

Abstract
Corynebacterineae is a specific suborder of Gram-positive bacteria that includes Mycobacterium tuberculosis and Corynebacterium glutamicum. The cell wall of these bacteria is composed of a heteropolymer of peptidoglycan (PG) linked to arabinogalactan (AG), which in turn is covalently associated with an atypical outer membrane, here called mycomembrane (M). The latter structure has been visualized by cryo-electron microscopy of vitreous sections, but its biochemical composition is still poorly defined, thereby hampering the elucidation of its physiological function. In this report, we show for the first time that the mycomembrane-linked heteropolymer of PG and AG (M-AG-PG) of C. glutamicum can be physically separated from the inner membrane on a flotation density gradient. Analysis of purified M-AG-PG showed that the lipids that composed the mycomembrane consisted almost exclusively of mycolic acid derivatives, with only a tiny amount, if any, of phospholipids and lipomannans, which were found with the characteristic lipoarabinomannans in the plasma membrane. Proteins associated with or inserted in the mycomembrane were extracted from M-AG-PG with lauryl-dimethylamine-oxide (LDAO), loaded on an SDS-PAGE gel, and analyzed by tandem mass spectrometry or by Western blotting. Sixty-eight different proteins were identified, 19 of which were also found in mycomembrane fragments released by the terminal-arabinosyl-transferase-defective ΔAftB strain. Almost all of them are predicted to contain a signal sequence and to adopt the characteristic β-barrel structure of Gram-negative outer membrane proteins. These presumed mycomembrane proteins include the already-known pore-forming proteins (PorA and PorB), 5 mycoloyltransferases (cMytA, cMytB, cMytC, cMytD, and cMytF), several lipoproteins, and unknown proteins typified by a putative C-terminal hydrophobic anchor.
INTRODUCTION
The cell envelope of mycobacteria is essential for virulence and forms a very efficient permeability barrier that contributes to their high resistance to hydrophilic drugs. Its ultrastructure and organization has been intensively studied, especially during recent decades. Considerable amounts of data have been accumulated, both from mycobacterial species and from Corynebacterium glutamicum, a very useful model organism. Our current view is still essentially based on the model of Minnikin (39), who was the first to propose the presence of an outer membrane-like structure (or mycomembrane [6]) covalently linked to an underlying polymer of arabinogalactan (AG) and peptidoglycan (PG). Several lines of evidence, and more particularly electrophysiological data combined with lipid analysis, have since supported this model. In 2008, cryo-electron microscopy of vitreous sections provided a definitive proof for the presence of the mycomembrane in these very atypical Gram-positive bacteria (24, 65).
Characterization of the mycomembrane is essential for understanding the physiology and the pathogenicity of these bacteria. Identification of the components of the uptake and secretion machineries is a challenging research avenue for the next decade. Despite tremendous efforts on deciphering the mycobacterial cell envelope, biochemical characterization of the mycomembrane is still far from being completed. The precise lipid composition is not known, and only a few proteins have been identified and characterized so far. In Gram-negative bacteria, such as Escherichia coli, there are about 60 outer membrane proteins (OMPs). Nearly all integral OMPs adopt a β-barrel conformation, and most of them function as open (porins) or gated channels that allow the uptake or the secretion of hydrophilic molecules. In mycobacteria, MspA is the archetype of mycomembrane proteins and represents the major diffusion pathway for small hydrophilic solutes in Mycobacterium smegmatis (58, 59). Its structure has been solved at atomic resolution (21) and has revealed a new protein architecture with an octameric goblet-like structure delimitating a 10-nm pore with a basal part reminiscent of the β-barrel organization of Gram-negative porins. Similarly, in C. glutamicum, it has been shown that PorH-PorA (5) is also a pore-forming protein complex isolated from cell envelopes, but in that case, its physiological function has not been clearly demonstrated (17). Its structure seems to be very different from that of MspA and consists of a hetero-oligomer of the two very small polypeptides PorH and PorA. Interestingly, PorH-PorA in vitro activity is strictly dependent on a posttranslational modification of both peptides by a mycolic acid residue (25). For Mycobacterium tuberculosis, OmpATb has been described as a pore-forming protein that contributes to the adaptation of cells to the acidic environment of the phagosome (40). However, Teriete and coworkers (62) have recently shown that the protein adopts a mixed α/β-structure, suggesting that it may not be inserted into the outer mycolate membrane as previously thought. Additional proteins are putatively anchored to or associated with the mycomembrane. These include mycoloytransferases (11), some glycosylated proteins (Rpf proteins) (23, 37), the S-layer protein of C. glutamicum (11), and some virulence factors, such as mycobacterial lipoproteins (31) or the Erp protein (29). However, their precise localization has not always been carefully examined and awaits future studies.
From in silico analysis, two independent studies have proposed a list of putative OMPs of M. tuberculosis based essentially on β-barrel computational predictions (46, 56). The two lists differ significantly, which suggests that a quite important number of false-positive proteins may have been predicted. More recently, Mah and coworkers have used the same basic approach but have extended their predictions by including genomic data from seven mycobacterial species (36). Although this is probably the most accurate prediction presently available, the proposed list is very large and some pitfalls are probably still expected. The difficulty of the in silico method is due mainly to the scarcity of experimental data concerning the structure of mycobacterial OMPs that can be used to benchmark the computational method.
Experimental methods could represent a promising alternative for identifying OMPs but also lipoproteins, OMPs that would not contain a classical signal peptide, and putative OMPs with large periplasmic or extracellular domains, which would be missed by in silico methods. Because of the covalent link between the mycolate layer and the arabinogalactan-peptidoglycan skeleton, isolation of the mycomembrane appears to be very challenging and only a few studies have been reported so far. Rezwan and coworkers (54) have described a quite efficient method to isolate the plasma membrane from the mycomembrane-containing cell wall in Mycobacterium smegmatis and Mycobacterium bovis by differential centrifugation. Because the fractions were slightly contaminated by the plasma membrane, the protein composition could not be determined. More recently, we reported the spontaneous shedding in the culture supernatant of mycomembrane fragments from a C. glutamicum ΔaftB strain (11). These fragments were virtually uncontaminated by other cell fractions, which allowed us to identify the most abundant mycomembrane-associated proteins by mass spectrometry. These include 5 mycoloyltransferases encoded by C. glutamicum that are major contributors to mycomembrane building. In this article, using fractionation on sucrose density gradients, we managed, for the first time, to isolate and to biochemically characterize a highly purified cell wall fraction corresponding to the native mycomembrane-arabinogalactan-peptidoglycan (M-AG-PG) complex of C. glutamicum. Proteins of the mycomembrane extracted either from the M-AG-PG complex or from ΔaftB outer membrane fragments were identified by tandem mass spectrometry analysis. Altogether, these results constitute the first complete set of biochemical data of the outer mycolate membrane of Corynebacterineae and represent a significant breakthrough in the functional characterization of this atypical structure.
MATERIALS AND METHODS
Materials.
High-performance liquid chromatography (HPLC)-grade acetonitrile (ACN), Coomassie brilliant blue R-250, dithiothreitol (DTT), formic acid (FA), iodoacetamide (IAM), and trifluoroacetic acid (TFA) were purchased from Sigma (St. Quentin Falavier, France). 4-(2-Aminoethyl)benzenesulfonyl fluoride (AEBSF) was from Acros Organics.
Bacterial strains and growth conditions.
All strains of C. glutamicum were derivatives of ATCC 13032 RES167 (20) and were grown in liquid brain heart infusion (BHI) medium at 30°C. ΔaftB (11) and ΔporA (16) mutant strains have been described elsewhere. E. coli DH5α was used for cloning manipulations and was grown in Luria-Bertani medium at 37°C. E. coli and C. glutamicum were prepared for electroporation as described previously (10). All plasmids introduced into C. glutamicum were constructed from the shuttle cloning vector pCGL482 (Cmr) (47). Chloramphenicol (Cm) was used at 25 μg/ml for E. coli and at 10 μg/ml for C. glutamicum.
Expression vectors, PCR, and nucleotide sequencing.
The porHA operon was amplified by PCR from C. glutamicum ATCC 13032 genomic DNA with the forward primer 5′-ATTAGGATCCCGGCGTGCCAAAGGGG-3′ and the reverse primer 5′-ATTACCCGGGCGAGCCGTTGTTAAGTAG-3′. The resulting PCR product contained the porHA open reading frame with 284 bp upstream of porH to direct transcription and translation (5). BamHI and XmaI restriction sites (underlined) were used to clone the PCR product into pCGL482. Vector pCGL1052 (55) was used to express FbpA (under its own promoter and sequence signal). After cloning, DNA fragments were sequenced at Eurofin MWG Operon.
Cell lysis and fractionation on sucrose density gradient.
Typically, 300 ml of 13032 RES167 cells were grown in BHI medium at 30°C and 200 rpm for 24 h, centrifuged at 5,000 × g for 15 min, and washed twice with 20 mM Tris-HCl buffer (pH 8.0). The pellet was frozen at −20°C before resuspension in 15 ml (final volume) of Tris buffer (optical density [OD] = 200) in the presence of 0.1 mg/ml of Pefabloc (AEBSF) and 1 mM EDTA. Bacterial cells were broken by three passages through a French press cell at 20,000 lb/in2 at 4°C. The resulting lysate (about 50% of lysis as determined by optical density) was centrifuged at very low speed (4,000 × g) for 15 min. The supernatant was carefully recovered and layered on a sucrose cushion (2 ml of 56% and 2 ml of 20% [wt/wt]) in SW41 12.5-ml tubes. After 2 h at 35,000 rpm, membranes were recovered at the 56%-20% interface, diluted with 25 mM HEPES (pH 7.4) buffer, and washed twice before resuspension in the same buffer. Membranes were finally layered on a sucrose step gradient (56% to 35%) in SW55Ti 4.8-ml tubes. After centrifugation for 42 h, two distinct bands (fraction 1 and fraction 2) were detected. For NADH oxidase measurements and anti-PorA Western blotting detections, the tubes content was collected as 250-μl aliquots from the top to the bottom. For other analysis, F1 and F2 were collected separately, washed twice in the absence of sucrose, and finally resuspended in 300 μl of HEPES buffer. In order to test F1 and F2 solubility in the presence of SDS, aliquots from both fractions were either directly centrifuged at 65,000 rpm for 30 min in a TLA 100.3 rotor or previously incubated in HEPES buffer containing 4% SDS and heated at 100°C for 30 min. The resulting pellets were visually compared.
Freeze fracture EM.
A small droplet of the purified F1 or F2 fraction recovered from the gradient was placed on the copper holder and quenched in liquid propane cooled with nitrogen. The frozen sample was fractured at −125°C in vacuo of about 10−5 Pa with a liquid-nitrogen-cooled knife in a Balzers 300 freeze-etching unit as described previously (1). The fractured sample was replicated with a 1.0- to 1.5-nm deposit of platinum-carbon and coated with a 20-nm carbon film. The Pt/C replica was cleaned with 2% SDS, washed with water, transferred onto a copper electron microscopy (EM) grid, and observed with a Philips CM100 electron microscope.
Lipid and sugar analysis from purified F1 or F2 fractions.
Lipids from the F2 fraction were extracted by the method of Bligh and Dyer (9). Briefly, wet F2 pellet from cell envelope fractionation was incubated in the one-phase solvent system CHCl3-CH3OH (1:2 [vol/vol]) for 16 h at room temperature. Then, CHCl3 and H2O (1:1 [vol/vol]) were added to obtain two phases: the organic lower phase, containing lipids, and the aqueous upper phase, containing polar compounds. The organic phase was dried under nitrogen, and lipids were resuspended in a minimal volume of CHCl3 and deposited on silica gel Durasil-25 plates (0.25 mm, 10 by 20 cm; Nacherey-Nagel), which were developed in CHCl3-CH3OH-H2O (65/25/4 [vol/vol/vol]). Glycolipids were revealed by spraying the thin-layer chromatography (TLC) plates with 0.2% (wt/vol) anthrone in concentrated H2SO4, followed by heating at 110°C. The aqueous phase of the partition was analyzed to determine the sugar composition of the F2 fraction by subjecting dry fraction to methanolysis with anhydrous 1.5 M HCl-CH3OH for 16 h at 80°C. The resulting methyl glycosides were then dried under nitrogen (traces of acids were removed by two coevaporations with methanol) and trimethylsilylated (61) before gas chromatography (GC) analysis. GC analysis was performed using a Hewlett-Packard 4890 chromatograph equipped with a fused silica OV1 capillary column (0.3 mm by 25 m). The temperature program was 60°C to 100°C with an increasing gradient of 20°C/min and then 100°C to 310°C with an increasing gradient of 5°C/min.
Lipoglycan identification.
Fractions F1 and F2 were solubilized in water and incubated with proteinase K (1 mg/ml) at 65°C for 3 h. Then, the different fractions were dialyzed using a membrane with a cutoff of 6,000 to 8,000 Da and lyophilized. The resulting materials were analyzed by SDS-PAGE gels with 15% acrylamide. After running, gels were fixed with a 50% methanol–12% trichloroacetic acid (TCA)–2% acetic acid solution during 20 min and washed with 10% ethanol–5% acetic acid for 10 min. Then, gels were immersed in 0.7% periodic acid–40% ethanol–5% acetic acid for 10 min. After successive washings for 10 min with 10% ethanol–5% acetic acid, 10% ethanol, and water, gels were incubated with 0.1% silver nitrate for 10 min and washed with water and 10% potassium carbonate. Finally, gels were treated with a reducer solution containing 0.01% formaldehyde in 2% potassium carbonate until staining.
NADH oxidase activity measurements.
All aliquots collected from the gradient were tested for NADH oxidase activity by diluting 10 μl of the fraction in 1 ml of 100 mM Tris-HCl (pH 7.5) containing 10 mM CaCl2, 5 mM MgCl2, and 0.25 mM NADH. Oxidation of NADH was monitored at 340 nm as a function of time for 10 min. Initial velocities were determined and plotted as a function of fraction number.
Amino acid and hexosamine analysis.
Twenty-microliter aliquots from F1 or F2 were hydrolyzed in 6 M HCl at 95°C for 16 h. After evaporation of the acid, hydrolysates were dissolved in 67 mM trisodium citrate-HCl (pH 2.2) and injected into a Hitachi L8800 analyzer equipped with a 2620MSC-PS column (ScienceTec). Quantification was carried out with respect to an amino acid standard (Ajinomoto-Takara Corporation) supplemented with muramic acid, glucosamine, galactosamine, and diaminopimelic acid, the three former compounds being present in peptidoglycan hydrolysates. For F2, the analysis was also made after extensive solubilization with SDS at 100°C for 30 min in order to evaluate the relative contribution of peptidoglycan amino acids in the protein composition of the fraction.
Extraction of mycomembrane proteins and separation on Q-Sepharose.
F2 fraction or ΔaftB fragments were first treated with urea to remove weakly associated proteins. Three hundred microliters of each fraction was diluted with 6 ml of 25 mM HEPES buffer (pH 7.4) containing 6 M freshly dissolved urea. After 15 min of incubation at room temperature, ΔaftB outer membrane fragments or F2 were recovered by centrifugation at 65,000 rpm for 30 min in a TLA 100.3 rotor at 10°C to avoid urea crystallization. The pellet was washed twice and resuspended in HEPES buffer containing 2% LDAO. After 30 min of incubation at room temperature, the insoluble material was discarded after centrifugation at 65,000 rpm for 30 min in a TLA 100.3 rotor at 10°C. The supernatant, containing LDAO-soluble proteins, was subjected to Q-Sepharose chromatography in HEPES buffer containing 0.5% (final concentration) LDAO. Proteins that were not retained on the column and proteins eluted with NaCl step gradient (6 steps from 50 mM to 1 M) were recovered, concentrated by TCA precipitation prior to separation by 12% SDS-PAGE, and visualized by Coomassie blue staining.
Tandem mass spectrometry.
Bands of interest were excised manually and subjected to in-gel trypsin digestion as previously described (50). Peptide mixtures were submitted to nano-liquid chromatography (nano-LC)-CHIP-IT (ion trap)-tandem mass spectrometry (MS/MS) using an Agilent 1200 nanoflow LC system coupled to a 6330 ion trap equipped with the Chip Cube orthogonal ionization system (Agilent Technologies, Santa Clara, CA). The commercially microfluidic chip contained a trap cartridge (Zorbax 300SB-C18; 40 nl, 5 μm) and an electrospray tip-connected analytical column (Zorbax 300SB-C18; 43 mm by 75 μm, 5 μm). In the enrichment mode, 5 μl of peptide extracts were loaded at a 3.5-μl/min flow rate onto the trap cartridge, which after loading was switched in-line with the analytical column. Peptides were separated at a 200-nl/min flow rate. The gradient profile consists of two linear gradients from 3 to 21% B in 4 min and from 21% to 75% B in 16 min, followed by an isocratic step to 85% B for 4 min and finally a 15-min equilibration step with 3% B (B is 0.1% formic acid in 95% acetonitrile).
The mass spectrometer was externally calibrated as recommended by the supplier, leading to typical mass errors lower than 100 ppm. The system was operated in data-dependent acquisition mode. A total of six scans were averaged to obtain a full-scan spectrum, while four scans were averaged for a fragmentation spectrum. The four most abundant ions, preferring doubly charged ions, were selected on each full-scan spectrum for collision-induced dissociation (CID) fragmentation (tap drive, 85; fragmentation amplitude, 1.30 V), and an active exclusion list was applied for 1 min after 2 consecutive fragmentation spectra. A permanent exclusion list was also used for typical background ions. All spectra were saved in profile mode, and other ion trap parameters were set as follow: mode, positive ion; ion charge control (ICC), 250,000; maximal accumulation time, 150 ms; MS scanning rate, 8,100 Thompson/s (Th/s); MS range, 250 to 2,000 Th; MS-MS scanning rate, 26,000 Th/s; MS-MS range, 100 to 2,000 Th; capillary, −2,030 V; skimmer, 30 V; cap exit, 100 V; smart parameter settings, on; drying gas, nitrogen at 300°C with a 4.0-liter/min flow rate. Helium was used as a cooling and fragmentation gas at a pressure of 6 × 10−7 mbar.
Protein identification by database search.
Raw MS/MS data were processed by Agilent data analysis software, version 3.4 (Agilent Technologies, Santa Clara, CA) and converted into *.mgf files. The Mascot MS/MS ions search engine (Matrix Science) was used, and searches were performed against the NCBI database (release 20081128; 7,415,798 sequences) with taxonomic specification to bacteria. Mass accuracy tolerance was set to 100 ppm on the parent ion mass and 0.5 Da in MS/MS mode. One missed cleavage per peptide was allowed, and some modifications were taken into account: carbamidomethylation for Cys as a fixed modification and methionine oxidation and Asn/Gln deamidation as variable modifications. No Mr and pI restriction was selected. The identifications were validated according to the established guidelines for proteomic data publication (12, 15), and notably, protein identification was validated only if at least two different sequences were identified with high-quality mass spectra (individual Mascot ion score > 47).
[3H]palmitate cell labeling.
Exponentially growing C. glutamicum 13032 and ΔmytA cells were incubated in the presence of 0.125 nCi of [3H]palmitate for 2 h at 30°C. After centrifugation, cell envelope proteins were extracted and loaded on an SDS-PAGE gel, which was fixed in 15% acetic acid and submitted to autoradiography for 7 days after treatment with an enhancing solution (Amersham) and dried.
Standard biochemical analysis.
C. glutamicum cell wall protein extracts were prepared as described by Joliff et al. (27). Briefly, cells were recovered by centrifugation and resuspended in Laemmli buffer (32) (100 OD units per ml) before the sample was heated for 5 min. After 15 min at room temperature, unbroken cells were discarded by centrifugation, and the supernatant was analyzed by SDS-PAGE.
Western blotting was done according to standard methods. The transfer of proteins was carried out for 1 h at 30 V on a Whatman Protran BA85 (0.45 μm) nitrocellulose membrane for FbpA analysis and for 30 min at 30 V on a Pall Life Science Biotrace NT (0.22 μm) nitrocellulose membrane for analysis of PorA. The membranes were probed with anti-PorA antibodies diluted at 1/1,000 (from R. Benz) or anti-FbpA diluted at 1/10 (from K. Huygen). Protein quantification was performed according to a standard adaptation of the Lowry (35) colorimetric procedure by using the Bio-Rad DC protein assay kit.
RESULTS
Fractionation of C. glutamicum cell envelope on a sucrose gradient.
The cell envelope of E. coli can be separated into two fractions, enriched in inner and outer membranes, by isopycnic sucrose gradient centrifugation of membranes obtained after breaking the cells with a French pressure cell or by spheroplast lysis. We adapted this technique to fractionate the cell envelope of C. glutamicum. Because C. glutamicum is resistant to lysozyme treatment, cells were disrupted in a French press, a method described as the best one to prepare the cell wall of M. smegmatis (54). Briefly, an overnight-grown culture of strain ATCC 13032 was submitted to French press lysis at high pressure as described in Materials and Methods. Unbroken cells were removed by low-speed centrifugation, and the clarified solution was layered on a sucrose cushion to recover all membrane fractions from the cytosolic material. After 2 h of ultracentrifugation (SW41, 35,000 rpm), membranes were stuck, as two superposed bands, at the interface between the 56% and 20% sucrose solutions (data not shown). These bands were collected, washed twice to remove sucrose, and further submitted to a step gradient ranging from 35 to 56% of sucrose (wt/wt). Isopycnic centrifugation resolved the mixture as two homogeneous fractions: a brownish low-density fraction (density [d] = 1.19 g cm−3), here called F1, and a high-density fraction (d = 1.23 g cm−3), here called F2 (Fig. 1A). As suggested by SDS-PAGE analysis and by electron microscopy images obtained after freeze fracture, F1 and F2 seemed to correspond to very distinct components of the cell envelope. Indeed, their protein patterns exhibited several apparent distinctive bands (Fig. 1C), and the physical aspects of the materials were dramatically different (Fig. 2). F1 contained rather small round vesicles with a lot of particles emerging at the surface and corresponding to the imprint of proteins (Fig. 2B), a picture expected for inner membrane vesicles. F2 contained almost exclusively very large unclosed fragments (Fig. 2C). Their sizes were often as large as that of the entire cell, and they display a rather smooth fracture plane like the one observed on intact bacteria (Fig. 2A). This material could correspond to large fragments of mycomembrane covalently associated to arabinogalactan and peptidoglycan (here called M-AG-PG), as expected from the Minnikin model. F1 and F2 fractions are obviously efficiently separated as shown by the clear-cut profile on the gradient (Fig. 1A) and did not seem to be cross-contaminated with each other as observed on freeze-fracture micrographs at a low magnitude in a large field (data not shown). The biochemical difference between F1 and F2 was also observed when both fractions were solubilized with SDS. As shown in Fig. 1B, F1 was completely solubilized, and no visible material could be recovered after high-speed centrifugation. In contrast, F2 was only partially solubilized by SDS, even after the sample was heated at 100°C for 30 min. An important white pellet was still observed after centrifugation, indicating the presence of an SDS-insoluble material (presumably arabinogalactan and peptidoglycan) in this fraction. In C. glutamicum, the biosynthesis of mycolic acids is not essential for growth (49, 64). More particularly, a Δpks13 strain, defective for mycolic acid biosynthesis, is still able to grow but lacks the typical mycomembrane structure as visualized by freeze fracture and by cryo-electron microscopy of vitreous sections (CEMOVIS) experiments (24, 49, 65). Interestingly, when the Δpks13 strain was analyzed by isopycnic sucrose gradient centrifugation (Fig. 1A), the high-density fraction F2 was completely absent. This strict correlation between the presence of the mycomembrane at the cell surface and the observation of F2 in the gradient is a strong indication that this fraction does contain the mycomembrane.
Fig 1.
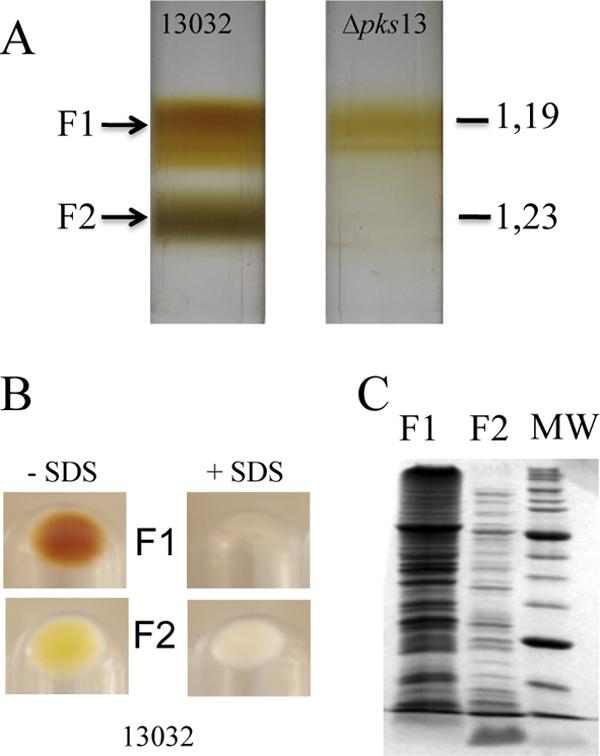
Fractionation of C. glutamicum cell envelope by isopycnic sucrose gradient centrifugation. After cell lysis, membrane-containing fractions were separated according to density on a sucrose step gradient. (A) Centrifugation tubes for the wild-type 13032 strain and for its isogenic Δpks13 mutant. Buoyant densities (d) are indicated in g cm−3. (B) Fractions that correspond to F1 and F2, isolated from the 13032 strain, were pooled, treated with 4% SDS, and heated at 100°C for 30 min or left untreated. Insoluble material was collected by ultracentrifugation, and the corresponding pellets are shown. (C) SDS-PAGE protein pattern of F1 and F2 isolated from strain 13032 after Coomassie blue staining. The same amounts of F1 and F2, corresponding to approximately 5 × 108 cells, were loaded on the gel. MW, molecular mass markers. The molecular masses of the bands with the highest intensities are 66 and 27 kDa, respectively.
Fig 2.
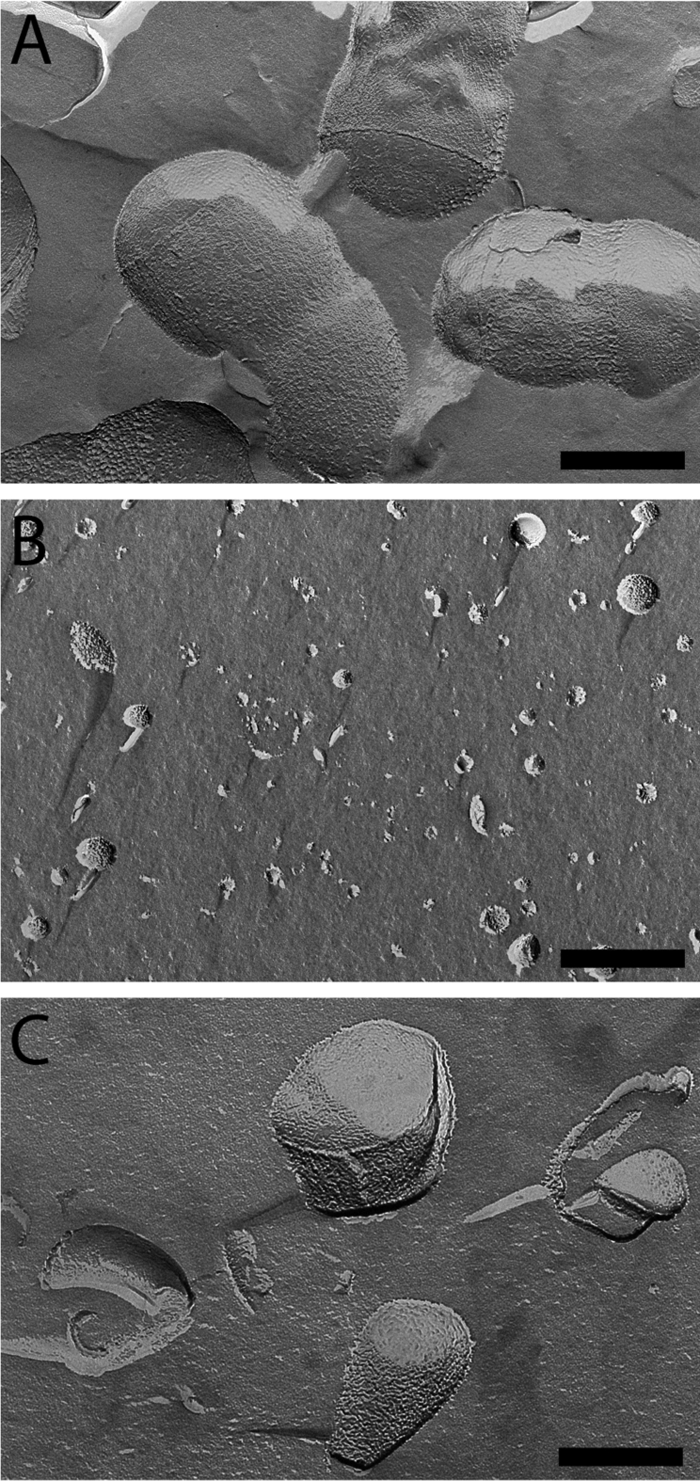
Visualization of F1 and F2 fractions by freeze fracture electron microscopy. (A) Micrograph of C. glutamicum 13032 cells grown in BHI medium. (B and C) Micrograph of F1 (B) or F2 (C). All images are at the same magnification. Scale bar = 200 nm.
Biochemical identification of membrane fractions.
The definitive identification of the isolated bands was based on the chemical composition and distribution of specific enzymes in the two fractions. Lipids were extracted with organic solvents from F1 or F2, separated by TLC, and revealed with the Dittmer reagent and anthrone for phospholipid and glycolipid detection, respectively. As shown in Fig. 3A, F1 contained phospholipids and some glycolipids (phosphatidyl-myo-inositol mannosides); in contrast, F2 showed high levels of trehalose dicorynomycolate (TDCM) and small amounts of trehalose monocorynomycolate (TMCM) but virtually no phospholipids. Indeed, phospholipids were not detected even after loading 5-fold more F2 material than F1 (data not shown). These results indicate a strict segregation of phospholipid and mycolate species between the two fractions. When this work was in progress, Bansal-Mutalik and Nikaido (4) reported a quantitative lipid analysis of the C. glutamicum cell envelope. Surprisingly, they detected large amounts of cardiolipin after solvent extraction of cell envelope fractions but not of fresh cells. Because cardiolipin and phosphatidylglycerol have very similar Rfs in TLC developed with CHCl3–CH3OH–H2O (65/25/4 [vol/vol/vol]), we reexamined the lipid composition of F1 and F2 by comparison with internal phosphatidylglycerol, phosphatidylinositol, and cardiolipin standards. Our results (data not shown) indicated that a compound with the same Rf as that of cardiolipin is the major phospholipid detected in F1 but that it is completely absent in F2 even when 5-fold more F2 material than F1 is spotted on the plate.
Fig 3.

Biochemical analysis of F1 and F2 fractions. (A) Polar lipids were isolated from F1 and F2 and separated on TLC plates. Phospholipids (phosphatidylglycerol [PG] and phosphatidylinositol [PI]) were visualized with the Dittmer reagent. PI derivatives, trehalose monocorynomyclate (TMCM) and trehalose dicorynomyclate (TMDM), were revealed by spraying anthrone. The T lane represents polar lipids extracted from whole cells. (B) NADH oxidase activity was monitored in all fractions collected from the gradient, and the initial velocity was plotted as a percentage of the highest activity (13.2 μmol · min−1). (C) Amino acids, sugars, and peptidoglycan (PG) markers were quantified from purified F1 and F2 fractions as described in Materials and Methods and expressed as nanomoles per milligram of dry material. Bars represent the standard errors of the means for 3 independent experiments. (D) Proteinase K-treated F1 and F2 fractions were loaded on a 15% SDS-polyacrylamide gel, and lipoglycans were revealed as described in Materials and Methods. According to dry weight, equal amounts of material were loaded for F1 and F2. The amount of material loaded in lane “F2 × 10” is 10 times higher than that in the two other lanes.
The presence of phospholipids and the absence of mycolate esters in F1 support the conclusion that F1 contains inner membrane vesicles and is not contaminated by the outer mycolate membrane. This is further strengthened by NADH oxidase activity measurement, which was used to benchmark the respiratory chain (54). The distribution of the NADH oxidase activity in each fraction of the gradient is shown in Fig. 3B. Fractions 7 to 10, corresponding to F1, contained over 95% of the total activity. Fractions 13 to 17, corresponding to F2, contained less than 3% of the NADH oxidase activity, demonstrating that this fraction was almost devoid of any contamination by the inner membrane.
In vivo, mycolic acids are covalently bound to the heteropolymer AG-PG; therefore, if F2 contained the mycomembrane, the fraction should also contain arabinogalactan and peptidoglycan. Accordingly, we determined the amounts of arabinose and galactose as AG markers, as well as muramic acid, glucosamine, and diaminopimelic acid as PG markers, in both F1 and F2 (Fig. 3C). First, F1 and F2 dry pellets were subjected to acidic methanolysis, and the generated methyl glycosides were trimethylsilylated to allow their identification by gas chromatography. Second, F1 and F2 were SDS treated for 30 min, and the SDS-insoluble material was hydrolyzed and analyzed for hexosamines as reported in Materials and Methods. Arabinose and galactose were found almost exclusively in F2 in a ratio of about 1:3.5 (Fig. 3C), which is in good agreement with the determined ratio in AG reported in previous studies of crude cell walls of C. glutamicum (38). Galactosamine was reported to be linked to the AG chain of slow-growing mycobacteria (19). Surprisingly, we detected small amounts of galactosamine in F2 (data not shown), and to our best knowledge, it is the first demonstration of its existence in the AG of C. glutamicum. Muramic acid, glucosamine, and diaminopimelic acid, in a ratio of about 1:1:1, typical of PG, were also found to hallmark F2. Altogether, these results are consistent with the conclusion that F2 corresponds to the M-AG-PG complex. The absence of any trace amount of glucosamine, muramic acid, or diaminopimelic acid in F1 further substantiates that it is not significantly contaminated by F2.
PIMs, LM, and LAM are embedded in the inner membrane.
Mannose is present in the mycobacterial cell envelope since it is an important constituent of phosphatidyl-myo-inositol mannosides (PIMs), mannans, lipomannans (LM), arabinomannans, and lipoarabinomannans (LAM). As shown in Fig. 3C, we detected significant amounts of mannose in F1, suggesting that these mannoconjugates are essentially associated with the inner membrane. In order to prove this hypothesis, we extracted soluble material from F1 or F2 and comparatively analyzed it after protease digestion. Lipids and lipoglycans represent at best 20% of F1, since proteolysis data showed that proteins represented more than 80% of the dry material. We estimated that the lipid and lipoglycan constituents of the mycomembrane should represent a comparable proportion, since AG and PG represent at best 80% of the M-AG-PG complex. Accordingly, aliquots corresponding to the same initial amounts (dry weight) of F1 and F2 were loaded on an SDS-PAGE gel and stained with silver nitrate after periodic cleavage. Consistently, PIMs and the lipoglycans LAM and LM were all found to be essentially associated with the F1 fraction (Fig. 3D). Of note, the detection of small amounts of arabinose and mannose in F1 (Fig. 3C) is in line with the presence of these lipoglycans in this fraction. Loading 10-fold more F2 material than F1 indicated the presence of some LM but no LAM in the M-AG-PG. Reasoning from that, one may speculate that LM is present in the mycomembrane only in small amounts. Besides, these data further demonstrated the purity of both F1 and F2.
PorH-PorA is a major integral protein of the mycomembrane.
Having established that our fractionation method is very efficient in isolating the M-AG-PG from the inner membrane, we turned to the protein composition of F2. PorH-PorA is the first identified porin of C. glutamicum and as such is expected to be inserted in the mycomembrane. Moreover, it has been shown to be accessible at the cell surface by immunogold labeling (33). Fractions collected from the bottom to the top of the gradient were analyzed by SDS-PAGE and transferred to a membrane before detection with anti-PorA antibodies. PorA could only be detected in fractions 13 to 17, corresponding to the F2 fraction (data not shown). In order to further confirm this result, we decided to perform a fractionation experiment on the wild-type strain, the PorH-PorA deletion strain, and the complemented strain. As expected, PorA was present in F2 of both the wild-type and complemented strains (Fig. 4B) and absent from the mutant strain. Surprisingly, the ΔporHA strain consistently had a slightly different behavior upon fractionation. Indeed, the apparent density of F2 was shifted to a lower value (d = 1.22 g cm−3), as visualized in Fig. 4, the normal density (1.23 g cm−3) being recovered in the complemented strain. That the decreased buoyant density of F2 from the ΔporHA mutant was not due to the loss of some dense material interacting with PorH-PorA was ruled out by showing that F2 from both the wild-type and ΔporHA mutant strains contained similar amounts of arabinose, galactose, muramic acid, glucosamine, and diaminopimelic acid but significantly smaller amounts of proteins per mg of extractible lipids (0.70 ± 0.14 mg for the ΔporHA strain instead of 2.13 ± 0.42 and 2.4 ± 0.48 for the wild-type and complemented strains, respectively). We thus hypothesize that PorA is a predominant integral protein of the mycomembrane and that its absence may influence the buoyant density of F2 according to our observations.
Fig 4.
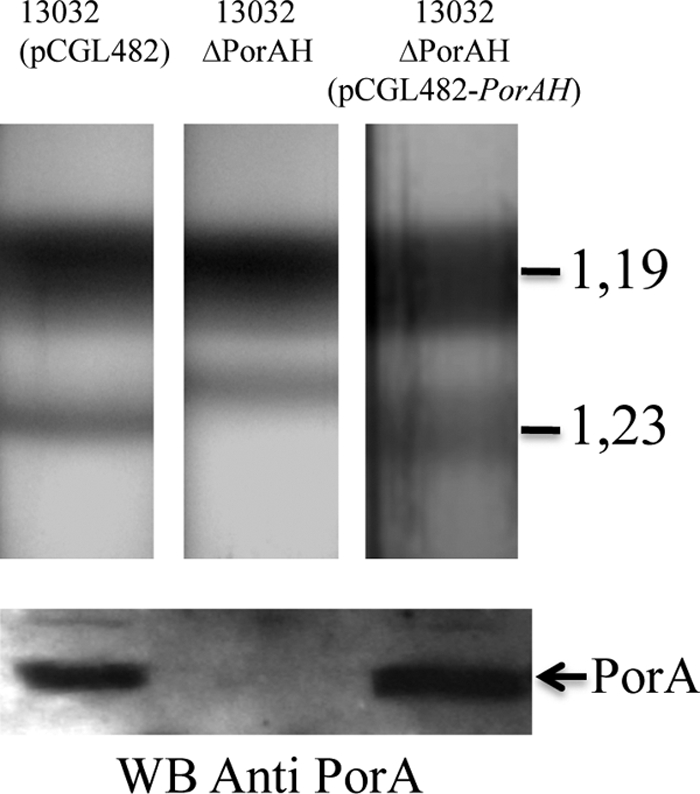
Effect of PorH-PorA deletion on C. glutamicum cell wall fractionation. Sucrose gradient centrifugation of the C. glutamicum strains. The upper panel shows ultracentrifugation tubes for wild-type ATCC 13032, its derivative PorH-PorA-deficient mutant (ΔporHA), and a complemented strain with pCGL482-porHA. The lower panel shows Western blot analysis of F2 fractions from strain ATCC 13032, the ΔporHA mutant, and the ΔporHA mutant containing pCGL482-PorHA incubated with anti-PorA antibodies. Proteins were visualized by Coomassie blue staining.
Identification of OMPs.
We have previously shown that the ΔaftB strain has an unstable mycomembrane whose fragments are shed in the culture medium (11). Here we show, with the wild-type strain, that it is possible to purify a fraction containing the mycomembrane associated with its underlying cell wall. These two complementary approaches appear to be extremely promising in order to perform a comprehensive proteomic analysis of OMPs by MS/MS analysis. Proteins inserted in or associated with either the ΔaftB fragments or the M-AG-PG fraction (F2) were extracted with LDAO (2%) after extensive washing with 6 M urea in order to remove contaminant soluble proteins, protein aggregates, proteins associated with the AG-PG polymer, and proteins that are loosely adhering to the membranes. All proteins that were not dissociated with urea but readily solubilized with LDAO were considered potential OMPs and were separated by ion-exchange chromatography. Flow-through and all eluted fractions were saved and separated by one-dimensional (1D) SDS-PAGE. Visible protein bands (50 for F2 and 34 for ΔaftB fragments) were cut and subjected to in-gel trypsin digestion. The resulting peptides were eluted and analyzed by nano-LC MS/MS. In total, 84 proteins were identified from F2 and 44 from the ΔaftB mycomembrane fragments. Identification was performed by using the standard criteria defined in reference 12. The complete lists of proteins are provided elsewhere (see Table S1 in the supplemental material). In the case of F2, there was clear contamination with ribosomal proteins that are very abundant proteins of the cell and are known to consistently contaminate the bottom of sucrose gradients. For clarity, these identified ribosomal proteins were clustered at the end of our list in Table S1. Of note, no ribosomal proteins were found in ΔaftB fragments. All other identified proteins (a list of 67 for F2 and 44 for ΔaftB fragments) are presented by increasing number of locus tag. For each protein, we included the annotation of the gene, the identification scores, and several relevant in silico predictions, such as the presence of a signal sequence (7, 44), the presence of a lipobox (2), or the prediction for β-barrel outer membrane proteins (3). On the basis of the annotated function, 20 proteins in the case of F2 and 7 in the case of ΔaftB fragments are likely to be cytosolic or inner membrane proteins. Interestingly, as many as 19 proteins were found both in the F2 fraction and in ΔaftB fragments (Table 1). This list represents mycomembrane proteins with a high degree of confidence. It includes proteins with a previously reported function (6 proteins) and a large number of uncharacterized or completely hypothetical proteins (12 proteins). Importantly, this list does not contain any predicted cytosolic or inner membrane proteins. It is composed of acidic proteins (94% of them have a pI below 5) with a high proportion of both putative exported proteins (89% do contain a sequence signal) and putative β-barrel proteins (84% are predicted by the TMββ Web server). Recently, Song et al. analyzed the genome of M. tuberculosis and found 144 putative OMPs on the basis of the presence of amphililic β-strands, the absence of hydrophobic α-helices, and the presence of an N-terminal signal peptide (56). Interestingly, 73% of the identified proteins of our list fit these criteria. Some of these proteins have a homolog in M. tuberculosis, and the corresponding gene names are reported in Table 1. Among known proteins is PorB, a pore-forming protein in C. glutamicum whose exact function has not been fully elucidated. Since its activity was initially shown in cell wall extracts of the PorH-PorA-deficient mutant, it may constitute an alternative anionic route for small solutes to enter the cell wall (17). PorH-PorA was not detected by our MS/MS analysis; however, this is not unexpected, since such small proteins (45 and 57 amino acids (aa) for PorA and PorH, respectively) were not resolved in our SDS-PAGE gel; furthermore, hydrophobic peptides are always difficult to recover. Thanks to the availability of specific PorA antibodies, this peptide was unambiguously detected by Western blotting in both the ΔaftB fragments (11) and the F2 fraction (Fig. 4). PorC was also detected in the F2 fraction. Altogether, all known pore-forming proteins of C. glutamicum were detected in our study, which is a clear validation of our approach. The 5 other known proteins all belong to the mycoloyltransferase family. This family is composed of 6 members in C. glutamicum, 5 of which were found in both mycomembrane fractions; the sixth member, namely, cMytE, was identified in the mycomembrane originating from ΔaftB fragments (see Table S1 in the supplemental material). The presence of these proteins is clearly significant; moreover, we were able to show that their FbpA ortholog in M. tuberculosis, expressed in C. glutamicum, was also located in both F2 (data not shown) and ΔaftB fragments (11). Among the putative and unknown mycomembrane proteins, 4 are predicted to contain a lipobox sequence and therefore could be posttranslationally acylated. In total, 14 putative lipoproteins were identified either in F2 or in ΔaftB fragments (see Table S1). Predicted lipoproteins represent about 3% of the genome of C. glutamicum (91 predicted open reading frames [ORFs]), and the presence of 14 of them in our lists suggests that at least some of them could be sorted to the mycomembrane, as is the case in Gram-negative bacteria (41). Finally, 2 unknown proteins (NCgl 0381 and NCgl 0513) display a specific hydrophobic sequence at their C terminus similar to that found in PS2 or in the Erp protein from M. tuberculosis. Five other examples are also detected in either F2 or ΔaftB fragments and could suggest the existence of a specific machinery for sorting and anchoring these proteins in the mycomembrane of C. glutamicum.
Table 1.
Putative mycomembrane proteins identified from both M-AG-PG and ΔaftB (defective in tranferring the terminal arabinosyl residue) fragmentsa
| Locus tagb | Gene annotation | SSc | Putative β−barrel protein (score)d | pI | M. tuberculosis homologf |
|---|---|---|---|---|---|
| NCgl0336 | Trehalose mycoloyltransferase cMytC | Sec (29) | No (2.975) | 4.56 | FbpABC |
| NCgl0987 | Trehalose mycoloyltransferase cMytD | Sec (26) | Yes (2.915) | 4.37 | FbpABC |
| NCgl2101 | Trehalose mycoloyltransferase cMytF | Sec (31) | Yes (2.959) | 4.25 | FbpABC |
| NCgl2777 | Trehalose mycoloyltransferase cMytA | Sec (43) | Yes (2.958) | 4.85 | FbpABC |
| NCgl2779 | Trehalose mycoloyltransferase cMytB | Sec (37) | No (3.014) | 5.41 | FbpABC |
| Cg3008e | PorA | No | Yes (2.893) | 3.77 | |
| NCgl0933 | PorB | Sec (27) | Yes (2.924) | 4.58 | |
| NCgl0329 | Putative ABC-type cobalamin/Fe3+-siderophore transport system, periplasmic component | Sec* (33) | Yes (2.952) | 4.06 | |
| NCgl0381 | Hypothetical protein | Sec (29) | Yes (2.897) | 4.18 | $ |
| NCgl0513 | Hypothetical protein | Sec (33) | Yes (2.863) | 4.23 | |
| NCgl0535 | Hypothetical protein | Sec (39) | Yes (2.880) | 5.04 | $ |
| NCgl0776 | Putative ABC-type cobalamin/Fe3+-siderophore transport system, periplasmic component | Sec* (35) | Yes (2.870) | 4.00 | |
| NCgl1048 | Putative secreted trypsin-like serine protease | Sec (32) | Yes (2.884) | 5.02 | |
| NCgl1337 | Hypothetical protein | Sec (29) | Yes (2.946) | 5.01 | |
| NCgl1480 | Putative cell wall-associated hydrolase | Sec (36) | Yes (2.829) | 4.34 | Rv1477 |
| NCgl2024 | Hypothetical protein | TAT* (34) | Yes (2.890) | 3.8 | |
| NCgl2375 | Putative protein ABC-type transporter, periplasmic component | Sec* (25) | No (3.011) | 4.36 | |
| NCgl2430 | Hypothetical protein | No | Yes (2.913) | 4.63 | |
| NCgl2789 | Hypothetical protein | Sec (31) | Yes (2.862) | 4.13 | Rv3811 |
| Totalg | |||||
| 19 proteins | 12 unknown proteins | 17/19 | 16/19 | >3.77–<5.41 | 7/19 |
LDAO-solubilized proteins from both fractions were identified by nano-LC electrospray ionization (ESI) MS/MS.
Locus tag according to the work of Ikeda and Nakagawa (26).
The length (aa) and the type of the signal sequence (SS) are indicated for each protein according to the SignalP server. Sec, general secretory pathway; TAT, twin arginine translocation pathway; No, no signal sequence; ∗, the presence of a lipobox is predicted according to the DOLOP database.
TMββ score is given according to the work of Bagos et al., 2004 (3). β-Barrel prediction is validated if a score is lower than 2.96.
Locus tag according to the work of Kalinowski et al. (28). PorA has been identified by Western blotting.
$, this protein has a hydrophobic C-terminal sequence similar to that of PS2 or Erp from M. tuberculosis.
No. of proteins with feature/total proteins or range of pIs is given in bottom row.
DISCUSSION
The presence of a specific outer mycolate membrane in the Corynebacterineae has been claimed for a long time but only clearly demonstrated in the last few years. Still, its biochemical composition is poorly documented because efficient methods for cell envelope fractionation are lacking. Initially, Niederweis (43) reported that a pore-forming protein (PorA) was preferentially located in a high-density fraction after C. glutamicum cell lysis. Later, Rezwan and collaborators (54) described a method to purify the cell wall from M. bovis and M. smegmatis. Although the latter method, relying on differential centrifugation of cell wall fragments after cell lysis, is an efficient one, the fractions were not pure enough to properly address their molecular composition. In this report, we adapted the standard procedure for membrane separation by isopycnic sucrose gradient centrifugation to set up a reproducible method for the isolation of relatively pure inner membrane (IM)- and mycomembrane-containing fractions from C. glutamicum. The low-density fraction, with a density of about 1.19 g cm−3, which contains phospholipids and NADH oxidase activity, represents the plasma membrane. The high-density fraction (1.23 g cm−3) is composed of mycolate derivatives, the PorH-PorA protein, arabinose, galactose, glucosamine, galactosamine, muramic acid, and diaminopimelic acid. It does not form round vesicles but rather large fragments that are almost insoluble in SDS. This material corresponds to the mycomembrane associated with the arabinogalactan and peptidoglycan polymer (M-AG-PG). Both fractions are only slightly cross-contaminated with each other as evaluated by the detection of specific markers. Neither muramic acid nor diaminopimelic acid can be detected in the inner membrane fraction, and conversely, less than 3% of NADH oxidase activity is found in the mycolate-containing fraction. The efficiency of the separation is further supported by the very different protein patterns associated with the two fractions and by electron microscopy images. Of note, however, some cytoplasmic and ribosomal proteins have been detected by mass spectrometry in the mycomembrane-containing fraction, suggesting a residual contamination with other compartments of the cell. Nevertheless, our fractionation procedure appeared to be the best separation reported so far for a member of the Corynebacterineae and a very promising starting point for addressing the biochemical composition of the C. glutamicum cell wall. Moreover, our results show for the first time that the C. glutamicum outer membrane is able to float on a sucrose gradient as large fragments associated with arabinogalactan and peptidoglycan, exactly as predicted by the Minnikin model (39).
Several studies suggested that the mycomembrane is not exclusively composed of mycolate derivatives but also includes a significant amount of phospholipids (18, 34, 53). More recently, the lipid composition and organization of the mycomembrane were indirectly studied in M. smegmatis by a combination of mild detergent solubilization and cryo-electron microscopy studies (24). In this organism, the authors proposed a model in which there is a symmetrical distribution of extractable lipids, including phospholipids, between both leaflets of the mycomembrane bilayer. Here we clearly showed that the mycomembrane-containing fraction of C. glutamicum does not contain significant amounts of phospholipids, in apparent conflict with the above data and with the general assumption formulated for mycobacteria and extended to other members of the Corynebacterineae suborder that mycomembrane extractable lipids comprise both mycolic acids and phospholipids. This assumption may be correct for mycobacteria but not for corynebacteria. Our past (54) and present attempts to fractionate mycobacterial cell walls with the method described herein to determine their composition were unsuccessful. The obtained fractions were not pure enough to convincingly address the question. During the submission of this work, Bansal-Mutalik and Nikaido presented new data about the lipid composition of the cell envelope of C. glutamicum. Interestingly, they detected the presence of large amounts of cardiolipin after chloroform-methanol-water (CMW) extraction of cell envelope preparations. Because cardiolipin is not released after CMW extraction of fresh cells unless they are incubated with lysozyme, they proposed that it could be tightly associated with the peptidoglycan or arabinogalactan complex. In our study, we have clearly confirmed the presence of cardiolipin in the cell envelope of C. glutamicum, but it is always associated with the inner membrane and not with the cell wall.
An important class of cell envelope constituents of the Corynebacterineae is composed of the biosynthetically related glycolipids, PIMs, and lipoglycans, LM and LAM. These glycolipids and lipoglycans are present within the cell envelope of both mycobacteria (13) and corynebacteria (51, 60). The structural features of PIMs and lipoglycans are well described: PIMs comprise phosphatidylinositol (PI)-mannosides with different degrees of acylation; LM and LAM are generated from a PIM intermediate by subsequent steps of mannosylation and arabinosylation. Although all the enzymes dedicated to the production of PIMs and lipoglycans are likely located in the IM, suggesting that the IM is the site of biosynthesis, several studies have shown that LM and LAM are (partly) exposed at the cell surface (45, 48, 51). Here, in C. glutamicum, our data showed that PIMs, LM, and LAM are most exclusively associated with the inner membrane. Small amounts of LM, but surprisingly no LAM, were also detected in F2 (Fig. 3C); thus, small amounts of LM are selectively sorted to the outer membrane by an unknown mechanism that could be analogous to lipopolysaccharide (LPS) transport at the cell surface of Gram-negative bacteria by the MsbA and Lpt machinery (57). Nevertheless, it seems clear that the plasma membrane represents the major localization of these molecules in C. glutamicum.
Until now, only cell wall channels have been localized in the mycomembrane in several species belonging to the Corynebacterineae, such as M. smegmatis and C. glutamicum (42). Since many unknown mycomembrane proteins are expected to have important functions in the mycobacterial physiology and virulence of pathogenic mycobacteria, there is an urgent need to develop methods to isolate mycomembranes and determine their protein content. Although it is quite surprising, the mycomembrane of C. glutamicum appears to contain small amounts of integral proteins, since we showed that M-AG-PG density is significantly modified by the presence of either PorA or PS2 (Fig. 4). This observation is consistent with the fact that M-AG-PG fragments have a rather smooth appearance after freeze fracture compared to the inner membrane vesicles (Fig. 2C). A low number of integral proteins, and more particularly porins, in the mycomembrane may indicate that this membrane is very impermeable to hydrophilic solutes, as has been proposed for M. smegmatis, whose exceptionally low permeability (in contrast to that of Gram-negative bacteria) is attributed to the presence of a low number of copies of MspA porins in its outer membrane (58, 59). In this work, and for the first time, we attempted to determine the mycomembrane proteome. We experimentally identified proteins of the mycomembrane from highly purified preparations of M-AG-PG or from ΔaftB fragments. Nineteen proteins were found in both materials. They represented 28% of the identified M-AG-PG proteins and 43% of the identified ΔaftB proteins (excluding ribosomal proteins), which is a significant degree of overlap and validates these candidates as putative mycomembrane proteins with a high degree of confidence. This group of proteins seems to be very homogeneous, since almost all proteins are very acidic and are predicted to be exported (presence of a signal sequence) and to adopt a β-barrel structure characteristic of Gram-negative outer membrane proteins. Among these proteins, a few are already known. These are the pore-forming proteins and the mycoloyltransferase family. Members of the latter are involved in mycomembrane biogenesis and have been shown to transfer a mycolic acid chain either from one molecule of trehalose monocorynomycolate to another one or from one molecule of trehalose monocorynomycolate to the terminal Araf of arabinogalactan. In C. glutamicum, 6 mycoloyltransferases have been previously identified and found to be either associated with the cell envelope or secreted in the culture medium. We demonstrated the presence of five of them in both ΔaftB and M-AG-PG fractions. In M. tuberculosis, 3 active mycoloyltransferases (Fbp) have been described, and as expected, we showed that FbpA is also targeted to the mycomembrane when expressed in C. glutamicum. We speculate that the interactions between these mycoloytransferases and the mycomembrane should be very strong, since they resist treatment with 6 M urea. This is surprising, since these proteins are known to be soluble proteins that do not contain any predicted hydrophobic segment. Because the catalytic transfer of a mycoloyl residue supposes the formation of an acyl-enzyme intermediate, we hypothesize that a mycoloyl chain could tether the enzyme at the surface of the mycomembrane. We consider this possibility very likely, since we were able to show that cMytC and cMytA are efficiently labeled when incubated with [3H]palmitate (our unpublished data). This situation resembles that of lipoprotein N-acyl transferases that have been shown recently to exist as extracytoplasmic thioester stable acyl-enzyme intermediates (14).
Among the putative and unknown proteins of the mycomembrane of C. glutamicum identified in this study, two proteins (NCgl 0381 and NCgl 0513) were found to contain a specific C-terminal hydrophobic sequence. In total, 7 proteins were found either in M-AG-PG or in ΔaftB fragments, suggesting that they may represent a new family of mycomembrane proteins anchored by a single transmembrane C-terminal domain. Interestingly, a comparable situation would be the Erp protein from M. tuberculosis, a virulence factor (8) that is secreted and also is found associated with the cell surface by a specific C-terminal sequence that is not required for virulence (29, 30). This sequence contains a hydrophobic region, relatively longer than standard transmembrane segments, rich in alanine, isoleucine, or valine and preceded by repetitive amino acids (serine or glycine). This sequence is quite similar to that of the S-layer protein PS2 of C. glutamicum, which has been shown to be essential to tether the protein at the cell surface, leading to the formation of two-dimensional paracrystalline arrays. This type of association is unusual and is not observed for Gram-negative outer membrane proteins, with the exception of the recently described PnlH protein from Dickeya dadantii, which is targeted to the outer membrane by a noncleavable TAT signal sequence (22).
Finally, among all mycomembrane proteins identified, there is an important class of putative lipoproteins. We found 17 proteins containing a lipobox sequence, and 4 of them were found both in M-AG-PG and ΔaftB fragments. To our best knowledge, this is the first report showing that the mycomembrane of a species of the Corynebacterineae contains lipoproteins, and it is therefore a strong indication that a lipoprotein sorting system must exist in the Corynebacterineae. In Gram-negative bacteria, all lipoproteins anchored in the inner leaflet of the outer membrane are initially acylated in the plasma membrane and then extracted from this inner membrane and eventually inserted in the periplasmic face of the outer membrane by the so called “Lol system” (63). This system has been largely characterized, and the critical sorting signal is located in the first amino acids following the N-terminal cysteine of all lipoproteins. In contrast, cell surface lipoproteins do not use the Lol system but are specifically translocated across the outer membrane by a dedicated system (52). To our knowledge, in the Corynebacterineae, evidence for the presence of an active Lol system or equivalent still awaits identification. This is a challenging task, but the fractionation method described in this work will probably provide an essential technical key to achieving this objective.
For a very long time, fractionation of Gram-negative cell envelopes on a sucrose gradient has been proved to be an invaluable tool for depicting cell envelope biogenesis. Undoubtedly, the present successful adaptation of this method to the very atypical cell envelope of C. glutamicum will greatly help in identifying lipoprotein sorting machinery and more generally fundamental mechanisms involved with mycomembrane function and biogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to Elodie Point and Christelle Lémy for their excellent technical assistance. We thank Céline Boursier and Florence Poirier (IFR141, Chatenay-Malabry, France) for providing access to their mass spectrometer. Roland Benz and Chris Huygens are kindly acknowledged for their generous gift of the PorA deletion strain, the anti-PorA polyclonal antibodies, and the anti-FbpA monoclonal antibodies, respectively. We are also very grateful to Andreas Engel and Henning Stahlberg (C-CINA, Biozentrum, University of Basel, Basel, Switzerland), Celia de Sousa, and Christine Houssin for constructive discussions and support.
This work was funded by Agence Nationale pour la Recherche grant ANR-07-BLAN-0363, the CNRS, and the University of Paris-Sud XI.
Footnotes
Published ahead of print 28 November 2011
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Aggerbeck LP, Gulik-Krzywicki T. 1986. Studies of lipoproteins by freeze-fracture and etching electron microscopy. Methods Enzymol. 128:457–472 [DOI] [PubMed] [Google Scholar]
- 2. Babu MM, et al. 2006. A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J. Bacteriol. 188:2761–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. 2004. PRED-TMBB: a web server for predicting the topology of beta-barrel outer membrane proteins. Nucleic Acids Res. 32:W400–W404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bansal-Mutalik R, Nikaido H. 2011. Quantitative lipid composition of cell envelopes of Corynebacterium glutamicum elucidated through reverse micelle extraction. Proc. Natl. Acad. Sci. U. S. A. 108:15360–15365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barth E, Barcelo MA, Klackta C, Benz R. 2010. Reconstitution experiments and gene deletions reveal the existence of two-component major cell wall channels in the genus Corynebacterium. J. Bacteriol. 192:786–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bayan N, Houssin C, Chami M, Leblon G. 2003. Mycomembrane and S-layer: two important structures of Corynebacterium glutamicum cell envelope with promising biotechnology applications. J. Biotechnol. 104:55–67 [DOI] [PubMed] [Google Scholar]
- 7. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795 [DOI] [PubMed] [Google Scholar]
- 8. Berthet FX, et al. 1998. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science 282:759–762 [DOI] [PubMed] [Google Scholar]
- 9. Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917 [DOI] [PubMed] [Google Scholar]
- 10. Bonamy C, Guyonvarch A, Reyes O, David F, Leblon G. 1990. Interspecies electro-transformation in Corynebacteria. FEMS Microbiol. Lett. 54:263–269 [DOI] [PubMed] [Google Scholar]
- 11. Bou Raad R, et al. 2010. A deficiency in arabinogalactan biosynthesis affects Corynebacterium glutamicum mycolate outer membrane stability. J. Bacteriol. 192:2691–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bradshaw RA, Burlingame AL, Carr S, Aebersold R. 2006. Reporting protein identification data: the next generation of guidelines. Mol. Cell Proteomics 5:787–788 [DOI] [PubMed] [Google Scholar]
- 13. Brennan PJ, Nikaido H. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29–63 [DOI] [PubMed] [Google Scholar]
- 14. Buddelmeijer N, Young R. 2010. The essential Escherichia coli apolipoprotein N-acyltransferase (Lnt) exists as an extracytoplasmic thioester acyl-enzyme intermediate. Biochemistry 49:341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carr S, et al. 2004. The need for guidelines in publication of peptide and protein identification data: Working Group on Publication Guidelines for Peptide and Protein Identification Data. Mol. Cell Proteomics 3:531–533 [DOI] [PubMed] [Google Scholar]
- 16. Costa-Riu N, Burkovski A, Kramer R, Benz R. 2003. PorA represents the major cell wall channel of the Gram-positive bacterium Corynebacterium glutamicum. J. Bacteriol. 185:4779–4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa-Riu N, et al. 2003. Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol. Microbiol. 50:1295–1308 [DOI] [PubMed] [Google Scholar]
- 18. Daffe M, Draper P. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131–203 [DOI] [PubMed] [Google Scholar]
- 19. Draper P, Khoo KH, Chatterjee D, Dell A, Morris HR. 1997. Galactosamine in walls of slow-growing mycobacteria. Biochem. J. 327(Pt. 2):519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dusch N, Puhler A, Kalinowski J. 1999. Expression of the Corynebacterium glutamicum panD gene encoding L-aspartate-alpha-decarboxylase leads to pantothenate overproduction in Escherichia coli. Appl. Environ. Microbiol. 65:1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faller M, Niederweis M, Schulz GE. 2004. The structure of a mycobacterial outer-membrane channel. Science 303:1189–1192 [DOI] [PubMed] [Google Scholar]
- 22. Ferrandez Y, Condemine G. 2008. Novel mechanism of outer membrane targeting of proteins in Gram-negative bacteria. Mol. Microbiol. 69:1349–1357 [DOI] [PubMed] [Google Scholar]
- 23. Hartmann M, et al. 2004. The glycosylated cell surface protein Rpf2, containing a resuscitation-promoting factor motif, is involved in intercellular communication of Corynebacterium glutamicum. Arch. Microbiol. 182:299–312 [DOI] [PubMed] [Google Scholar]
- 24. Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 105:3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huc E, et al. 2010. O-mycoloylated proteins from Corynebacterium: an unprecedented post-translational modification in bacteria. J. Biol. Chem. 285:21908–21912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ikeda M, Nakagawa S. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99–109 [DOI] [PubMed] [Google Scholar]
- 27. Joliff G, et al. 1992. Cloning and nucleotide sequence of the csp1 gene encoding PS1, one of the two major secreted proteins of Corynebacterium glutamicum: the deduced N-terminal region of PS1 is similar to the Mycobacterium antigen 85 complex. Mol. Microbiol. 6:2349–2362 [DOI] [PubMed] [Google Scholar]
- 28. Kalinowski J, et al. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25 [DOI] [PubMed] [Google Scholar]
- 29. Kocincova D, et al. 2004. The hydrophobic domain of the mycobacterial Erp protein is not essential for the virulence of Mycobacterium tuberculosis. Infect. Immun. 72:2379–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kocincova D, Sonden B, de Mendonca-Lima L, Gicquel B, Reyrat JM. 2004. The Erp protein is anchored at the surface by a carboxy-terminal hydrophobic domain and is important for cell-wall structure in Mycobacterium smegmatis. FEMS Microbiol. Lett. 231:191–196 [DOI] [PubMed] [Google Scholar]
- 31. Kovacs-Simon A, Titball RW, Michell SL. 2011. Lipoproteins of bacterial pathogens. Infect. Immun. 79:548–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 33. Lichtinger T, Burkovski A, Niederweis M, Kramer R, Benz R. 1998. Biochemical and biophysical characterization of the cell wall porin of Corynebacterium glutamicum: the channel is formed by a low molecular mass polypeptide. Biochemistry 37:15024–15032 [DOI] [PubMed] [Google Scholar]
- 34. Liu J, Barry CE, III, Besra GS, Nikaido H. 1996. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J. Biol. Chem. 271:29545–29551 [DOI] [PubMed] [Google Scholar]
- 35. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 36. Mah N, Perez-Iratxeta C, Andrade-Navarro MA. 2010. Outer membrane pore protein prediction in mycobacteria using genomic comparison. Microbiology 156:2506–2515 [DOI] [PubMed] [Google Scholar]
- 37. Mahne M, Tauch A, Puhler A, Kalinowski J. 2006. The Corynebacterium glutamicum gene pmt encoding a glycosyltransferase related to eukaryotic protein-O-mannosyltransferases is essential for glycosylation of the resuscitation promoting factor (Rpf2) and other secreted proteins. FEMS Microbiol. Lett. 259:226–233 [DOI] [PubMed] [Google Scholar]
- 38. Meniche X, et al. 2008. Partial redundancy in the synthesis of the D-arabinose incorporated in the cell wall arabinan of Corynebacterineae. Microbiology 154:2315–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Minnikin DE. 1982. Lipids: complex lipids, their chemistry, biosynthesis and roles, p 95–184 In Ratledge C, Stanford J. (ed), The biology of the mycobacteria, vol 1 Academic Press, London, United Kingdom [Google Scholar]
- 40. Molle V, et al. 2006. pH-dependent pore-forming activity of OmpATb from Mycobacterium tuberculosis and characterization of the channel by peptidic dissection. Mol. Microbiol. 61:826–837 [DOI] [PubMed] [Google Scholar]
- 41. Narita S, Matsuyama S, Tokuda H. 2004. Lipoprotein trafficking in Escherichia coli. Arch. Microbiol. 182:1–6 [DOI] [PubMed] [Google Scholar]
- 42. Niederweis M. 2003. Mycobacterial porins—new channel proteins in unique outer membranes. Mol. Microbiol. 49:1167–1177 [DOI] [PubMed] [Google Scholar]
- 43. Niederweis M, Maier E, Lichtinger T, Benz R, Kramer R. 1995. Identification of channel-forming activity in the cell wall of Corynebacterium glutamicum. J. Bacteriol. 177:5716–5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nielsen H, Engelbrecht J, Brunak S, von Heijne G. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10:1–6 [DOI] [PubMed] [Google Scholar]
- 45. Ortalo-Magne A, et al. 1996. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J. Bacteriol. 178:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pajon R, Yero D, Lage A, Llanes A, Borroto CJ. 2006. Computational identification of beta-barrel outer-membrane proteins in Mycobacterium tuberculosis predicted proteomes as putative vaccine candidates. Tuberculosis (Edinb.) 86:290–302 [DOI] [PubMed] [Google Scholar]
- 47. Peyret JL, et al. 1993. Characterization of the cspB gene encoding PS2, an ordered surface-layer protein in Corynebacterium glutamicum. Mol. Microbiol. 9:97–109 [DOI] [PubMed] [Google Scholar]
- 48. Pitarque S, et al. 2008. The immunomodulatory lipoglycans, lipoarabinomannan and lipomannan, are exposed at the mycobacterial cell surface. Tuberculosis (Edinb.) 88:560–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Portevin D, et al. 2004. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. U. S. A. 101:314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Preger V, et al. 2009. Auxin-responsive genes AIR12 code for a new family of plasma membrane b-type cytochromes specific to flowering plants. Plant Physiol. 150:606–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Puech V, et al. 2001. Structure of the cell envelope of corynebacteria: importance of the non-covalently bound lipids in the formation of the cell wall permeability barrier and fracture plane. Microbiology 147:1365–1382 [DOI] [PubMed] [Google Scholar]
- 52. Pugsley AP, Kornacker MG, Ryter A. 1990. Analysis of the subcellular location of pullulanase produced by Escherichia coli carrying the pulA gene from Klebsiella pneumoniae strain UNF5023. Mol. Microbiol. 4:59–72 [DOI] [PubMed] [Google Scholar]
- 53. Rastogi N. 1991. Recent observations concerning structure and function relationships in the mycobacterial cell envelope: elaboration of a model in terms of mycobacterial pathogenicity, virulence and drug-resistance. Res. Microbiol. 142:464–476 [DOI] [PubMed] [Google Scholar]
- 54. Rezwan M, Laneelle MA, Sander P, Daffe M. 2007. Breaking down the wall: fractionation of mycobacteria. J. Microbiol. Methods 68:32–39 [DOI] [PubMed] [Google Scholar]
- 55. Salim K, Haedens V, Content J, Leblon G, Huygen K. 1997. Heterologous expression of the Mycobacterium tuberculosis gene encoding antigen 85A in Corynebacterium glutamicum. Appl. Environ. Microbiol. 63:4392–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song H, Sandie R, Wang Y, Andrade-Navarro MA, Niederweis M. 2008. Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 88:526–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sperandeo P, et al. 2008. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 190:4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stahl C, et al. 2001. MspA provides the main hydrophilic pathway through the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 40:451–464 [DOI] [PubMed] [Google Scholar]
- 59. Stephan J, et al. 2005. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol. Microbiol. 58:714–730 [DOI] [PubMed] [Google Scholar]
- 60. Sutcliffe IC. 1995. Identification of a lipoarabinomannan-like lipoglycan in Corynebacterium matruchotii. Arch. Oral Biol. 40:1119–1124 [DOI] [PubMed] [Google Scholar]
- 61. Sweeley CC. 1963. Purification and partial characterization of sphingomyelin from human plasma. J. Lipid Res. 4:402–406 [PubMed] [Google Scholar]
- 62. Teriete P, et al. 2010. Mycobacterium tuberculosis Rv0899 adopts a mixed alpha/beta-structure and does not form a transmembrane beta-barrel. Biochemistry 49:2768–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tokuda H, Matsuyama S. 2004. Sorting of lipoproteins to the outer membrane in E. coli. Biochim. Biophys. Acta 1694:IN1–IN9 [PubMed] [Google Scholar]
- 64. Tropis M, et al. 2005. The crucial role of trehalose and structurally related oligosaccharides in the biosynthesis and transfer of mycolic acids in Corynebacterineae. J. Biol. Chem. 280:26573–26585 [DOI] [PubMed] [Google Scholar]
- 65. Zuber B, et al. 2008. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190:5672–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.