions for gastric cancer (i.e. atrophic gastritis and intestinal metaplasia) and therefore the cancer itself. Multiple signaling pathways may be involved since particular alleles of H. pylori genes are associated with increased risk of cancer development (Batista et al., 2011). Particular host polymorphisms that relate to innate immunity (e.g. affecting IL-1, IL-1RA, and TNF-α expression) enhance the risk of gastric cancer associated with H. pylori positivity (El-Omar et al., 2001; El-Omar et al., 2003; Figueiredo et al., 2002). H. pylori-induced gastric inflammation recruits stem cells from the bone marrow, which may also participate in tumorigenesis (Houghton et al., 2004). The traditional model of gastric carcinogenesis focuses on host, strain, or interaction differences in explaining H. pylori-induced gastric adenocarcinoma (Peek and Blaser, 2002). An alternative model is shown in Figure 4 in which the H. pylori-host interaction causes progressive degradation of the gastric niche over decades, H. pylori is essentially lost (Karnes et al., 1992) and the changes lead to the success of new competing microbiome populations that are cancer-promoting.
Figure 4. Multi-decade development of gastric adenocarcinoma initiated by H. pylori: ecologic model.
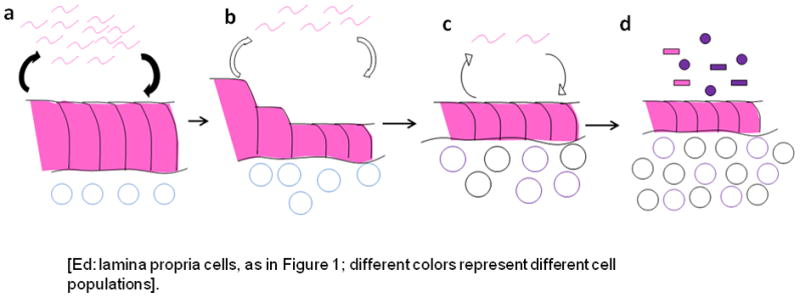
The equilibrium relationship of H. pylori and its host involves recruitment of a population of immune and inflammatory cells in the gastric lamina propria (Panel a). Over time (decades), the conjunction of the organism and its host response results in continued injury to the epithelium with progressive loss of normal architecture and function (Panel b). This leads to the development of atrophic gastritis (Panel c), with permanently altered architecture, and a reduction in acid secretory function. With hypo-chlorhydria, the gastric niche now is dominated by competing microbiome members that have pathogenic properties leading to further inflammation and tissue injury (Panel d). Over this decades-long (essentially life-long) progression, H. pylori bacterial populations gradually decline. In the final stage, the H. pylori-induced atrophic gastritis lowers gastric acidity which then is a reduced barrier to the intrusion of adventitious pathogenic oro-pharyngeal and intestinal bacteria.
Gastric lymphoma
The presence of H. pylori is strongly associated with primary gastric lymphoma (Parsonnet et al., 1994; Wotherspoon et al., 1993). The presence of H. pylori is important in the proliferation of the tumors, since when H. pylori is eliminated, they often regress (Yamamoto et al., 2008; Zullo et al., 2010). The tumors are clonal, but a spectrum encompassing benign polyclonal lymphoid expansion, emergence of a dominant clone, and malignant transformation of the clone can be observed (Thiede et al., 1999). This scenario is consistent with the Class A model for microbiome-mediated hematopoietic and lymphoid malignancies consistent as outlined in Figure 3.
Esophageal adenocarcinoma (EAC)
The incidence of GE junction adenocarcinomas (namely EAC and gastric cardia adenocarcinoma) is rapidly rising in Western countries (Devesa et al., 1998) just as H. pylori is disappearing (Chen and Blaser, 2008; Roosendaal et al., 1997). The development of these cancers (and those on the gastric cardia side of the GE junction) follows gastro-esophageal reflux disease (GERD), and its metaplastic sequela (Barrett’s esophagus). The presence of H. pylori, especially cagA+ strains, is inversely associated with all three lesions (Peek and Blaser, 2002). These findings have been confirmed (Islami and Kamangar, 2008), and are consistent with the hypothesis that a change in the gastric microbiome resulting from the absence of H. pylori is contributing to this epidemic of GE junction adenocarcinomas. This is biologically plausible based on emerging trends, and on the knowledge that loss of these highly interactive (especially cagA+) organisms change gastric physiology, affecting acid secretion (Moss & Calam, 1992), hormone interactions (Francois et al., 2008; Francois et al., 2011), and T-cell populations (Robinson et al., 2008). Further, the loss of H. pylori is associated with changes in the composition of the gastric microbiome involving many other microbial taxa (Maldonado-Contreras et al., 2011). That change in the microbiome in one location (gastric) may affect cancer risk in an adjacent, but separate, compartment (distal esophagus) suggests an important paradigm for other cancers: the causal agents need not be residents of the affected tissues (Figure 3, Class C). Nevertheless, there is an esophageal microbiota (Pei et al., 2004), that it is clearly perturbed in pre-malignant lesions of the esophagus (Yang et al., 2009). Whether this is cause or effect is uncertain at present, but is a promising area for exploration.
Paradigm 2. Metabolic effects of residential organisms leading to distant malignancies (Estrobolome)
The human estrobolome
We postulate that an important contribution of the human gut microbiota to host physiology is a functional estrobolome, the aggregate of enteric bacterial genes whose products are capable of metabolizing estrogens. Especially important are bacterial species possessing β-glucuronidases and β-glucuronides, enzymes involved in estrogen de-conjugation and conjugation (Cole et al., 1985; Dabek et al., 2008; Gadelle et al., 1985; Gloux et al., 2011; McBain and Macfarlane, 1998). The estrobolome is predicted to impact endogenous estrogen metabolism by modulating the enterohepatic circulation of estrogens, thus affecting circulating and excreted estrogen levels (Figure 5). A woman’s lifetime burden of estrogen exposure may reflect in part the metabolic functioning of her estrobolome. An estrobolome enriched in gene products promoting estrogen metabolite deconjugation reactions may result in greater reabsorption of free estrogens. Estrobolome variation in levels of functional deconjugative ability may thus influence development of estrogen-driven neoplasia (Figure 3, Class C).
Figure 5. The estrobolome and its switch.
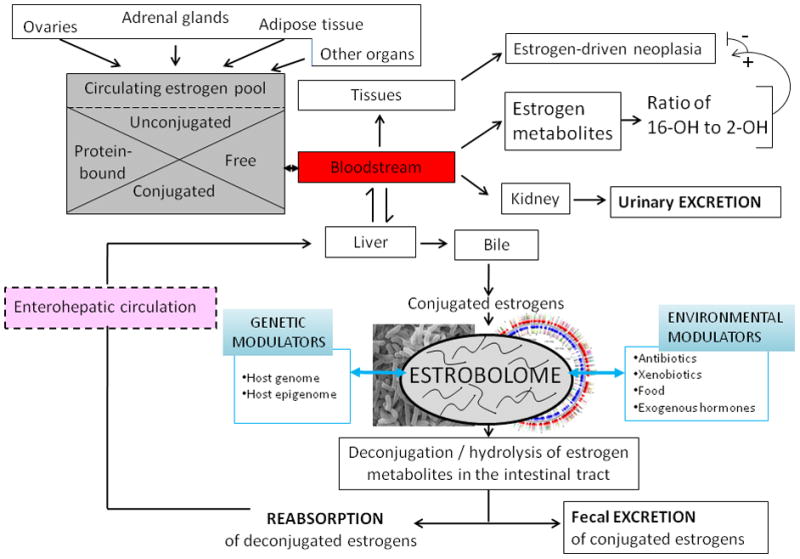
Estrogens are steroids hormones derived from the step-wise reduction of C21 cholesterol. The ovaries are the only organs capable of full C21 (cholesterol) → C18 (estrogen) synthesis; at all other sites of estrogen synthesis (e.g. adrenals, adipose tissue), the availability of C19 androgens as substrates and aromatase are limiting factors. Estrogens circulate in the bloodstream free or protein-bound, and are conjugated or unconjugated molecules that may enter target tissues or be eliminated by the kidneys. Circulating estrogens undergo Phase I hepatic metabolism. In the liver, estrogens and their resultant estrogen metabolites (EMs) then may be conjugated, through methylation, glucuronidation, or sulfonation reactions. Conjugated estrogens are subject to biliary excretion. The estrobolome, the aggregate of enteric bacterial genes whose products are capable of metabolizing estrogens, acts on conjugated estrogens and estrogen metabolites, with downstream physiologic effects. An estrobolome enriched in genes encoding enzymes favoring deconjugation promotes reabsorption of free estrogens that contribute to the host’s total estrogen burden. Suppression of deconjugation, that may follow antibiotic exposure, leads to increased estrogen excretion (Martin et al., 1975). Estrobolomes varying in functional activity lead to different host-estrogen equilibria, via enterohepatic circulation of varied proportions of conjugated to unconjugated estrogens. The composition of the estrobolome can be modulated by host-specific and/or environmental drivers (e.g. antibiotics) exerting selective pressure on its parental bacterial populations. Shown here for illustration is the ratio of 2-OH/16-OH hydroxylated EMs, that may serve as urinary or serum markers of risk for certain estrogen related cancers (Bradlow et al., 1995; Gupta et al., 1998; Kabat et al., 1997; Meilahn et al., 1998; Muti et al., 2000)
Estrogens and estrogen-driven cancer
Estrogens, steroid hormones derived from the progressive reduction of C21 cholesterol, may act locally (intracrine function), or circulate to exert effects on target organs (endocrine function). The three major forms of endogenous estrogen, estradiol (E2, dominant during reproductive years), estrone (E1, dominant after menopause), and estriol (E3, dominant during pregnancy) are 4-ring C18 molecules. Estrogens appear in circulation as free or protein-bound entities, and in both conjugated and unconjugated states. Estrogen exposure begins prenatally and is lifelong. The multiple beneficial effects (cardiovascular, metabolic, bone, fertility, cognition), contrast with roles in estrogen-driven cancers. Women with the highest circulating estrogen levels are at increased risk for developing postmenopausal endometrial (Lukanova et al., 2004) and breast cancers (Hankinson et al., 1998; Kaaks et al., 2005; Key et al., 2002; Lukanova et al., 2004; Toniolo et al., 1995; Woolcott et al., 2010). Estrogen metabolism varies between women with the full physiologic repertoire of its metabolites being unknown. Estrogens and estrogen-like molecules classically exert their cellular effects by binding to and activating estrogen receptors, although other receptor-independent mechanisms do exist.
Healthy and cancerous cells express receptors for estrogen
The two recognized estrogen receptors (ERα, ERβ) are homologous ligand-modulated transcription factors. Both are present in many healthy tissues (including breast, ovary, testis, prostate, bone, brain, vascular system) and in certain cancers of the breast, endometrium, prostate, bone, and lung (Kuiper et al., 1996). Ligands other than E2, E1 and E3 can bind with varying affinities to both ERα and ERβ (Kuiper et al., 1998) with activation increasing or repressing host cell transcription with consequent biologic activity (Jeyakumar et al., 2011). The centrality of estrogen to malignancy is reflected in therapeutic interventions that target estrogen receptor signaling, including the selective estrogen receptor antagonists (SERMs), widely used to treat ER(α)-receptor-expressing breast cancer.
Hepatic estrogen metabolism and the enterohepatic circulation of estrogens
Both E1 and E2 undergo Phase I oxidative hepatic metabolism leading to the formation of catechol estrogens, (e.g. 2-OH, 4-OH, and 16-OH estrogens) (Yager et al., 2009) that then are conjugated or transformed to semiquinones, which have been implicated in oncogenesis (Cavalieri et al., 2006). The ratio of circulating and urinary 2-OH to 16-OH estrogen pathway metabolites may be a marker of breast and endometrial cancer risk (Bradlow et al., 1995; Gupta et al., 1998; Kabat et al., 1997; Meilahn et al., 1998; Muti et al., 2000). Phase II hepatic conjugation reactions include methylation, via catechol-O-methyltransferase; glucuronidation via uridine 5′-diphospho-glucuronosyltransferase; or sulfonation, via sulphotransferase (Raftogianis et al., 2000). Some methylated estrogens, such as 2-methoxyestradiol, exhibit proapototic, antiangiogenic, and antiproliferative activities (Lakhani et al., 2003) and are currently being studied as anticancer agents. Conjugated estrogens are not important ligands for the estrogen receptors (Raftogianis et al., 2000) and are subject to biliary excretion (Adlercreutz and Martin, 1980; Sandberg and Slaunwhite, 1957). Most importantly, there is an enterohepatic circulation of estrogens (Figure 5) with repeated re-circulation circuits (Adlercreutz, 1962; Sandberg and Slaunwhite, 1957). Recent refinements in analytic methodologies now permit accurate measurements of conjugated and unconjugated estrogens and estrogen metabolites in serum and urine (Zeigler et al, 2010), and should stimulate investigations of estrogen metabolism in malignancies.
Estrogen metabolism requires a functional estrobolome
Gut microbial functions driving estrogen metabolism and contributing to the proportions of recirculated and excreted estrogens and estrogen metabolites has long been considered (Adlercreutz and Jarvenpaa, 1982; Eriksson, 1970). Reduction in populations of specific gut bacteria in humans, as occurs with exposure to antibiotics, causes increased fecal excretion of conjugated estrogens and decreases in urinary estrogens (Martin et al., 1975), highlighting the modulatory role of the gut microbiome’s deconjugating machinery (Adlercreutz and Jarvenpaa, 1982; Adlercreutz et al., 1979; Adlercreutz et al., 1978; Martin et al., 1975). Human fecal extracts metabolize estrogens ex vivo, as would be expected of the estrabolome. The reactions include reduction and oxidation, the generation E2 from E1 as well as from estradiol 3-glucuronide; E1 from E2 and from estrone 3-sulfate; and E3 from 16α-hydroxyesterone (Lombardi et al., 1978). Similarly, in-vitro incubation of E1, E2, and 16α-hydroxyestrone with human feces leads to the interconversion of E1 and E2, the reduction of 16α-hydroxyestrone to E3, 16-oxoestradiol to 16-epiestriol, and 15α-hydroxyestrone to 15α-hydroxyestradiol (Järvenpää et al., 1980). Finally, germ-free mice provide additional evidence for the centrality of host-microbe interactions in estrogen metabolism (Shimizu et al., 1998).
Interventions that target the estrobolome affect estrogens
Bacterial composition of the human estrobolome likely reflects host factors, such as delivery mode at birth as well as lifetime environmental influences, including antibiotic use and dietary composition. Vegetarians have increased fecal excretion of conjugated estrogens (Goldin et al., 1982; Gorbach and Goldin, 1987) and dietary manipulations affect overall gut microbiome composition and function (Turnbaugh et al., 2009; Faith et al., 2011; Muegge et al., 2011). Such pressures exerted over time on the bacterial communities that constitute the estrobolome may lead to emergence of functionally distinct patterns, analogous to the broader categorization of the gut phylogenetic enterotypes (Arumugam et al., 2011). The confluence of both genetic and environmental modulators may shape estrobolome functionality that affects cancer risk (Figure 5); as such, estrobolome analysis could be harnessed to reduce emergence of estrogen-driven malignancies, such as Type I (endometrioid) endometrial cancer, estrogen receptor-positive breast cancer, and some ovarian cancers. Interventions that modify the bacterial constituents of the estrobolome also could modulate functional activity. Manipulations that specifically target species with β-glucuronidase and β-glucuronide activities could aid in reducing estrogen-related cancer risk. Changing bacterial populations to diminish hydroxylation and reductive functions can be accomplished with of antimicrobial agents, pre-biotics, or probiotics. If such interventions proved successful, estrobolome status might inform future risk of malignancy and measures could target restoration and maintenance of a “healthy” estrobolome for that host.
Paradigm 3. Abrogation of clinical latency leading to malignant outcomes
Epstein-Barr virus (EBV)
EBV is an ancient (Lacoste et al., 2010; McGeoch et al., 1995), ubiquitous, and persistent virus that obligately infects humans, leading to life-long latency. By adulthood, nearly all humans have acquired EBV (Henle et al., 1969). As such, EBV should be considered part of the human virome and it is with this premise that we discuss EBV and associated malignancies in this review.
EBV is a 172 kb, double-stranded DNA virus of the γ-herpesvirus subfamily of Lymphocryptovirus, present as a characteristic (Penkert et al., 2011) circular episome within the human memory B-lymphocyte nucleus (Niederman et al., 1970). EBV is associated with relatively low short-term cost and even possible benefit to the host (Barton et al., 2007), but harbors long-term malignant potential. The emergence of an EBV-associated cancer, often decades after EBV acquisition, reflects complex interactions between the virus and its host that culminate in the loss of latency and highlights how a nearly universal member of our commensal microbiota contributes to malignancy. In hosts who develop Burkitt’s lymphoma (BL), post-transplantation lymphoproliferative disorders (PTLD), or nasopharygeal carcinoma (NC), EBV has a bi-phasic life cycle. However, most EBV positive individuals do not develop EBV-associated cancers, but instead just harbor latent virus. EBV’s presence is thus necessary, but not sufficient for the emergence of an EBV-associated malignancy.
Interactions of Epstein-Barr Virus with its human host
EBV is transmitted between humans through saliva. Children become susceptible to EBV transmission after maternal antibody protection wanes; infection is either asymptomatic or may lead to non-specific symptoms. EBV acquisition in adolescence or in young adulthood, as seen in Western societies, results in illness termed infectious mononucleosis (IM) in about half of the infected individuals. The intensity of the host’s T-cell responses to primary EBV is responsible for this acute illness (Callan et al., 1996).
After the initial replicative lytic phase, EBV undergoes a shift to one of a series of tightly regulated latency states, each characterized by transcriptional repression to evade immune surveillance (Figure 6). Differential regulation of promoter utilization underlies EBV latency gene expression, possibly involving chromatin insulator protein (Tempera et al., 2011). Compared to the >80 genes expressed in the lytic cycle, in a Latency III program EBV principally expresses 6 nuclear antigens (EBNAs) and all 3 integral latent membrane proteins (LMPs) whereas in Latency II, only EBNA 1, and LMP 1 & 2 are expressed, and in Latency I, only EBNA1 is expressed (Figure 6). EBNA1 is crucial for EBV episome replication and maintenance, and Latency I represents the most restrictive form of latency in dividing cells. A state of “Latency 0” in which there is no viral antigen expression is postulated to occur in vivo, consistent with the life-long persistence of the EBV genome in memory B-cells in healthy hosts (Babcock et al., 1998; Miyashita et al., 1995). Even in latency, EBV is capable of regulating B-cell protein expression and proliferation and the viral genome can replicate as an episome, in tandem with host cell division.
Figure 6. Schematic of EBV-host interactions.
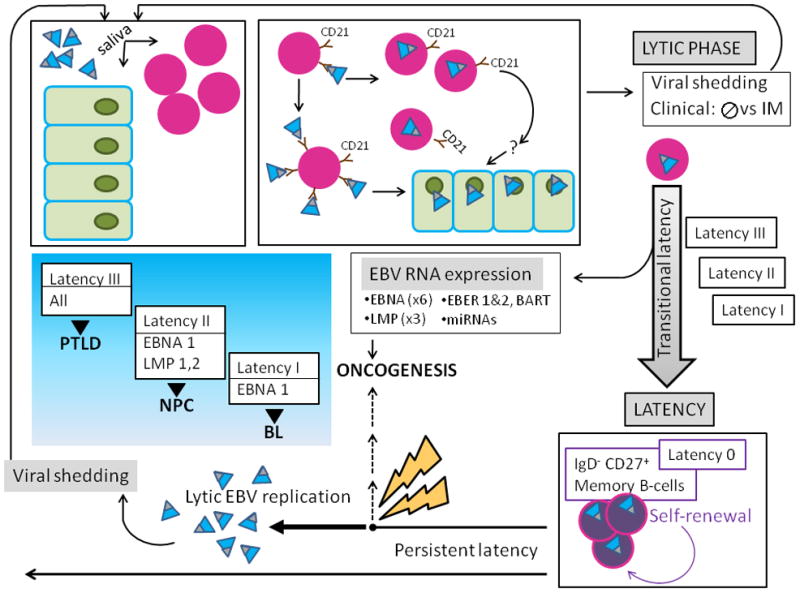
A) EBV (teal virions with black tip) is acquired orally, and targets B-cells (lavender nucleated circles) and permissive epithelial cells of the oral pharynx (green). The major EBV envelope glycoproteins gp350 and gp220 (black tip) interact with complement receptor CD 21 (brown) on the surface of naïve resting B-lymphocytes, leading to viral binding. Additional EBV B-cell interactions involving fusion proteins and HLA class II molecules (not shown) lead to virus-cell fusion and EBV internalization. EBV bound to the B-cell surface likely allows for its transfer to orophangeal epithelial cells. The initial lytic viral reproductive phase may be asymptomatic (usually) or may manifest clinical symptoms and is termed infectious mononucleosis (IM). During the lytic phase, virus is shed via the saliva and can infect naïve hosts.
B) After the initial lytic phase, EBV evades host immunosurveillance to achieve persistence though “translational latency”, the tightly regulated selective expression of viral latent proteins and of non-coding RNAs. The former include six Epstein-Barr virus Nuclear Antigens (EBNAs) (1, 2, 3A, 3B, 3C, and LP) and three integral Latent Membrane Proteins (LMPs) (1, 2A, and 2B). The latter include EBERs (1 and 2), small non-coding RNAs abundantly expressed in latently infected EBV cells and multiple microRNAs, encoded by two transcripts (in the BART and BHRF1 loci), that contribute to EBV-associated cellular transformation. Three distinct “transitional” EBV latency programs (Latency I, II, and III) are characterized by specific gene expression profiles that allow for establishing latency and enhancing cell survival and proliferation. After the initial lytic phase, EBV replicates as an episome, in tandem with the host cell genome. EBV employs host cell-driven DNA genomic methylation and modulation of NF-κβ activity, and Notch signaling pathway manipulations (not shown) to establish true latency (Latency 0) in resting memory B-cells (purple circles), with highly restricted EBV gene expression. Non-pathogenic and invisible to the host immune system, Latency 0 EBV persistently populates memory B-lymphocytes. In the course of the latently infected hosts’ life, episodic disruptions of latency occur (depicted as ‘STRESS’ and yellow bolt), resulting in EBV replication and viral shedding with potential spread to other hosts. Latent EBV also can contribute to several cancers (dashed line), including lymphomas such as Burkitt’s Lymphoma (BL), and Nasopharyngeal Carcinoma (NPC). Exogenous immunosuppression may result in Post-Transplantation Lymphoproliferative Disorders (PTLD). The emergence of malignancy appears to require interactions of co-factors, for example P. falciparum in BL, and individual host characteristics, including HLA type in NPC.
EBV manipulates host innate immune signaling pathways
EBV exploits fundamental host innate signaling pathways including NF-κB, TNF-alpha, and Notch receptor pathways, throughout its life cycle. It does so to its advantage, beginning when viral envelope glycoprotein (gp350) binds to host B-cell surface CD21 and TLR 2, leading to persistent NF-κB classical pathway activation (Gaudreault et al., 2007). Later, EBV achieves immortalization via activation of both the alternative and classical NF-κB pathways (Kung and Raab-Traub, 2010; Mosialos et al., 1995; Song and Kang, 2010). EBV is able to convert host B lymphocytes into lymphoblastoid cell lines by expressing EBV nuclear and membrane proteins, EBNAs, and LMPs (in Latency III) that regulate transcription through the Notch and TNF-alpha receptor pathways (Cahir-McFarland et al., 2004). EBV is able to reroute the TNF receptor family signaling pathway (Izumi and Kieff, 1997; Le Clorennec et al., 2008; Liebowitz, 1998; Mosialos et al., 1995). Such interactions favor EBV persistence, yet EBV latency may also provide mutualistic benefit to its hosts as an immune adjuvant (White et al., 2010), protecting against lethal Listeria monocytogenes and Yersinia pestis infections (Barton et al., 2007). Over a host’s lifetime, incompletely characterized stimuli occasionally induce EBV to emerge from latency and initiate a lytic state via sequential expression of genes responsible for replication and whole virion assembly. This (generally clinically silent) replication results in viral shedding and potential spread to other EBV-naïve hosts.
EBV-associated cancer as a consequence of persistence
EBV plays a part in neoplastic transformation when latency is breeched.. EBV LMP 1 and LMP2A can activate the mTOR, AKT, and PI3K pathways regulating functions relevant to tumorigenesis, including cellular proliferation, growth, survival, and mobility (Fukuda and Longnecker, 2007; Moody et al., 2005; Swart et al., 2000), whereas LMP1 miRNA inhibits the tumor suppressor p53 (Fukuda and Longnecker, 2007; Liu et al., 2005; Moody et al., 2005; Swart et al., 2000). Signaling of tumor suppression pathways that initiate cell cycle arrest and priming of apoptotic pathways occur when latency is lost. EBV-associated neoplasms show three distinct patterns of latency-associated gene expression (Rowe et al., 1992). Burkitt’s lymphoma (BL) exemplifies the Latency I program in which the Qp promoter-induced EBNA1 is expressed with small EBV-encoded RNAs (EBERs) and BamHI-A rightward transcript (BART). Latency II, characteristic of nasopharyngeal carcinoma (NPC), EBV-positive Hodgkin’s lymphoma, EBV-positive gastric carcinoma, and T and NK-cell lymphomas, involves expression of EBERs, BART, and Qp promoter-induced EBNA, with added LMP 1, 2A, and/or 2B expression (Brooks et al., 1992). Post-transplantation lymphoproliferative diseases (PTLD) are associated with the Latency III program in which all EBV latent gene products are expressed. EBV Latency III-regulated gene products mediate cell migration, antigen presentation, MAP kinase pathway, and interferon (IFN) signaling (Cahir-McFarland et al., 2004).
Latency I program: EBV-associated Burkitt’s lymphoma, exogenous cofactors, and c-Myc
Burkitt’s lymphoma (BL), a high grade B-cell malignancy, has distinct clinical-epidemiological variants: endemic Burkitt’s lymphoma (eBL), sporadic (sBL) and HIV-associated (HIVBL). Each of these tumors have reciprocal chromosomal translocations involving immunoglobulin loci on chromosomes 14, 22, or 2, and c-myc (MYC) on chromosome 8 (Manolov and Manolov, 1972; Dalla-Favera et al., 1982), but only eBL tumors incorporate EBV DNA. Typical of Latency I, only EBNA-1 is expressed in eBL, which is the most common pediatric tumor in Sub-Saharan Africa where EBV acquisition occurs very early in life. Its distribution in regions of Africa and Papua New Guinea where malaria is holo-endemic has implicated P. falciparum as co-factor, promoting lymphoma through immunosuppressive effects on EBV-specific T-cell immunity (Njie et al., 2009; Whittle et al., 1984) and B-cell proliferation involving P. falciparum erythrocyte membrane protein 1 (PfEMP1) interactions with host memory B-cells harboring latent EBV (Chene et al., 2007). An alternative mechanism implicates malaria and initial EBV-induced B-cell proliferation, with consequent activation-induced deaminase (AID) dysregulation leading to the characteristic c-myc translocation and lymphoma (Thorley-Lawson and Duca, 2007). In that schema, emergence of sBL reflects spontaneous (non-microbe driven) c-myc translocation whereas, in eBL, an EBV-driven series of events, abetted by P. falciparum, culminates in the characteristic oncogenic c-myc.
Latency II program: Host HLA types and EBV-associated nasopharyngeal carcinoma
EBV is present in epithelial nasopharyngeal carcinoma (NPC) cells where it establishes a latency pattern with EBNA 1 and either LMP 1 or LMP 2 protein expression (Latency II) (Brooks et al., 1992). The highest incidence of NPC is in Southeast China, particularly in Guangxi Province, where it occurs in association with specific host HLA haplotypes (Tang et al., 2010). The underlying pathogenesis of EBV-induced nasopharyngeal carcinoma remains obscure.
Latency III program: Host immune suppression and EBV-associated PTLD
Post-transplantation lymphoproliferative disorders (PTLD) represent heterogeneous diseases that vary from reactive polyclonal B-cell hyperplasia, to monoclonal tumors, to fatal, aggressive non-Hodgkin’s lymphomas (Penn et al., 1969). PTLD may occur after hematopoietic stem cell or solid organ transplantation. Most cases of PTLD are of B-cell origin and EBV-associated (Young et al., 1989). Intensive immunosuppressive regimens to avoid graft rejection affect risk for PTLD development. The highest incidence of PTLD after small bowel transplantation compared to other organs, (e.g. kidney) may relate to the quantity of transplanted lymphoid tissue, with more EBV+ B-cell populations. (Cohen, 2000). EBV latency becomes disrupted in PTLD, diseases related to the intersection of 20th Century medical technologies with an ancient virome constituent. PTLD lesions express EBV Latency III genes, the encoded proteins involved in signal transduction, transcription, protein catabolism, and cell motility, shape, and adhesion characteristics (Carter et al., 2002; Delecluse et al., 1995; McKnight et al., 1994; Rea et al., 1994). Impaired α-EBV T-cell function permits EBV-driven B-cell proliferation resulting in PTLD.
Future therapies
Hanahan and Weinberg outline 10 therapeutic approaches to target the central functional and enabling pathways involved in cancer (Hanahan and Weinberg, 2011). We envision that microbes can be harnessed (Blaser, 1997, 2010) to perform many of these therapeutic functions. For example, colonization of specific niches in the gut lumen (stomach, distal esophagus, rectum) with probiotic bacterial strains capable of modulating local inflammation and immunity, could help control luminal gastro-intestinal tract neoplasia. Similarly, establishment and maintenance of an estrobolome (with diminished deconjugation activity) that favors estrogen excretion pathways may reduce risk of developing estrogen-driven malignancy. Knowledge of the microbiome can be applied to targeting both specific anatomic sites and functional capabilities. Members of the human microbiota can suppress aspects of cellular immunity (e.g.: B. fragilis (Mazmanian et al., 2008; Round et al., 2011) whereas others (e.g. cagA+ H pylori) induce pro-inflammatory cytokines (Backert and Selbach, 2008). In the future, we can either beneficially target particular effector microbes to the desired location or engineer the desired genes into microbes that naturally colonize those sites.
Conclusions
The examples provided are not comprehensive, but rather indicate several conserved and paradigmatic mechanisms; microbiome constituents likely will have roles in other human cancers (Garrett et al., 2009; Wu et al., 2009). As recently discussed (Dominguez-Bello et al., 2011), nature must remove senescent individuals to liberate resources for younger members of the same species. A role for our ancient, interactive residential microbes in clock-like functions (Blaser, 1997) is intuitive. In that manner, our residents may be true symbionts, improving host fitness early in life through metabolic and pathogen-defense functions, and leading to demise through neoplasia in the post-reproductive period. If the hypothesis that malignancy is part of mammalian selection is correct, then other human cancers whose rates are log-linear with age will predictably be found to reflect microbiome-induced pathogenesis.
Finally, the microbiome affects the metabolism of xenobiotics (Figure 2), such as pharmaceutical agents (Clayton et al., 2009), including those used to treat cancer. Responses to particular chemotherapeutic agents have been linked to specific gut microbiome metabolic activities (Wallace et al., 2010). Better knowledge of microbiome composition and metabolic activities will ultimately improve therapeutic choices, with new agents, dosing regimens, and monitoring strategies. Using such knowledge should improve therapeutic/toxic ratios, and represents another exciting frontier in cancer research.
Acknowledgments
Supported by R01GM63270, R01DK090989, UH2 AR057506, 5 P30 CA016087, and 1UL1RR029893 from the National Institutes of Health, by the Diane Belfer Program for Human Microecology, and by the Margaret Q. Landenberger Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adlercreutz H. Studies on oestrogen excretion in human bile. Acta Endocrinol Suppl (Copenh) 1962;42(Suppl 72):1–220. [PubMed] [Google Scholar]
- Adlercreutz H, Jarvenpaa P. Assay of estrogens in human feces. J Steroid Biochem. 1982;17:639–645. doi: 10.1016/0022-4731(82)90565-9. [DOI] [PubMed] [Google Scholar]
- Adlercreutz H, Martin F. Biliary excretion and intestinal metabolism of progesterone and estrogens in man. J Steroid Biochem. 1980;13:231–244. doi: 10.1016/0022-4731(80)90196-x. [DOI] [PubMed] [Google Scholar]
- Adlercreutz H, Martin F, Jarvenpaa P, Fotsis T. Steroid absorption and enterohepatic recycling. Contraception. 1979;20:201–223. doi: 10.1016/0010-7824(79)90094-5. [DOI] [PubMed] [Google Scholar]
- Adlercreutz H, Martin F, Lindstrom B. Gas chromatographic and mass spectrometric studies on oestrogens in bile--2. Men and non-pregnant women. J Steroid Biochem. 1978;9:1197–1205. doi: 10.1016/0022-4731(78)90013-4. [DOI] [PubMed] [Google Scholar]
- Ames BN, Gold LS. Too many rodent carcinogens: mitogenesis increases mutagenesis. Science. 1990;249:970–971. doi: 10.1126/science.2136249. [DOI] [PubMed] [Google Scholar]
- Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011 doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity. 1998;9:395–404. doi: 10.1016/s1074-7613(00)80622-6. [DOI] [PubMed] [Google Scholar]
- Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10:1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, et al. DNA sequence and expression of the B95–8 Epstein-Barr virus genome. Nature. 1984;310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- Bardhan PK. Epidemiological features of Helicobacter pylori infection in developing countries. Clin Infect Dis. 1997;25:973–978. doi: 10.1086/516067. [DOI] [PubMed] [Google Scholar]
- Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HWt. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ. Ecology of Helicobacter pylori in the human stomach. J Clin Invest. 1997;100:759–762. doi: 10.1172/JCI119588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ. Understanding microbe-induced cancers. Cancer Prev Res (Phila) 2008;1:15–20. doi: 10.1158/1940-6207.CAPR-08-0024. [DOI] [PubMed] [Google Scholar]
- Blaser MJ. Harnessing the power of the human microbiome. Proc Natl Acad Sci U S A. 2010;107:6125–6126. doi: 10.1073/pnas.1002112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- Bradlow HL, Davis DL, Lin G, Sepkovic D, Tiwari R. Effects of pesticides on the ratio of 16 alpha/2-hydroxyestrone: a biologic marker of breast cancer risk. Environ Health Perspect. 1995;103(Suppl 7):147–150. doi: 10.1289/ehp.95103s7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks L, Yao QY, Rickinson AB, Young LS. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol. 1992;66:2689–2697. doi: 10.1128/jvi.66.5.2689-2697.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol. 2004;78:4108–4119. doi: 10.1128/JVI.78.8.4108-4119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan MF, Steven N, Krausa P, Wilson JD, Moss PA, Gillespie GM, Bell JI, Rickinson AB, McMichael AJ. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med. 1996;2:906–911. doi: 10.1038/nm0896-906. [DOI] [PubMed] [Google Scholar]
- Carter KL, Cahir-McFarland E, Kieff E. Epstein-barr virus-induced changes in B-lymphocyte gene expression. J Virol. 2002;76:10427–10436. doi: 10.1128/JVI.76.20.10427-10436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008;198:553–560. doi: 10.1086/590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chene A, Donati D, Guerreiro-Cacais AO, Levitsky V, Chen Q, Falk KI, Orem J, Kironde F, Wahlgren M, Bejarano MT. A molecular link between malaria and Epstein-Barr virus reactivation. PLoS Pathog. 2007;3:e80. doi: 10.1371/journal.ppat.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- Cole CB, Fuller R, Mallet AK, Rowland IR. The influence of the host on expression of intestinal microbial enzyme activities involved in metabolism of foreign compounds. J Appl Bacteriol. 1985;59:549–553. doi: 10.1111/j.1365-2672.1985.tb03359.x. [DOI] [PubMed] [Google Scholar]
- Dabek M, McCrae SI, Stevens VJ, Duncan SH, Louis P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol. 2008;66:487–495. doi: 10.1111/j.1574-6941.2008.00520.x. [DOI] [PubMed] [Google Scholar]
- Delecluse HJ, Kremmer E, Rouault JP, Cour C, Bornkamm G, Berger F. The expression of Epstein-Barr virus latent proteins is related to the pathological features of post-transplant lymphoproliferative disorders. Am J Pathol. 1995;146:1113–1120. [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devesa SS, Blot WJ, Fraumeni JF., Jr Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer. 1998;83:2049–2053. [PubMed] [Google Scholar]
- Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412:99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–1201. doi: 10.1016/s0016-5085(03)00157-4. [DOI] [PubMed] [Google Scholar]
- Eriksson H. Steroids in germfree and conventional rats. Unconjugated metabolites of [4–14C]pregnenolone and [4–14C]corticosterone in faeces from female rats. Eur J Biochem. 1970;16:261–267. doi: 10.1111/j.1432-1033.1970.tb01080.x. [DOI] [PubMed] [Google Scholar]
- Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, Capelinha AF, Quint W, Caldas C, van Doorn LJ, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, de Perez AZ, Perez-Perez GI, Tseng CH, Blaser MJ. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut. 2008;57:16–24. doi: 10.1136/gut.2007.131672. [DOI] [PubMed] [Google Scholar]
- Francois F, Roper J, Joseph N, Pei Z, Chhada A, Shak JR, de Perez AZ, Perez-Perez GI, Blaser MJ. The effect of H. pylori eradication on meal-associated changes in plasma ghrelin and leptin. BMC Gastroenterol. 2011;11:37. doi: 10.1186/1471-230X-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol. 2007;81:9299–9306. doi: 10.1128/JVI.00537-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Gadelle D, Raibaud P, Sacquet E. beta-Glucuronidase activities of intestinal bacteria determined both in vitro and in vivo in gnotobiotic rats. Appl Environ Microbiol. 1985;49:682–685. doi: 10.1128/aem.49.3.682-685.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, Lord GM, Glickman JN, Glimcher LH. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–219. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault E, Fiola S, Olivier M, Gosselin J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol. 2007;81:8016–8024. doi: 10.1128/JVI.00403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloux K, Berteau O, El Oumami H, Beguet F, Leclerc M, Dore J. A metagenomic beta-glucuronidase uncovers a core adaptive function of the human intestinal microbiome. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4539–4546. doi: 10.1073/pnas.1000066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, McDougal A, Safe S. Estrogenic and antiestrogenic activities of 16alpha- and 2-hydroxy metabolites of 17beta-estradiol in MCF-7 and T47D human breast cancer cells. J Steroid Biochem Mol Biol. 1998;67:413–419. doi: 10.1016/s0960-0760(98)00135-6. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Manson JE, Colditz GA, Hunter DJ, Spiegelman D, Barbieri RL, Speizer FE. Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1998;90:1292–1299. doi: 10.1093/jnci/90.17.1292. [DOI] [PubMed] [Google Scholar]
- Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res (Phila) 2008;1:329–338. doi: 10.1158/1940-6207.CAPR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, Tham KT, Camorlinga M, Blaser MJ, Falkow S, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611–620. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvenpää P, Kosunen T, Fotsis T, Adlercreutz H. In vitro metabolism of estrogens by isolated intestinal micro-organisms and by human faecal microflora. Journal of Steroid Biochemistry. 1980;13:345–349. doi: 10.1016/0022-4731(80)90014-x. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J Biol Chem. 2011;286:12971–12982. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaaks R, Berrino F, Key T, Rinaldi S, Dossus L, Biessy C, Secreto G, Amiano P, Bingham S, Boeing H, et al. Serum sex steroids in premenopausal women and breast cancer risk within the European Prospective Investigation into Cancer and Nutrition (EPIC) J Natl Cancer Inst. 2005;97:755–765. doi: 10.1093/jnci/dji132. [DOI] [PubMed] [Google Scholar]
- Kabat GC, Chang CJ, Sparano JA, Sepkovie DW, Hu XP, Khalil A, Rosenblatt R, Bradlow HL. Urinary estrogen metabolites and breast cancer: a case-control study. Cancer Epidemiol Biomarkers Prev. 1997;6:505–509. [PubMed] [Google Scholar]
- Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- Kung CP, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF- kappaB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J Virol. 2010;84:6605–6614. doi: 10.1128/JVI.00344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste V, Lavergne A, de Thoisy B, Pouliquen JF, Gessain A. Genetic diversity and molecular evolution of human and non-human primate Gammaherpesvirinae. Infect Genet Evol. 2010;10:1–13. doi: 10.1016/j.meegid.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23:165–172. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- Le Clorennec C, Ouk TS, Youlyouz-Marfak I, Panteix S, Martin CC, Rastelli J, Adriaenssens E, Zimber-Strobl U, Coll J, Feuillard J, et al. Molecular basis of cytotoxicity of Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J Virol. 2008;82:6721–6733. doi: 10.1128/JVI.02250-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J, McCray AT. ‘Ome sweet’ omics-- A genealogical treasury of words. The Scientist. 2001;17:8–8. [Google Scholar]
- Lee IO, Kim JH, Choi YJ, Pillinger MH, Kim SY, Blaser MJ, Lee YC. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem. 2010;285:16042–16050. doi: 10.1074/jbc.M110.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science. 2008a;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008b;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Wei H, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105:2117–2122. doi: 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- Liebowitz D. Epstein-Barr virus and a cellular signaling pathway in lymphomas from immunosuppressed patients. N Engl J Med. 1998;338:1413–1421. doi: 10.1056/NEJM199805143382003. [DOI] [PubMed] [Google Scholar]
- Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Chang YT, Chen SC, Chuang YC, Chen YR, Lin CS, Chen JY. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene. 2005;24:2635–2646. doi: 10.1038/sj.onc.1208319. [DOI] [PubMed] [Google Scholar]
- Lombardi P, Goldin B, Boutin E, Gorbach SL. Metabolism of androgens and estrogens by human fecal microorganisms. J Steroid Biochem. 1978;9:795–801. doi: 10.1016/0022-4731(78)90203-0. [DOI] [PubMed] [Google Scholar]
- Lukanova A, Lundin E, Micheli A, Arslan A, Ferrari P, Rinaldi S, Krogh V, Lenner P, Shore RE, Biessy C, et al. Circulating levels of sex steroid hormones and risk of endometrial cancer in postmenopausal women. Int J Cancer. 2004;108:425–432. doi: 10.1002/ijc.11529. [DOI] [PubMed] [Google Scholar]
- Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Peltonen J, Laatikainen T, Pulkkinen M, Adlercreutz H. Excretion of progesterone metabolites and estriol in faeces from pregnant women during ampicillin administration. J Steroid Biochem. 1975;6:1339–1346. doi: 10.1016/0022-4731(75)90363-5. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- McBain AJ, Macfarlane GT. Ecological and physiological studies on large intestinal bacteria in relation to production of hydrolytic and reductive enzymes involved in formation of genotoxic metabolites. J Med Microbiol. 1998;47:407–416. doi: 10.1099/00222615-47-5-407. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- McKnight JL, Cen H, Riddler SA, Breinig MC, Williams PA, Ho M, Joseph PS. EBV gene expression, EBNA antibody responses and EBV+ peripheral blood lymphocytes in post-transplant lymphoproliferative disease. Leuk Lymphoma. 1994;15:9–16. doi: 10.3109/10428199409051672. [DOI] [PubMed] [Google Scholar]
- Meilahn EN, De Stavola B, Allen DS, Fentiman I, Bradlow HL, Sepkovic DW, Kuller LH. Do urinary oestrogen metabolites predict breast cancer? Guernsey III cohort follow-up. Br J Cancer. 1998;78:1250–1255. doi: 10.1038/bjc.1998.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita EM, Yang B, Lam KM, Crawford DH, Thorley-Lawson DA. A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell. 1995;80:593–601. doi: 10.1016/0092-8674(95)90513-8. [DOI] [PubMed] [Google Scholar]
- Moody CA, Scott RS, Amirghahari N, Nathan CO, Young LS, Dawson CW, Sixbey JW. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol. 2005;79:5499–5506. doi: 10.1128/JVI.79.9.5499-5506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R, Gordon JI. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schunemann HJ, Stanulla M, Yang J, Sepkovic DW, Trevisan M, et al. Estrogen metabolism and risk of breast cancer: a prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11:635–640. doi: 10.1097/00001648-200011000-00004. [DOI] [PubMed] [Google Scholar]
- Niederman JC, Evans AS, Subrahmanyan L, McCollum RW. Prevalence, incidence and persistence of EB virus antibody in young adults. N Engl J Med. 1970;282:361–365. doi: 10.1056/NEJM197002122820704. [DOI] [PubMed] [Google Scholar]
- Njie R, Bell AI, Jia H, Croom-Carter D, Chaganti S, Hislop AD, Whittle H, Rickinson AB. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J Infect Dis. 2009;199:31–38. doi: 10.1086/594373. [DOI] [PubMed] [Google Scholar]
- Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, Orentreich N, Vogelman JH, Friedman GD. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004;101:4250–4255. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn I, Hammond W, Brettschneider L, Starzl TE. Malignant lymphomas in transplantation patients. Transplant Proc. 1969;1:106–112. [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr. 2000:113–124. doi: 10.1093/oxfordjournals.jncimonographs.a024234. [DOI] [PubMed] [Google Scholar]
- Rea D, Fourcade C, Leblond V, Rowe M, Joab I, Edelman L, Bitker MO, Gandjbakhch I, Suberbielle C, Farcet JP, et al. Patterns of Epstein-Barr virus latent and replicative gene expression in Epstein-Barr virus B cell lymphoproliferative disorders after organ transplantation. Transplantation. 1994;58:317–324. [PubMed] [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57:1375–1385. doi: 10.1136/gut.2007.137539. [DOI] [PubMed] [Google Scholar]
- Roosendaal R, Kuipers EJ, Buitenwerf J, van Uffelen C, Meuwissen SG, van Kamp GJ, Vandenbroucke-Grauls CM. Helicobacter pylori and the birth cohort effect: evidence of a continuous decrease of infection rates in childhood. Am J Gastroenterol. 1997;92:1480–1482. [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe M, Lear AL, Croom-Carter D, Davies AH, Rickinson AB. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J Virol. 1992;66:122–131. doi: 10.1128/jvi.66.1.122-131.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg AA, Slaunwhite WR., Jr Studies on phenolic steroids in human subjects. II. The metabolic fate and hepato-biliary-enteric circulation of C14-estrone and C14-estradiol in women. J Clin Invest. 1957;36:1266–1278. doi: 10.1172/JCI103524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Muranaka Y, Fujimura R, Ishida H, Tazume S, Shimamura T. Normalization of reproductive function in germfree mice following bacterial contamination. Exp Anim. 1998;47:151–158. doi: 10.1538/expanim.47.151. [DOI] [PubMed] [Google Scholar]
- Song YJ, Kang MS. Roles of TRAF2 and TRAF3 in Epstein-Barr virus latent membrane protein 1-induced alternative NF-kappaB activation. Virus Genes. 2010;41:174–180. doi: 10.1007/s11262-010-0505-4. [DOI] [PubMed] [Google Scholar]
- Swart R, Ruf IK, Sample J, Longnecker R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. J Virol. 2000;74:10838–10845. doi: 10.1128/jvi.74.22.10838-10845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Zeng Y, Poisson A, Marti D, Guan L, Zheng Y, Deng H, Liao J, Guo X, Sun S, et al. Haplotype-dependent HLA susceptibility to nasopharyngeal carcinoma in a Southern Chinese population. Genes Immun. 2010;11:334–342. doi: 10.1038/gene.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley-Lawson DA, Duca KA. Pathogens cooperate in lymphomagenesis. Nat Med. 2007;13:906–907. doi: 10.1038/nm0807-906. [DOI] [PubMed] [Google Scholar]
- Toniolo PG, Levitz M, Zeleniuch-Jacquotte A, Banerjee S, Koenig KL, Shore RE, Strax P, Pasternack BS. A prospective study of endogenous estrogens and breast cancer in postmenopausal women. J Natl Cancer Inst. 1995;87:190–197. doi: 10.1093/jnci/87.3.190. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DW, Keppel CR, Schneider SE, Reese TA, Coder J, Payton JE, Ley TJ, Virgin HW, Fehniger TA. Latent herpesvirus infection arms NK cells. Blood. 2010;115:4377–4383. doi: 10.1182/blood-2009-09-245464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle HC, Brown J, Marsh K, Greenwood BM, Seidelin P, Tighe H, Wedderburn L. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature. 1984;312:449–450. doi: 10.1038/312449a0. [DOI] [PubMed] [Google Scholar]
- Woolcott CG, Shvetsov YB, Stanczyk FZ, Wilkens LR, White KK, Caberto C, Henderson BE, Le Marchand L, Kolonel LN, Goodman MT. Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: the Multiethnic Cohort Study. Endocr Relat Cancer. 2010;17:125–134. doi: 10.1677/ERC-09-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de Boni M, Isaacson PG. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342:575–577. doi: 10.1016/0140-6736(93)91409-f. [DOI] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynder EL, Gori GB. Contribution of the environment to cancer incidence: an epidemiologic exercise. J Natl Cancer Inst. 1977;58:825–832. doi: 10.1093/jnci/58.4.825. [DOI] [PubMed] [Google Scholar]
- Yager EJ, Szaba FM, Kummer LW, Lanzer KG, Burkum CE, Smiley ST, Blackman MA. gamma-Herpesvirus-induced protection against bacterial infection is transient. Viral Immunol. 2009;22:67–72. doi: 10.1089/vim.2008.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Nakamura T, Matsuo K, Tajika M, Kawai H, Ohmiya N, Niwa Y, Goto H, Nakamura S. Significance of CXCR3 expression in gastric low-grade B-cell lymphoma of mucosa-associated lymphoid tissue type for predicting responsiveness to Helicobacter pylori eradication. Cancer Sci. 2008;99:1769–1773. doi: 10.1111/j.1349-7006.2008.00883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137:588–597. doi: 10.1053/j.gastro.2009.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L, Alfieri C, Hennessy K, Evans H, O’Hara C, Anderson KC, Ritz J, Shapiro RS, Rickinson A, Kieff E, et al. Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease. N Engl J Med. 1989;321:1080–1085. doi: 10.1056/NEJM198910193211604. [DOI] [PubMed] [Google Scholar]
- Zullo A, Hassan C, Cristofari F, Andriani A, De Francesco V, Ierardi E, Tomao S, Stolte M, Morini S, Vaira D. Effects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol. 2010;8:105–110. doi: 10.1016/j.cgh.2009.07.017. [DOI] [PubMed] [Google Scholar]