

Abstract
There is a general need for engineering of protein-like molecules that organize into geometrically-specific superstructures on molecular surfaces, directing further functionalization to create richly textured, multi-layered assemblies. Here we describe a computational approach whereby the surface properties and symmetry of a targeted surface define the sequence and superstructure of surface-organizing peptides. Computational design proceeds in a series of steps that encode both surface recognition and favorable inter-subunit packing interactions. This procedure is exemplified in the design of peptides that assemble into a tubular structure surrounding single-walled carbon nanotubes (SWNTs). The geometrically-defined, virus-like coating created by these peptides converts the smooth surfaces of SWNTs into highly textured assemblies, with long-scale order, capable of directing the assembly of gold nanoparticles into helical arrays along the SWNT axis.
De novo protein design has historically been used to test the principles governing protein folding and assembly(1–3). These principles have also been extended to the design of structures capable of binding metal ions(4, 5), peptides(6–8), DNA(9, 10), inorganic materials(11), and proteins that catalyze reactions similar to those found in nature(12–15). However, protein design might have greater impact when applied to the engineering of controllable, structurally-defined molecular assemblies(16). A solution to this problem would enable the manipulation and organization of objects on the molecular and atomic levels – a major challenge of modern nanoscience.
We describe a general approach for designing molecules that assemble along geometrically-specific surfaces into a pre-defined superstructure. Earlier studies focused on amphiphilic peptides that encourage binding and assembly at soft interfaces(17–19), but without explicit consideration of interpeptide packing geometry that defines the nano- to macrostructure of the overall complex. A good design strategy for encoding a specific mode of assembly is to engineer a protein structural unit that presents a functional group compatible with the targeted surface and associates into a periodic superstructure with a geometric repeat matching that of the targeted substrate (Fig. 1A). However, an infinite continuum of such symmetry-matching arrangements can be generated out of common protein structural units. Thus, the most challenging aspect of designing such a surface-organizing assembly is the identification of a reasonable superstructure geometry, a problem we address in this study. Here, we apply our approach to design peptides that wrap SWNTs in a structurally-specific manner, creating a richly textured molecular surface. Previously studied biomolecules that interact with SWNTs include single-stranded DNA molecules (20, 21), nanotube-binding peptides selected by phage-display (22), and synthetic peptides with chemical features that favor SWNT binding (23, 24). Beyond interacting with and solubilizing SWNTs, a unique and relatively unexplored potential offered by biomolecules is the ability to program structurally-specific modes of surface assembly, enabling nucleation of further superstructure, functionalization, and manipulation (25).
Figure 1.

A) A simplified diagram of the surface assembly process(35). The design goal is to achieve ordered arrays of peptide subunits on the target surface (global order; top outcome in figure) and to avoid kinetic traps of surface binding (local order; bottom outcome), which can be caused by overly strong interactions between individual subunits and the surface. B)-D) illustrate the general design framework on the example of decorating a graphene sheet. B) Selection rule 1: Cβ methyl group of Ala and the α-helix are picked as the surface-contacting functional group and structural unit, respectively. C) Selection rule 2: two possible unit cells, a single helix and an antiparallel dimer, are shown along with the corresponding Bravais-lattice vectors defining unit-cell images. D) Assemblies containing undesignable interfaces are discarded in selection rule 3 (differences in designability are usually much more subtle than illustrated here for clarity). E) Optimal template geometry designed to target the surface of an (3,8) SWNT surface (helical pattern of the SWNT shown with black benzenoid rings).
The design process consists of three selection rules, which successively restrict the space of possible peptide-surface assemblies, and ultimately dictate peptide sequence (see Fig. 1). Selection rule 1 identifies groups compatible with the target surface, as well as a protein structural unit capable of displaying such groups in a productive manner (see Fig. 1B). Selection rule 2 defines the intersubunit packing of these units on the target surface. Symmetry operations are used to create an elementary unit cell, which is then replicated to match the geometric repeat of the surface (see Fig. 1C). A continuum of assemblies remains possible at this point, each creating new protein-protein interfaces, within the unit cell and between neighboring unit cells. The key insight is provided by selection rule 3, which ensures that these interfaces are designable – that is, they can be accommodated, in a stable and specific manner (see Fig. 1D). Designable protein structural motifs occur frequently in nature, such that a structural database search can be used to assess the feasibility of specific intersubunit packing in addition to revealing sequence features that encode it(26). In summary, the three selection rules define the intrinsic recognition motif, and its packing into a higher-order assembly in accord with the long-range order of the underlying surface.
These selection rules emerged from our efforts to engineer peptides targeting common species of SWNTs. In picking a functional group for contacting the SWNT (selection rule 1), we avoided strong hydrophobic recognition motifs employed in earlier studies(23), instead relying on weaker protein-SWNT interactions to encourage the cooperative formation of the intended higher-order assembly (see Fig. 1A). We therefore chose the Cα methylene of Gly or the Cβ methyl of Ala, presented in a repeating manner on an α-helix as the elementary structural unit.
Selection rule 2 stipulates that the arrangement of protein structural units should match the symmetry of the underlying surface. The cylindrical shape of a SWNT suggested an assembly with rotational or rotational screw symmetry, so we considered α-helical coiled coils forming a supercoil along the SWNT axis (Fig. 1E). Common SWNTs have relatively hydrophobic surfaces, and radii in the range of ~3.75–4.1 Å (for the (5,6), (5,7), and (3,8) chiralities). This, together with the choice of a small sidechain for surface recognition defined the radius of the coiled coil to be around 9 Å, restricting the stoichiometry of the bundle to between 5 and 7 units (26). We chose an antiparallel hexamer over a parallel α-helical bundle to exploit the additional degree of freedom (axial shift), available to antiparallel interfaces (26). Although SWNTs are relatively smooth, their electronic surface is not entirely homogeneous and we considered that it may be advantageous in design to match the pitch angle of the helices formed by overlapping benzenoid rings down SWNT surfaces (see Fig. 1E) (27).
Although the first two selection rules identified a specific topology, a large number of possible bundles with reasonable interfaces could be generated based on the four remaining parameters: the inter-helical separation, starting helical phase, superhelical pitch, and helical axial shift. Allowing fifty discrete values for each parameter within geometrically feasible ranges results in 6,250,000 possible design templates. We had previously found that no more than 1 in 100 α-helical coiled coils constructed using geometrically feasible parameter values are in fact designable with natural amino acids (26). Therefore, in selection rule 3, we searched for assembly parameters that optimized the designability of the modeled interfaces, leading to a single most designable template for each targeted SWNT.
To assess designability, we used a rapid distance-matrix based method for searching tertiary motifs in the Protein Data Bank (PDB) that are geometrically similar to the query interface (Fig. 2A). The number of matches within a given cutoff of the query interface amounts to a metric of its designability, and sequences of the matches help define features encoding intersubunit packing. Since this information is gathered from a wide range of structural contexts, sequences of the matches should be highly divergent at all positions except those that are particularly critical to the stability and structural specificity of the motif. The conserved positions are held constant in design, while the variable positions provide handles for encoding additional features, such as interaction with SWNTs, modulation of solubility, stability and specificity, or recruitment of additional functionality.
Figure 2.

Designability analysis in selection rule 3. The two unique interfaces of an antiparallel homo-hexamer are designated here as AA′ and A′A and illustrated in the left and right columns, respectively. A) Number of matches as a function of the Cα RMSD cutoff. B) Structural variation in the top 100 best matches. Tube thickness equals to the mean square deviation of the corresponding atom within the ensemble of top 100 matches. Blue to red indicates the N-to-C terminal direction. C) Sequence logo diagrams for the two interfaces of the (3,8)-targeted template derived from unique matches with Cα RMSD below 0.5 Å and 0.6 Å for AA′ and A′A, respectively(36). Heptad assignments, in the context of the full hexamer, are indicated for each sequence position.
The selection rules were implemented into an automated procedure and applied to design of assemblies on the surfaces of SWNTs (3,8), (5,7) and (5,6), matching both size and pitch angles to each SWNT (corresponding pitch angles were −14.7°, −5.5°, and −3°, respectively (27)). An antiparallel hexamer has two geometrically distinct helix-helix interfaces (Fig. 2A inset). The designability of these interfaces in the optimal template was starkly different among the three pitch angles (Fig. 2A-B). For example, the optimal −14.7° template identified 119 and 89 natural motifs that were within 0.6 Å Cα RMSD of the two helix-helix interfaces comprising this assembly. The corresponding values for the best −5.5° structure were 4 and 7, and none were found within this cutoff for the −3° structure. Thus, the −14.7° template would be considered a much more designable target using common, genetically encoded amino acids.
Profiles of residue propensities in aligned sequences (Fig. 2C) show that optimal designability is reached when the two unique interfaces of the hexamer are quite different – one should be a “tight” Alacoil-like interface, while the other should resemble an antiparallel leucine zipper-like motif. Note that this information is obtained automatically, without resorting to extensive sidechain repacking calculations on candidate backbone structures.
Having chosen the −14.700B0 structure as the target, we followed two paths to complete the design process. In the first, a sequence was computationally optimized to adopt this hexameric antiparallel bundle around the (3,8) SWNT, constraining the strongly conserved positions from propensity profiles (positions d and e; Fig. 2C). Standard computational design techniques were applied to select the remaining variable positions (section 1.2 of Supporting Material (27)) producing two sequences, HexCoil-Gly and HexCoil-Ala (see Fig. 3A), differing only in the identity of the SWNT-contacting position (Gly or Ala, respectively).
Figure 3.

A) Sequences of designed peptides, native DSD and control peptides. B) Crystal structure of HexCoil-Ala (left; asymmetric unit; mesh represents electron density contoured at 1.5σ; see Table S3) and its comparison to the asymmetric unit of the designed oligomer (right; gray structures corresponds to the design). C) The Ala-rich surface of the asymmetric unit of the HexCoil-Ala crystal structure is well poised to interact with a SWNT. D) Model structure of HexCoil-Gly with a (3,8) SWNT. Blue-to-red indicates N-to-C terminal direction. E) Crystal structure of native DSD. F) Model of DSD-Ala with a (3,8) SWNT. Van der Waals surfaces are shown semi-transparently in C), D), E), and F).
In a second approach we searched the PDB for a more complex scaffold that embedded the full −14.7 00B0 hexameric bundle within it and would be amenable to further design. A structural-similarity search identified a remarkably similar bundle (0.9 Å Cα RMSD over 156 residues) in the inner ring of helices of a domain-swapped helical bundle (called DSD; PDB code 1G6U; Fig. 3E, S4-S5)(28). Additionally, the strong sequence features discovered for the (3,8)-optimal template (Fig. 2C) were also present in DSD. Therefore, the central pore-lining Glu and Lys residues of DSD were converted to Gly or Ala to accommodate a SWNT in peptides designated DSD-Gly and DSD-Ala.
The hierarchic principles of our design approach suggest that a large portion of the driving force for assembly should originate from modestly favorable helix-helix interactions, which should stabilize the basic antiparallel dimeric unit, even in the absence of SWNTs. Without the underlying solid substrate, the hexameric bundle structure might not be the most stable one formed, but we expected to see assembly into related bundles in which the dimeric interface was preserved. Indeed, sedimentation equilibrium analytical ultracentrifugation (AUC) showed DSD-Gly and DSD-Ala to exist in a dimer-hexamer equilbirium between 10 µM to 100 µM peptide concentration (Fig. S7). HexCoil-Ala associated into tetramers (Fig. S8), whose structure was solved using diffraction data extending to 2.44 Å resolution by X-ray crystallography (see Fig. 3). The asymmetric unit consists of an antiparallel dimer, whose structure is within 1.2 Å of the designed model (calculated over the backbone of 20 central residues per monomer). The designed Ala-rich face is well-situated to interact with the surface of the SWNT (Fig. 3C). Finally, far UV circular dichroism spectroscopy (CD) of these peptides confirmed their helical content in solution and when bound to SWNT (Fig. S9). Interestingly, HexCoil-Gly, which contains multiple helix-destabilizing Gly residues, assembled only in the presence of SWNTs (Fig. S9) similar to previously designed surface-binding peptides(29, 30).
The peptides formed water-soluble assemblies of SWNTs, producing aqueous suspensions that were stable for months. Two-dimensional photoluminescence (2D-PL) spectra were used to identify individual SWNT chiralities through their characteristic resonances (31), and to rule out aggregation of SWNTs, which induces energy transfer between different species (32). Designed peptides produce SWNT suspensions with 2D-PL peaks corresponding to (5,6), (5,7), and (3,8) chiralities (Fig. 4A–C). The de novo designed peptides HexCoil-Ala and HexCoil-Gly sequester significantly more SWNTs into solution, compared to DSD variants (Fig. 4B). Interestingly, though the (3,8) species is a minor product in the mixture of SWNTs used in our experiments, HexCoil-Ala and HexCoil-Gly show a dominant peak corresponding to this chirality (Fig. 4C). This is of particular significance given that the target substrate for these designs was indeed the (3,8) species SWNT.
Figure 4.
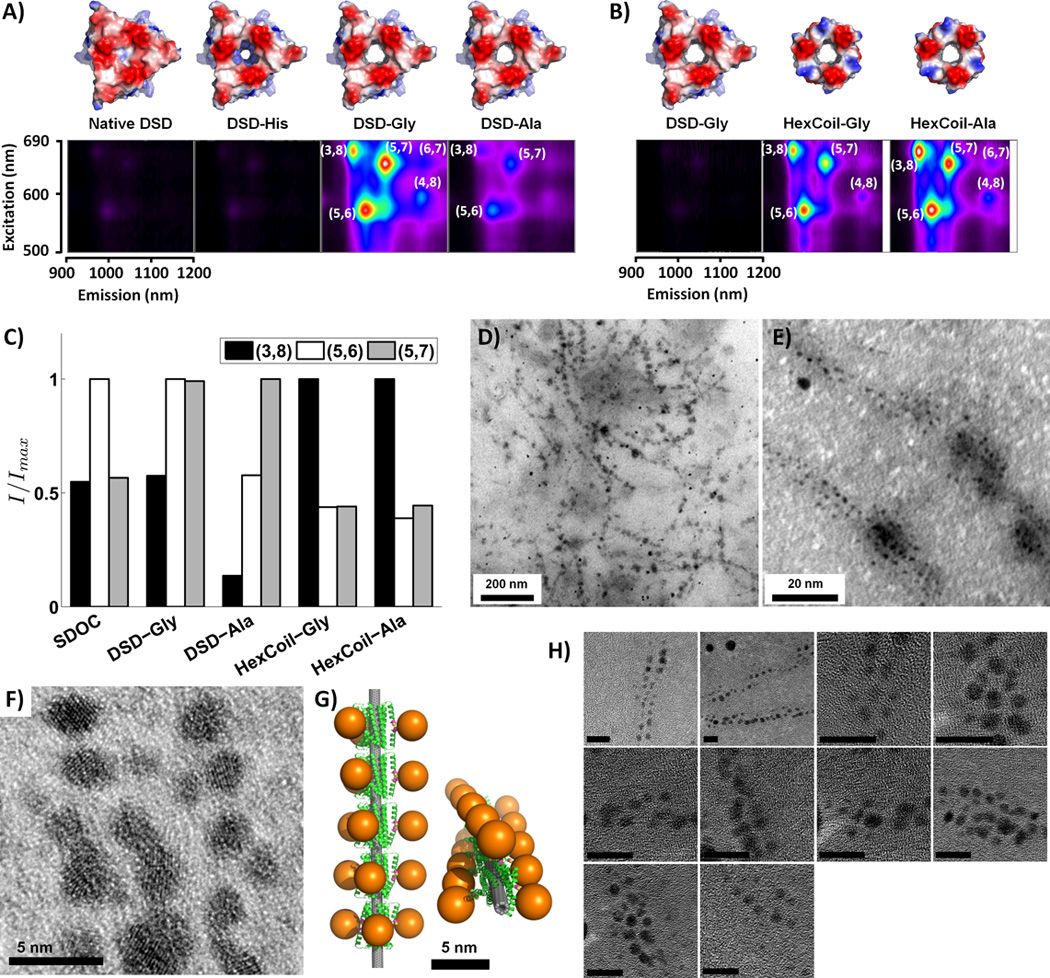
2D-PL and TEM analysis of SWNT/peptide complexes. A)-B) 2D-PL spectra of SWNT suspensions produced by A) DSD-based peptides and B) de novo designed peptides (pseudocolor scale is internally consistent within each section). C) Fitting photoluminescence maps to a sum of 2-dimensional Lorentzians provides the total intensity weight contribution of each SWNT species, I. Shown are relative contributions of the three SWNT types with the strongest signals (e.g. (3,8), (5,6) and (5,7)) in suspensions produced by four peptides and, as reference, the common surfactant sodium deoxy cholate (SDOC; spectrum in Fig. S14). In each case, weights are normalized to the most contributing species. D)-F) and H) show TEM images of gold nanoparticles grown on Cys-modified DSD-Gly hexamers wrapped around individual SWNTs. E) is a higher magnification version of D) and F) contains a high-resolution TEM image. Scale bars are 10 nm in H). G) Computational model of the complex. Gold particles are represented with 30-Å diameter spheres attached to the Cys Sγ atoms of the Cys-modified DSD-Gly.
A number of control peptides were prepared to evaluate the structural mode of SWNT/peptide assembly. To probe the role of the small Ala and Gly residues contacting the SWNT, native DSD and an analog of DSD-Gly with two of its Gly residues changed to His were studied. Furthermore, to test the role of helix-helix packing in the HexCoil-Gly and HexCoil-Ala, the apolar residues at the “d” and “e” positions that pack at the two distinct helix-packing interfaces, and the SWNT-contacting “a” position, were interchanged (Fig. S12). The resulting peptides, cHexCoil-Gly and cHexCoil-Ala (Fig. 3A), have identical amino-acid compositions, hydrophobicity, and helical faces, and nearly identical hydrophobic moments (a measure of amphiphilicity) as their parents, but differ in their abilities to engage in the detailed packing interactions intended to stabilize surface assemblies. These negative control peptides (DSD, DSD-His, cHexCoil-Gly, or cHexCoil-Ala) were very inefficient at solubilizing SWNTs (Fig. 4A, S12), verifying the intended mode of SWNT contact and suggesting that the success of our designs rests upon the ability to form favorable inter-subunit interactions and a higher-order assembly.
Once SWNTs are wrapped by peptides in a structurally determined way, their solvent-exposed surfaces can be further elaborated to direct the assembly – or even the synthesis – of a third biological or non-biological layer. To illustrate this, we used the peptide/SWNT assembly to direct nucleation and assembly of gold nanoclusters in a geometrically-defined manner. The DSD-Gly peptide appeared advantageous for these studies, as its peripheral helices packing against the central hexameric ring allow for the construction of independent outward-facing binding sites along a larger-radius superhelix, facilitating microscopic imaging. A single Cys was introduced near the N-terminus of DSD-Gly, such that pairs of symmetry-related helices created convergent gold-binding sites (Figs. 4G and S11). Addition of Au(III) under reducing conditions led to the appearance of 2 to 4 nm gold clusters visible by TEM (Fig. 4, S3). Consistent with the design model, the pattern of spots is linear and systematically in-phase, and the observed inter-particle spacing of 47 Å in very good agreement with the model’s prediction of 52 Å (Figs. S1–3) (27).
The selection rules described here provide an objective reproducible method to design surface-binding peptides. Their aim is to assure that all effects are favorable for the formation of the intended assembly. Optimal interaction geometry between protein units, physicochemical compatibility between the surface and the protein, and matching between the geometry of the assembly and the symmetry of the substrate are all encoded at the same time in a “minimally frustrated” design. In applying this strategy to SWNT surfaces, we expected that the dominant surface features would be radius and the water-repellant nature, thus the driving force for assembly would originate primarily from matching the size and hydrophobicity of the SWNT, as well as inter-subunit packing. Indeed this strategy worked. The intended SWNTs were bound, thereby converting the very short-scale periodicity of a SWNT surface to long-scale periodicity of a SWNT/protein assembly, as illustrated by using the complex to further direct the nucleation of an additional layer of gold nanoparticles.
SWNTs present a challenging case for organizing structurally-specific assemblies due to their relatively featureless surfaces. Other molecular surfaces, such as ionic structures or boron nitride nanotubes(33), are likely to have much higher heterogeneity in presented atomic groups, leading to better potential for anisotropy with respect to surface interactions. In such cases, we would expect the orientation of the coating assembly relative to the crystal lattice would be a very important discriminator and director of order. It is encouraging that even with the rather simple and smooth surfaces of SWNTs we have already achieved a significant level of success. The DSD versus HexCoil series of peptides illustrate different endpoints of the design process. Whereas the DSD scaffold was serendipitously discovered to approximately match the assembly geometry optimized via our approach, HexCoil-Ala and HexCoil-Gly were designed de novo to bind the (3,8) SWNT. Thus, it is encouraging that the latter peptides are more efficient and significantly more selective agents for solubilizing the desired target, showing a strong preference for solubilizing this tube type despite it being a minor component in a mixture of SWNTs. It is possible that the interfaces in the HexCoil peptides, which are unencumbered by the presence of a more involved tertiary packing, are sufficiently preorganized to allow selective binding, but not so rigid as to require a perfect fit for selective recognition to take place.
In summary, biological systems specialize in assembly, and hybrid nano-bio structures provide a powerful way to direct the assembly and tune the properties of nanomaterials. Computational protein design provides the means to do so in a highly directed and functionally relevant manner (34).
Supplementary Material
References
- 1.Cordes MH, Davidson AR, Sauer RT. Curr Opin Struct Biol. 1996 Feb;6:3. doi: 10.1016/s0959-440x(96)80088-1. [DOI] [PubMed] [Google Scholar]
- 2.Dahiyat BI, Mayo SL. Science. 1997 Oct 3;278:82. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlman B, et al. Science. 2003 Nov 21;302:1364. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh D, Pecoraro VL. Curr Opin Chem Biol. 2005 Apr;9:97. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun JR, et al. Biopolymers. 2005;80:264. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 6.Ghirlanda G, Lear JD, Lombardi A, DeGrado WF. J Mol Biol. 1998 Aug 14;281:379. doi: 10.1006/jmbi.1998.1912. [DOI] [PubMed] [Google Scholar]
- 7.Reina J, et al. Nat Struct Biol. 2002 Aug;9:621. doi: 10.1038/nsb815. [DOI] [PubMed] [Google Scholar]
- 8.Grigoryan G, Reinke AW, Keating AE. Nature. 2009 Apr 16;458:859. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashworth J, et al. Nature. 2006 Jun 1;441:656. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JS, Pabo CO. Proc Natl Acad Sci U S A. 1998 Mar 17;95:2812. doi: 10.1073/pnas.95.6.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masica DL, Schrier SB, Specht EA, Gray JJ. J Am Chem Soc. 2010 Sep 8;132:12252. doi: 10.1021/ja1001086. [DOI] [PubMed] [Google Scholar]
- 12.Rothlisberger D, et al. Nature. 2008 May 8;453:190. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 13.Jiang L, et al. Science. 2008 Mar 7;319:1387. [Google Scholar]
- 14.Haring D, Distefano MD. Bioconjug Chem. 2001 May–Jun;12:385. doi: 10.1021/bc000117c. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan J, DeGrado WF. Proc Natl Acad Sci U S A. 2004 Aug 10;101:11566. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairman R, Akerfeldt KS. Curr Opin Struct Biol. 2005 Aug;15:453. doi: 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Degrado WF, Lear JD. J Am Chem Soc. 1985 December;107:7684. 1985. [Google Scholar]
- 18.Segman S, Lee MR, Vaiser V, Gellman SH, Rapaport H. Angew Chem Int Ed Engl. 2010;49:716. doi: 10.1002/anie.200904566. [DOI] [PubMed] [Google Scholar]
- 19.Rapaport H. Supramolecular Chemistry. 2006;18:445. [Google Scholar]
- 20.Zheng M, et al. Science. 2003 Nov 28;302:1545. doi: 10.1126/science.1091911. [DOI] [PubMed] [Google Scholar]
- 21.Tu X, Manohar S, Jagota A, Zheng M. Nature. 2009 Jul 9;460:250. doi: 10.1038/nature08116. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, et al. Nat Mater. 2003 Mar;2:196. doi: 10.1038/nmat833. [DOI] [PubMed] [Google Scholar]
- 23.Dieckmann GR, et al. J Am Chem Soc. 2003 Feb 19;125:1770. doi: 10.1021/ja029084x. [DOI] [PubMed] [Google Scholar]
- 24.Ortiz-Acevedo A, et al. J Am Chem Soc. 2005 Jul 6;127:9512. doi: 10.1021/ja050507f. [DOI] [PubMed] [Google Scholar]
- 25.Katz E, Willner I. Chemphyschem. 2004 Aug 20;5:1084. doi: 10.1002/cphc.200400193. [DOI] [PubMed] [Google Scholar]
- 26.Grigoryan G, Degrado WF. J Mol Biol. 2010 January;405:1079. doi: 10.1016/j.jmb.2010.08.058. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Materials and methods are available as supporting material on Science Online
- 28.Ghirlanda G, Lear JD, Ogihara NL, Eisenberg D, DeGrado WF. J Mol Biol. 2002 May 24;319:243. doi: 10.1016/S0022-2836(02)00233-4. [DOI] [PubMed] [Google Scholar]
- 29.Capriotti LA, Beebe TP, Jr, Schneider JP. J Am Chem Soc. 2007 Apr 25;129:5281. doi: 10.1021/ja070356b. [DOI] [PubMed] [Google Scholar]
- 30.Nygren P, Lundqvist M, Broo K, Jonsson BH. Nano Lett. 2008 Jul;8:1844. doi: 10.1021/nl080386s. [DOI] [PubMed] [Google Scholar]
- 31.O'Connell MJ, et al. Science. 2002 Jul 26;297:593. doi: 10.1126/science.1072631. [DOI] [PubMed] [Google Scholar]
- 32.Torrens ON, Milkie DE, Zheng M, Kikkawa JM. Nano Lett. 2006 Dec;6:2864. doi: 10.1021/nl062071n. [DOI] [PubMed] [Google Scholar]
- 33.Golberg D, Bando Y, Tang CC, Zhi CY. Adv. Mater. 2007;19:2413–2432. [Google Scholar]
- 34.This work was supported by the NSF MRSEC DMR05-20020 grant to J.M.K., M.D. and W.F.D, the NIH grant number GM54616 to W.F.D., NSF NSEC grant to W.F.D., and NIH grant number 5F32GM084631-02 to GG. K.A. acknowledges support from the Roy and Diana Vagelos Program in the Molecular Life Sciences and L.W. acknowledges funding from the NSF- IGERT program (Grant DGE-0221664). We would like to thank Dr. Amy E. Keating for comments on the manuscript.
- 35.Chakraborty AK, Golumbfskie AJ. Annu Rev Phys Chem. 2001;52:537. doi: 10.1146/annurev.physchem.52.1.537. [DOI] [PubMed] [Google Scholar]
- 36.Crooks GE, Hon G, Chandonia JM, Brenner SE. Genome Res. 2004 Jun;14:1188. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.