

Abstract
The aim of this study was to identify the antiviral mechanism of a novel compound, BPR3P0128. From a large-scale screening of a library of small compounds, BPR3P compounds were found to be potent inhibitors of influenza viral replication in Madin–Darby canine kidney (MDCK) cells. BPR3P0128 exhibited inhibitory activity against both influenza A and B viruses. The 50% inhibitory concentrations were in the range of 51 to 190 nM in MDCK cells, as measured by inhibition-of-cytopathic-effect assays. BPR3P0128 appeared to target the viral replication cycle but had no effect on viral adsorption. The inhibition of cap-dependent mRNA transcription by BPR3P0128 was more prominent with a concurrent increase in cap-independent cRNA replication in a primer extension assay, suggesting a role of BPR3P0128 in switching transcription to replication. This reduction in mRNA expression resulted from the BPR3P-mediated inhibition of the cap-dependent endoribonuclease (cap-snatching) activities of nuclear extracts containing the influenza virus polymerase complex. No inhibition of binding of 5′ viral RNA to the viral polymerase complex by this compound was detected. BPR3P0128 also effectively inhibited other RNA viruses, such as enterovirus 71 and human rhinovirus, but not DNA viruses, suggesting that BPR3P0128 targets a cellular factor(s) associated with viral PB2 cap-snatching activity. The identification of this factor(s) could help redefine the regulation of viral transcription and replication and thereby provide a potential target for antiviral chemotherapeutics.
INTRODUCTION
Influenza viruses are respiratory pathogens that affect humans and are responsible for substantial morbidity and mortality. The viral genome (viral RNA [vRNA]) comprises eight segments of negative-sense RNA that encode up to 12 proteins (43, 60). Each segment of RNA is encapsidated into a ribonucleoprotein (RNP) complex containing a trimeric RNA-dependent RNA polymerase complex comprising PA, PB1, and PB2 and multiple copies of a nucleocapsid protein (NP). The viral polymerase activity resides in the RNP complexes, whose replication and transcription take place in the nucleus of the infected cells. The newly synthesized viral RNPs (vRNPs) must be transported out of the nucleus, and this export requires cellular and viral proteins (4).
The influenza virus polymerase transcribes cap- and poly(A)-dependent mRNA using a cap-dependent endoribonuclease (cap-snatching) mechanism (45). The host pre-mRNAs are bound to the cap-binding domains of the PB2 subunit by their 5′ cap. A fragment of the first 10 to 13 nucleotides of the host mRNA is cleaved by the endoribonuclease located in the N terminus of the PA subunit (11, 43, 63). The production of primers is activated only when the 5′ and 3′ end sequences of vRNA bind sequentially to the PB1 subunit (33). vRNA has been used as a template to transcribe the mRNA joined by the PB1 subunit (33, 43). Transcription of influenza virus can thus be divided into the following steps: (i) binding of the 5′ and 3′ vRNA sequences to the PB1 subunit, which is likely to cause a conformational change in the polymerase complex (6, 33); (ii) binding of the 5′ cap (m7GTP) of a host pre-mRNA to the PB2 subunit (22); (iii) cleavage of a phosphodiester bond 10 to 13 nucleotides downstream of the cap by the PA subunit; and (iv) activation of the transcription of viral mRNAs at the cleaved 3′ end of the capped fragment. This polymerase complex catalyzes both mRNA transcription and replication of negative-strand vRNAs, which contrasts with the primer (cap)-independent process and produces a full-length replicative intermediate cRNA. This cRNA is then replicated to produce more vRNA. The timing of mRNA and cRNA/vRNA synthesis differs because replication follows mRNA transcription and de novo protein synthesis (43). However, the tuning mechanism for the balance between transcription and replication has remained elusive. Hypotheses based on pieces of vital evidence of the factors controlling the viral switch to replication have been proposed. The switch is thought to be regulated by the availability of an NP, the stability of cRNA mediated by the vRNP complex, and NS2/NEP (nonstructural protein 2/nuclear export protein) (35, 48, 56). A more recent finding identified the mechanism through which influenza virus-specific small viral RNAs regulate the switch (44).
The regions within the PB2 subunit of the influenza virus RNA polymerase involved in cap binding have also been studied in great detail. Early studies showed that cap binding is a function of PB2 (43). In addition, mutagenesis and cross-linking studies show that other regions of PB2, PB1, and, possibly, PA are required for cap binding (14, 16, 23). Attempts have been made to map the region of PB2 involved in cap binding. Two aromatic amino acids, Phe363 and Phe404, are required for cap binding and for transcriptional activity. These have been proposed to sandwich a methylated guanosine, as in other cap-binding proteins (13, 14). The location of the cap-binding site on PB2 has been determined at the atomic level by crystal structure and functional analyses, although the direct involvement of Phe363 was not confirmed (22).
A search for influenza virus-inhibiting drugs is particularly important in the face of new pandemic strains and the high rate of emergence of influenza virus strains resistant to several existing influenza virus antivirals (10, 32, 59). We conducted a drug-screening study using a large-scale anti-influenza virus cell-based assay based on the inhibition of the virally induced cytopathic effect (CPE) (52, 53, 62). An initial hit, CSV0C019002, with a 50% effective (inhibitory) concentration (EC50) of 4.27 μM toward A/WSN/33 was identified after screening 20,800 randomly selected small compounds (molecular masses, <500 Da) from a library. This lead compound was effective against a panel of influenza A and B viruses with similar potency (data not shown). The activity of this novel class of compounds was improved using structure-activity relationship studies to explore the pharmacophore (molecular framework responsible for a drug's biological activity) requirements for anti-influenza virus activity. The most potent compound, BPR3P0128, was identified biochemically as having potent anti-cap-snatching activity, and its chemical structure was distinct from that of two compounds identified previously in this screening (Fig. 1) (52, 53, 62) and two other viral polymerase inhibitors, T-705 (favipiravir) and compound A3 (19, 27). Here, we characterized BPR3P0128 and identified its underlying antiviral mechanism.
Fig 1.

Chemical structures of CSV0C019002 and BPR3P0128. More than 200 analogues were synthesized on the basis of CSV0C019002 as an initial hit. BPR3P0128 was identified as a potent anti-influenza virus agent after intensive lead optimization, including variation of substituents, ring expansions/contractions, extension of the structure, and application of ring variations.
MATERIALS AND METHODS
Viruses, cell culture, and viral infection.
Influenza virus strain A/WSN/33 (H1N1) virus stocks purchased from the American Type Culture Collection (ATCC) were used in this study unless stated otherwise. The clinical influenza virus strains listed in Table 1 were from the Clinical Virology Laboratory of Chang Gung Memorial Hospital, Taiwan, except A/CA/07/2009 (H1N1pdm), which was obtained from the Taiwanese Centers for Disease Control. The information for other viruses is as follows: enterovirus 71 (EV71/Tainan/4643/98/MP4) was obtained from Jen-Ren Wang, National Cheng Kung University (Tainan, Taiwan), coxsackievirus B3 (CVB3; clinical lab quality control [QC] no. CB3-PC-0811; collection date, November 2008; validated by antibody; catalog no. 3306, from Millipore, Billerica, MA), herpes simplex virus type 2 (HSV-2; clinical lab QC no. H201; collection date, January 2003; validated by antibodies, catalog no. 206-84, from Argene), and adenovirus (Ad; clinical lab QC no. Ad-PC-0812; collection date, December 2008; validated by antibodies, catalog no. 5009, from Millipore). The human rhinovirus 2 (HRV2) strain used was clinical lab QC no. HR201. Madin-Darby canine kidney (MDCK), human embryonic kidney 293 (HEK293), and rhabdomyosarcoma (RD) cells were from ATCC. MDCK cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco, Invitrogen), 2 mM l-glutamine (Gibco), and a nonessential amino acid mixture (NEAA; 0.1 mM; Gibco). HEK293 and RD cells were cultivated in DMEM containing 10% heat-inactivated FBS. Human lung carcinoma A549 cells were cultivated in minimal essential medium (Invitrogen) supplemented with 10% heat-inactivated FBS and 0.1 mM NEAA. Influenza viruses and enteroviruses were propagated using MDCK and RD cells, respectively, and viral titers were measured using a plaque assay (52, 61).
Table 1.
Antiviral activities of BPR3P0128 against various viruses
| Cell line or virus strain | CC50 (μM) | EC50 (μM)a |
|---|---|---|
| MDCK | >20 | |
| A549 | >20 | |
| RD | >20 | |
| HEK293 | >20 | |
| A/WSN/33 (H1N1) | 0.083 ± 0.022 | |
| A/TW/3355/97 (H1N1) | 0.09 ± 0.02 | |
| A/CA/07/2009 (H1N1pdm) | 0.145 ± 0.027 | |
| A/TW/3446/02 (H3N2) | 0.190 ± 0.100 | |
| B/TW/99/07 | 0.051 ± 0.015 | |
| B/TW/710/05 | 0.110 ± 0.051 | |
| B/TW/70325/05 | 0.054 ± 0.038 | |
| EV71/Tainan/4643/98/MP4 | 0.029 ± 0.001 | |
| Coxsackievirus B3 | 0.062 ± 0.002 | |
| Human rhinovirus 2 | 0.079 ± 0.002 | |
| HSV-2 | >20 | |
| Adenovirus | >20 |
Influenza virus was tested in MDCK cells; coxsackievirus B3, enterovirus 71, and human rhinovirus 2 were tested in RD cells; and adenovirus and HSV-2 were tested in A549 cells. Data are presented as means ± SDs of the results of at least two independent experiments.
Chemicals.
Oseltamivir carboxylate (GS4071), the active metabolite of oseltamivir, was prepared by Kak-Shan Shia (National Health Research Institute, Miaoli, Taiwan), and a stock solution of 10 mM was prepared in dimethyl sulfoxide (DMSO). BPR3P0128 (6-bromo-2-[1-(2,5-dimethyl-phenyl)-5-methyl-1H-pyrazol-4-yl]-quinoline-4-carboxylic acid), 5-bromoisatin (1.0 mmol), 1-[1-(2,5-dimethyl-phenyl)-5-methyl-1H-pyrazol-4-yl]-ethanone (1.0 mmol), and KOH (4 mmol) were dissolved in ethanol (2 ml), and the mixture was refluxed for 48 h. Removal of the solvent under vacuum produced a residue, which was dissolved in H2O (5 ml), and the aqueous solution was washed twice with ethanol (5 ml). The aqueous phase was cooled and acidified to pH 1 with 3 N HCl, and the precipitate formed was collected by suction filtration, washed with H2O, and vacuum dried to give the title compound in 38% yield.
Cytotoxicity and antiviral effectiveness assays.
The compound library was obtained from ChemDiv, Inc. (San Diego, CA). The cytotoxic effect of BPR3P0128 on cells was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based assay. Cells (2 × 104/well) were seeded into 96-well culture plates and incubated overnight. The medium was then removed, and the cells were incubated with various concentrations of BPR3P0128 in the medium for 72 h. Triplicate wells were used for each treatment. The medium was then removed, MTT was added at a concentration of 5 mg/ml, and the cells were incubated for 3 h at 37°C. The medium was then withdrawn carefully without touching the cells. DMSO (200 μl/well) was added to dissolve the crystals, and the absorbance of each well was measured at 570 nm in a microplate reader (VICTOR3; PerkinElmer).
The anti-influenza virus activity (EC50) exerted by BPR3P0128, measured as the inhibition of virus-induced cell death (CPE), was measured using the crystal violet staining method. Briefly, 96-well tissue culture plates were seeded with 2.4 × 104 cells/well and incubated for 16 to 20 h at 37°C under 5% CO2. The cells were washed once with Dulbecco's phosphate-buffered saline (DPBS), infected with virus at 9 50% tissue culture infective doses, and then overlaid with various concentrations of compounds in a total volume of 200 μl of E0 medium (DMEM with penicillin [100 U/ml], streptomycin [100 μg/ml], l-glutamine [2 mM], NEAA mixture [0.1 mM], and 2.5 μg/ml trypsin). After incubation at 37°C under 5% CO2 for 72 h, the cells were treated with 100 μl of 4% paraformaldehyde overnight and then stained with 0.1% crystal violet for 30 min at room temperature. The density of the cells was measured at 570 nm on a VICTOR3 microplate reader. The concentration of compounds necessary to reduce the virus-induced cell death by 50%, relative to the virus control, was calculated according to the Reed-Muench method and was expressed as the EC50.
For the plaque assay, MDCK cells were seeded into six-well tissue culture plates and incubated overnight. The cells were incubated with influenza virus at about 60 PFU/well with serial dilutions of the test compounds. After adsorption of the virus for 1 h at 37°C, the virus suspension was removed and the cells were washed with Hanks' balanced salt solution (HBSS). The cells were then overlaid with DMEM containing 0.3% agarose with the indicated concentrations of compounds. After incubation for 48 h at 37°C under 5% CO2, the cells were fixed with 10% formaldehyde and then stained with 1% crystal violet. The numbers of plaques were counted, and the antiviral activity of the compounds was calculated with respect to the virus control.
Immunofluorescence microscopy and Western blotting.
MDCK cells (1 × 105 cells) were seeded onto a glass coverslip and incubated overnight. The cells were infected with influenza virus A/WSN/33 at a multiplicity of infection (MOI) of 1 for 1 h, washed three times with DPBS, and then treated with DMEM with or without BPR3P0128 (1 μM). The cells were harvested 9 h after infection, fixed with 4% paraformaldehyde for 20 min, and permeabilized with 0.5% Triton in PBS for 3 min at room temperature. The coverslips were incubated with blocking solution (0.5% bovine serum albumin in PBS) for 1 h at room temperature and then reacted with goat anti-NP primary antibody (ViroStat, Portland, ME) diluted in blocking solution for 1.5 h. The coverslips were then washed three times with blocking solution and incubated with the appropriate Alexa-Fluor-488-labeled secondary antibody, and the fluorescence was evaluated using a Zeiss Axiovert 200 M microscope.
For Western blotting, MDCK cells (5 × 105) were seeded into each well of a six-well plate and incubated overnight. The cells were then infected with influenza virus A/WSN/33 at an MOI of 1 for 1 h, washed with PBS, treated with DMEM with or without BPR3P0128 (1 μM), and harvested at the indicated times after infection. Total cell lysates were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were transferred to polyvinylidene difluoride membranes, which were incubated with primary antibody for 1 h and then with the appropriate secondary antibody. The proteins were detected using an enhanced chemiluminescence Western blotting detection system (Millipore). Antibodies directed against NP and M1 of influenza A virus were from Abcam (Cambridge, United Kingdom) and ViroStat, respectively. Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Santa Cruz Biotechnology (Santa Cruz, CA).
Monoclonal antibody against double-stranded RNA (dsRNA) was prepared to characterize the replicative RNA by immunofluorescence microscopy. Briefly, methylated bovine serum albumin (Calbiochem) was mixed with an equal volume of poly(I:C) (Sigma) dissolved in SSC buffer (150 mM NaCl and 15 mM sodium citrate, pH 7.2), and the resultant insoluble mixture was emulsified with one part Freund's adjuvant (18). The mixture was then injected subcutaneously into BALB/c mice. The supernatants of hybridoma cells to poly(I:C) and the dsRNA of influenza virus (A/WSN/33) were characterized using an enzyme-linked immunosorbent assay and immunofluorescence microscopy, respectively. Our anti-dsRNA antibodies thus have the specificity to detect dsRNA in influenza virus-infected cells, which contrasts with the commercially available J2 monoclonal antibodies that were screened against L-dsRNA from Saccharomyces cerevisiae killer virus (50, 58).
Measurement of virus RNA synthesis by qRT-PCR.
MDCK cells were infected with A/WSN/33 virus at an MOI of 1, and BPR3P0128 was added during the infection period as described in the scheme shown in Fig. 3. Total RNA was extracted from cells at the times indicated using TRIzol reagent (Invitrogen). The first strand of cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase and random hexamers (Invitrogen). Real-time quantitative reverse transcription-PCR (qRT-PCR) was performed in a 20-μl reaction mixture containing 50 nM forward and reverse primers (M1 forward primer, 5′-GAC CAA TCC TGT CAC CTC-3′; reverse primer, 5′-GAT CTC CGT TCC CAT TAA GAG-3′), 1× SYBR green master mix (Protech Technology Enterprise, Taipei, Taiwan), and various amounts of template. The fluorescence emitted by SYBR green was detected using the ABI StepOne Plus sequence detection system (Applied Biosystems, Foster City, CA). To quantify the changes in gene expression, the change in threshold cycle (ΔCT) method was used to calculate the relative changes normalized against the GAPDH gene (forward primer, 5′-AAG AAG GTG GTG AAG CAG GC-3′; reverse primer, 5′-TCC ACC ACC CTG TTG CTG TA-3′). The CT was defined as the cycle at which fluorescence was determined to be significantly greater than the background. The ratio of viral RNA to the internal control was normalized to the control level 0 h after infection, which was arbitrarily set equal to 1.0.
Fig 3.

Inhibition of viral protein and vRNA synthesis by BPR3P0128. MDCK cells were infected with A/WSN/33 virus at an MOI of 1 at −1 h. BPR3P0128 (1 μM) was added from 0 h, and cell lysates were harvested at the indicated times after infection (hours postinfection [hpi]). The data are presented as means ± SEMs of the results from at least two independent experiments. (A) Inhibition of viral protein synthesis by BPR3P0128. Aliquots of 50 μg lysate per lane were subjected to SDS-PAGE separation, followed by Western blotting using anti-M1 and anti-NP antibodies. GAPDH was used as a loading control. The immunoblot results were quantified by densitometric analysis and at 3 h after infection were normalized to those for the virus control, whose level was arbitrarily set equal to 1. The data are presented as means ± SEMs of the results of two independent experiments. ∗, P < 0.05 compared with the respective control. (B) BPR3P0128 (3P0128) inhibits viral protein synthesis, as indicated by immunofluorescence staining. MDCK cells were infected with A/WSN/33 at an MOI of 1, and the cells were harvested 9 h after infection and stained with anti-NP and 4′,6-diamidino-2-phenylindole (DAPI). (C) Inhibition of replicative RNA synthesis by BPR3P0128. The cells were harvested 9 h after infection and observed with immunofluorescence microscopy. DAPI and mouse anti-dsRNA antibody staining show the locations of the nucleus and replicative viral RNAs, respectively. The infected cells are indicated by arrows. The right column shows the overlaid images of dsRNA immunofluorescence and DAPI staining. (D) Inhibition of viral RNA synthesis by BPR3P0128. Viral RNA for M1 was detected by qRT-PCR. The ratio of gene expression to that for its respective internal control (GAPDH) was normalized to the control level at 0 h after infection, which was arbitrarily set equal to 1.0. This is a representative result from two independent experiments.
Inhibition of viral polymerase activity by BPR3P0128 using a reporter assay and primer extension assay.
The production of viral RNA was measured using a plasmid-based reverse genetics (minireplicon or RNP reconstitution) system, followed by a luciferase assay (42) or primer extension (38) assay.
For the luciferase assay, about 5 × 104 HEK293 cells/well were transfected with plasmid pPOLI-firefly luciferase (Fluc) (0.1 μg), pHW2000-PB1 (0.1 μg), pHW2000-PB2 (0.1 μg), pHW2000-PA (0.1 μg), or pHW2000-NP (0.1 μg) or with pTK-Renilla luciferase (Rluc; driven by the thymidine kinase promoter; 5 ng; Promega, Madison, WI) as the transfection control using the calcium phosphate method. pPOLI-Fluc is a plasmid which expresses the influenza virus-like RNA driven by a truncated human RNA polymerase I promoter. These polymerase complex proteins originated from A/PR/8/34 virus. Tenfold serial dilutions of BPR3P0128 were added 8 h after transfection, and the cells were incubated for a further 48 h. Cells were then harvested and subjected to the luciferase assay using a dual-luciferase assay system (Promega) and a VICTOR3 plate reader (PerkinElmer). Values at each dose were analyzed in triplicate, and the means and standard deviations (SDs) from three experiments were graphed. The y axis in Fig. 5A is the ratio of Fluc to Rluc control, which was normalized to the mock treatment (arbitrarily set equal to 1.0).
Fig 5.

Inhibition of influenza virus RNP complex activity by BPR3P0128 by minireplicon (RNA reconstitution) assay (A and B) and in virus-infected MDCK cells (C). (A) Detection of RNP activity by a reporter assay. Various concentrations of BPR3P0128 were incubated with HEK293 cells previously transfected with pPOLI-Fluc (Fluc), pHW2000 expression vectors for influenza virus PB1, PB2, PA, and NP proteins, and pTK-Rluc (Rluc) as a transfection control. (Bottom) NP is a protein expression control, and DMSO was included as the vehicle control. This is a representative result of at least three independent experiments. The data are presented as means ± SEMs. (B) Inhibition of vRNA species synthesis by BPR3P0128 in a minireplicon system using a primer extension assay. HEK293 cells were seeded overnight and transfected with plasmids for 8 h, followed by BPR3P0128 treatment for 2 days. Total cellular RNA was then isolated and analyzed by primer extension using γ-32P-end-labeled CAT-specific primers and hybridization at 50°C for 3 h. The resulting reactions were terminated with formamide and the mixtures were separated on 8% polyacrylamide gels containing urea. The mRNA is shown as a wide-ranging band 10 to 13 nucleotides longer than cRNA because of its 5′-cap-snatched sequence. The expected sizes of mRNA, cRNA, and vRNA were 98 to 106, 89, and 158 bp, respectively. (C) Inhibition of RNA species synthesis by BPR3P0128 in virus-infected cells assessed in primer extension assays. Monolayers of MDCK cells were seeded overnight and infected with influenza virus A/WSN/33 at an MOI of 1. After adsorption for 1 h, the culture medium containing the unbound viruses was replaced with DMEM in the presence of BPR3P0128. Total cellular RNA was isolated 8 h after infection, and the viral RNA species were analyzed by hybridizing with viral M-gene-specific γ-32P-end-labeled primers for 3 h. This primer extension reaction was then subjected to reverse transcription, and the resulting mixtures were separated on 8% polyacrylamide gels containing urea. The results shown here are single representatives of at least three independent experiments.
For the minireplicon assay in transfected cells, 1 μg of each of pHW2000-PB1, pHW2000-PB2, pHW2000-PA, pHW2000-NP, and pPOLI-CAT-RT was cotransfected into HEK293 cells in a six-well plate. pPOLI-CAT-RT possesses the chloramphenicol acetyltransferase (CAT) open reading frame in negative polarity surrounded by the untranslated regions of the NS gene of influenza virus A/WSN/33. BPR3P0128 was added 8 h after transfection, and the cells were incubated for a further 48 h. Total cellular RNA was then isolated using an RNeasy minikit (Qiagen, Hilden, Germany) and hybridized with γ-32P-end-labeled primers at 50°C for 3 h. This primer extension reaction was performed using avian myeloblastosis virus (AMV) reverse transcriptase (Primer Extension System; Promega) at 42°C for 2 h. The resulting mixtures were terminated by denaturing at 90°C for 10 min and then separated on 8% polyacrylamide gels containing 7 M urea. The expected sizes of mRNA, cRNA, and vRNA were 98 to 106, 89, and 158 bp, respectively. Two CAT-specific primers were used in the reverse transcription reactions: primer CAT(+) (5′-ATG TTC TTT ACG ATG CGA TTG GG-3′) for detecting the extension products of CAT mRNA (98 to 106 bp) and cRNA (89 bp) and primer CAT(−) (5′-CGC AAG GCG ACA AGG TGC TGA-3′) for vRNA (158 bp). Primer extension was performed on MDCK cells infected with A/WSN/33 at an MOI of 1, and the cells were incubated with 0 to 10 μM BPR3P0128. Cells were harvested 8 h after infection, and total RNA extraction and reverse transcription were performed as described above. The primers derived from segment 7 (M gene) were used in these reactions: 5′-ACC GTC GCT TTA AAT ACG G-3′ (for detecting vRNA) and 5′-TCA AGT CTC GGT GCG ATC TCG-3′ (for detecting mRNA and cRNA). Primer extension assays were performed at least three times with independently transfected or infected cells.
Inhibition of capped RNA binding and 5′ vRNA binding to polymerase complexes by BPR3P0128.
Assays to measure the binding of specific RNAs to polymerase complexes were performed as described previously (33, 34). Briefly, HeLa cells were cotransfected with Vac-PB1, Vac-PB2, and Vac-PA, and nuclear extracts of the influenza virus polymerase complex were prepared for subsequent experiments. For cap-binding assays, following a 30-min reaction for the binding of nuclear extract (2.5 μg) with an unlabeled oligoribonucleotide containing the chemically synthesized 5′-terminal sequence of vRNA, the 13-nucleotide capped alfalfa mosaic virus (ALMV) RNA 4 RNA fragment containing an m7G32pppGm 5′ end (10 ng, 1 × 105 cpm) was added, and the mixture was incubated in the presence of BPR3P0128 at 30°C for 30 min. For the 5′ vRNA binding assays, nuclear extracts were incubated in binding buffer with 32P-labeled oligonucleotide containing the 5′-terminal sequence of vRNA (10 ng, 1 × 105 cpm). These protein-RNA complexes were separated from free RNA by electrophoresis on 4% nondenaturing gels.
Inhibition of polymerase endoribonuclease assay by BPR3P0128.
Polymerase endoribonuclease assays were performed as described previously (33). The β-eliminated ALMV RNA 4 (4.56 μg) was capped and methylated in vitro with vaccinia virus capping enzyme (guanyltransferase and 7-methyltransferase) and 2′-O-methyltransferase in the presence of [α-32P]GTP (45). mRNAs labeled with m7G32pppGm at the 5′ end were gel purified, extracted by phenol-chloroform, filtered through G-25 Sephadex spin columns, and then precipitated with ethanol. Following binding of 5′ and 3′ vRNAs, the radiolabeled cap (0.25 ng, 1 × 105 cpm) was added along with 0.1 ng of tRNA as an inhibitor of the nonspecific inhibition of nuclease activity. The mixture was incubated with or without BPR3P0128 at 30°C for 60 min. Reactions were terminated with the addition of formamide stop solution, and the mixture was analyzed on 20% polyacrylamide-urea gels.
RESULTS
Antiviral activity of BPR3P0128.
CSV0C019002 was one of the initial lead compounds identified from hit screening of 20,800 compounds by measuring inhibition of virus-induced CPE (Fig. 1) (52, 53, 62). The influenza virus strain A/WSN/33 was used at an MOI of 3 × 10−5 to infect MDCK cells and to induce a CPE. Compounds that inhibited at least 50% of the CPE at a concentration of 10 μM were selected for lead optimization and for further investigation of their antiviral activities and the underlying mechanism. Using an iterative structure-activity relationship analysis, BPR3P0128 with a core structure of quinoline was then identified to be the most potent analogue against A/WSN/33 and had an EC50 of 0.083 ± 0.022 μM and a 50% cell cytotoxic concentration (CC50) of >20 μM (Table 1). A plaque reduction assay was performed to verify the antiviral activity (Fig. 2). BPR3P0128 dose-dependently reduced the yield of viral progeny produced by infection of MDCK cells with influenza virus A/WSN/33 (Fig. 2). The EC50 for viral plaque formation was estimated to be 68.4 ± 8.9 nM, similar to that obtained in the anti-CPE assay (Table 1).
Fig 2.

Inhibition of progeny virus production measured by a plaque reduction assay. MDCK cells were incubated with influenza virus at about 60 PFU/well and with serial dilutions of BPR3P0128. After adsorption of the virus for 1 h at 37°C, the viral suspension was removed and the cells were washed with HBSS. The cells were then overlaid with DMEM containing 0.3% agarose with the indicated compound concentrations (micromolar). After incubation for 48 h, the numbers of plaques were counted and the antiviral activity of the compound was calculated in relation to the virus-only control.
We then investigated whether BPR3P0128 inhibits viral protein and RNA synthesis within the viral infectious (replication) cycle. In a time course experiment, MDCK cells were infected with influenza virus A/WSN/33 at an MOI of 1 and were then treated with or without BPR3P0128 at a concentration of 1 μM, which inhibits virus-induced CPEs. Cells were harvested at the indicated times after infection and subjected to Western blotting using antibodies specific to M1 and NP proteins. The viral proteins produced by the influenza virus were evident 3 h after infection, and their expression persisted thereafter. The amounts of NP and M1 proteins produced by the influenza virus were reduced in BPR3P0128-treated cells by about 2-fold (Fig. 3A, lanes 3, 6, and 9) compared with untreated cells (lanes 2, 5, and 8) at 6 and 9 h after infection. This suggests that the reduction in viral protein production could be attributed to inhibition of viral translation by BPR3P0128.
To investigate whether BPR3P0128 might have an effect on the subcellular distribution of viral proteins, as seen in nucleozin treatment, in which the nuclear import of NP is abrogated and NP accumulates in the cytoplasm (30), we performed detailed fluorescence microscopy analysis. In the presence of BPR3P0128, the immunofluorescence staining intensity declined, indicating a decrease in the viral protein level; this result was consistent with the immunoblotting data (Fig. 3B).
We next used immunofluorescence microscopy and qRT-PCR to examine the effect of BPR3P0128 on the synthesis of viral RNA. MDCK cells were infected with influenza virus A/WSN/33 at an MOI of 1 and incubated with or without 1 μM BPR3P0128 during the postinfection stages. The cells were fixed 9 h after infection, reacted with a primary anti-dsRNA antibody, and evaluated by immunofluorescence microscopy. The dsRNA was distributed exclusively in the nuclei of the infected cells, where viral RNA replicates actively (Fig. 3C; compare the middle row with the top row). The expression of dsRNA was reduced markedly in the presence of BPR3P0128, indicating that viral replication was impaired (Fig. 3C, bottom row). We also used qRT-PCR to investigate total viral RNA synthesis in MDCK cells infected with virus and treated with or without BPR3P0128 at 3, 6, and 9 h after infection. Random hexamers were used in the reverse transcription reactions, and all three RNA species were detected. The total viral RNA level was reduced significantly by 55 to 68% when cells were treated with this compound (Fig. 3D). In summary, BPR3P0128 inhibited viral replication in infected cells, and this inhibition reduced the production of viral protein and RNA.
Mechanism-of-action studies of BPR3P0128.
We investigated the specificity of the antiviral activity of BPR3P0128 by subjecting it to bioevaluation against a variety of strains and viruses, including different strains of influenza A and B viruses, EV71, CVB3, HRV2, and two DNA viruses, adenovirus and HSV-2. BPR3P0128 exhibited potency against both influenza viruses A and B (Table 1). Interestingly, this compound also inhibited three other RNA viruses listed in Table 1, EV71, CVB3, and HRV2. BPR3P0128 did not show activity toward the two DNA viruses. BPR3P0128 was also effective against recent clinical isolates of oseltamivir-resistant viruses in an assay using oseltamivir carboxylate as a control (Table 2). As expected, these strains were resistant to oseltamivir carboxylate. BPR3P0128 showed similar inhibitory efficacy against these resistant viruses, strongly suggesting that its inhibitory mechanism differs from that of oseltamivir (Table 2).
Table 2.
Susceptibility to BPR3P0128 of recent oseltamivir-resistant influenza virus H1N1 isolates
| Influenza virus | EC50 (μM)a |
||
|---|---|---|---|
| BPR3P0128 | Amantadine | Oseltamivir carboxylate | |
| A/TW/147/09 | 0.060 ± 0.001 | 1.47 ± 0.60 | >25 |
| A/TW/70058/09 | 0.075 ± 0.005 | 0.762 ± 0.28 | >25 |
| A/TW/70066/09 | 0.074 ± 0.003 | 0.96 ± 0.32 | >25 |
Susceptibility was determined by inhibition of CPE in MDCK cells. Data are presented as means ± SDs of the results of at least two independent experiments.
To investigate its possible time-dependent inhibitory effects on influenza virus replication, BPR3P0128 was added to monolayers of infected cells during or after viral adsorption, and the viral yield in one infectious cycle was assessed using a plaque formation assay (Fig. 4). In this time-of-addition assay, even when BPR3P0128 was added 4 h after viral adsorption, there was a marked inhibition of viral yield. There remained about a 40% reduction when the compound was added 6 h after infection. This suggests that BPR3P0128 affects an early step of the replication cycle after viral entry, such as vRNA synthesis or vRNA export out of the nucleus, although the underlying mechanism requires further investigation (Fig. 4). This compound did not inhibit the phosphatidylinositol 3-kinase/Akt signaling pathway, which is activated at an early stage of infection to support influenza virus replication (data not shown) (12).
Fig 4.

Mode of action of BPR3P0128 by time-of-addition experiments. MDCK cells were infected with A/WSN/33 at an MOI of 0.001 at −1 h. The timing of treatment with the compound (1 μM, at 37°C) is indicated at the top. BPR3P0128 was added at the time of infection (−1 h) and 0, 2, 4, and 6 h after infection, as indicated. After treatment, the culture medium was replaced with fresh medium, and the culture supernatant was harvested for the plaque assay 12 h after infection. Virus control, controls with no compound added. Plaque numbers for each treatment were normalized to the virus control, which was arbitrarily set equal to 100%. The data are presented as means ± SEMs of the results of two independent experiments.
BPR3P0128 inhibition of viral polymerase activity.
We used a plasmid-based minireplicon (RNP reconstitution) assay to identify the mechanism underlying the inhibition of influenza viral polymerase by BPR3P0128 (42). Plasmids for the expression of RNP complex of A/PR/8/34 were transfected together with pPOLI-Fluc into HEK293 cells (Fig. 5A). Treatment with BPR3P0128 decreased the ratio of Fluc to Rluc activity, indicating that BPR3P0128 inhibited the RNP complex-mediated transcription of pPOLI-Fluc, resulting in decreased Fluc activity in this system (Fig. 5A). No activity was detected when PA or Fluc was omitted from the minireplicon system, indicating that the inhibition was specific. This inhibitory effect of BPR3P0128 might not result from reduced viral protein expression because the level of NP was not altered in any sample (Fig. 5A, bottom).
We used a primer extension assay to study further whether BPR3P0128 can inhibit the synthesis of specific vRNA species (38) (Fig. 5B). RNA synthesis in transfected cells was subjected to primer extension using radiolabeled oligonucleotides complementary to positive- or negative-sense CAT RNAs. The vRNA detected in the absence of PA (Fig. 5B, lane 2) represents POLI-derived template RNA. BPR3P0128 strongly inhibited mRNA synthesis in a dose-dependent fashion (Fig. 5B). Surprisingly, synthesis of cRNA was not inhibited but was increased in a dose-dependent manner, suggesting that transcription but not replication of the polymerase is the primary molecular target of BPR3P0128. This plasmid-based reverse genetics RNP reconstitution assay contains only minimal components of the transcription and replication machinery and might not fully recapitulate typical viral replication inside cells. We confirmed this primer extension result with virally infected MDCK cells (Fig. 5C). Cells were infected with A/WSN/33 at an MOI of 1 and were then incubated with BPR3P0128. Cells were harvested 8 h after infection, and total RNA extraction and reverse transcription were performed using a primer specific for the viral M gene (segment 7) as an indication of RNA synthesis. No vRNAs were detected in mock infection controls, and the three RNA species were clearly identifiable in virus-infected cells, demonstrating that the bands observed correspond to bona fide influenza virus replication (Fig. 5C, lanes 1 and 2). The solvent for the compound (DMSO) did not cause any inhibition, but the synthesis of total RNA was reduced considerably, a finding that is consistent with the minireplicon assay data (Fig. 5B). Nevertheless, similar to the qRT-PCR data (Fig. 3D), total RNA synthesis was reduced in the presence of BPR3P0128 (Fig. 5C, lanes 4 to 6). Interestingly, similar to the reconstituted reverse genetics assay results given above (Fig. 5B), mRNA was severely inhibited with a parallel increase in vRNA production in the presence of BPR3P0128, indicating that this compound specifically targets polymerase-associated functions. The reduction in the amount of cRNA by this compound might indicate that it had gone through the second cycle of replication to make more vRNA (Fig. 5C, lanes 4 to 6).
BPR3P0128 inhibition of viral polymerase cap-snatching activity.
The inhibition of viral mRNA synthesis by BPR3P0128 led us to investigate the effect of this compound on the cap-dependent mRNA transcription activity of RNP. Synthesis of mRNAs is capped primer dependent: that is, primed by short capped RNA fragments generated from cellular pre-mRNAs by endonucleolytic cleavage (45). In contrast, the synthesis of cRNA and vRNA molecules is initiated in an unprimed manner de novo during influenza virus replication (43). Thus, the proposed mode of action of BPR3P0128 involves targeting the viral mRNA synthesis machinery, such as cap-snatching-related functions during replication. Cap-binding activity has been shown to reside in the PB2 subunit, and capped RNA binding can be activated only by 5′ vRNA binding to the PB1 subunit (3, 33, 55). We performed assays to measure the cap-binding, endoribonuclease, and 5′-vRNA-binding activities of the polymerase complex prepared from nuclear fractions expressing recombinant PA, PB1, and PB2 in the presence of BPR3P0128. The assay for the binding of capped RNA fragments to polymerase complexes was performed using a gel-shift assay (33). The nuclear extracts containing viral polymerase complexes were incubated with an RNA fragment of a 13-nucleotide capped RNA from ALMV, containing a cap structure, in the presence of increasing concentrations of BPR3P0128. The DMSO control had no inhibitory effect on capped RNA-polymerase complex formation (Fig. 6A, lane 1). No capped RNA-polymerase complex was formed when the polymerase complex was missing from the reaction (negative control) (data not shown). These results indicate that the binding between polymerase and capped RNA was genuine. The inhibition became apparent at a BPR3P0128 concentration of 0.125 μM and was dose dependent. Because capped RNA binding can be stimulated only by 5′ vRNA binding to the RNP complex, we explored whether BPR3P0128 can affect the 5′ vRNA binding activity of the polymerase complex, which is one step before cap binding. As shown in Fig. 6B, 5′ vRNA binding to the polymerase complex was not prevented by BPR3P0128 when the same concentrations of this compound were used to inhibit capped RNA binding.
Fig 6.
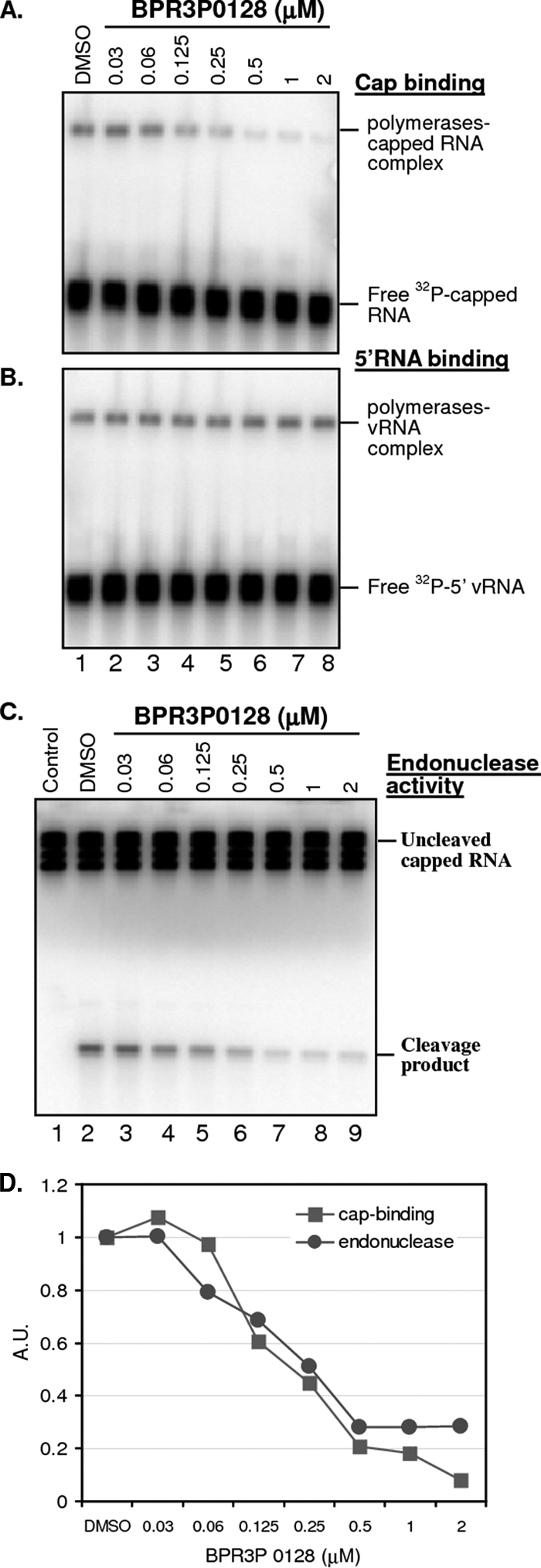
Inhibition of viral cap binding and endoribonuclease activity by BPR3P0128. (A) Cap binding shown in a gel shift assay. Following a 30-min reaction for the binding of an unlabeled oligoribonucleotide containing the 5′-terminal sequence of vRNA, the 13-nucleotide-long capped ALMV RNA 4 fragment containing an m7G32pppGm 5′ end (10 ng, 1 × 105 cpm) was added, and the mixture was incubated in the presence of BPR3P0128 for 30 min at 30°C. The protein-RNA complexes were separated from free RNA by electrophoresis on 4% nondenaturing gels. (B) Binding of 5′ vRNA to RNP. Nuclear extracts containing polymerase complexes (2.5 μg) were incubated at 25°C for 30 min in the binding buffer with a 32P-end-labeled oligoribonucleotide containing the 5′-terminal sequence of vRNA (10 ng). The protein-RNA complexes were resolved from free RNA by electrophoresis on 4% nondenaturing gels. (C) Endoribonuclease assays. Following binding of the oligoribonucleotide containing the 5′-terminal sequence of vRNA and the subsequent binding of the oligoribonucleotide containing the 3′-terminal sequence of vRNA, the full-length ALMV RNA 4 fragment containing an m7G32pppGm 5′ end (0.25 ng) was added along with 0.1 μg of tRNA as an additional inhibitor of nonspecific nucleases. The mixtures were incubated at 30°C for 60 min in the presence of BPR3P0128 and separated on 20% polyacrylamide-urea gels. The results shown here are single representatives of three independent experiments. Control, the labeled RNA was incubated in the absence of a polymerase complex. (D) Quantification of the cap-binding and endoribonuclease experiments. The levels of viral polymerase-cap complex and the cleavage product were normalized to the level of the vehicle control (DMSO), which was arbitrarily set equal to 1.0. The 50% inhibitory concentration (IC50), calculated by Reed-Muench method, was about 0.25 μM for both assays. A.U., absorbance units.
We further determined whether cap binding inhibited by BPR3P0128 affects the subsequent step of endoribonuclease activity that cleaves the bound capped RNAs 10 to 13 nucleotides from their 5′ ends (45). After sequential binding of the 5′-terminal sequence of vRNA and 3′-terminal oligoribonucleotides, full-length ALMV RNA 4 containing an m7G32pppGm 5′ end was added. No cleavage product was detected in the absence of the polymerase complex (Fig. 6C, lane 1). The endoribonuclease activity was specifically and dose-dependently inhibited by BPR3P0128 but not by the DMSO vehicle control. This suggests that this inhibition results from the loss of cap-binding activity exerted by BPR3P0128 because both activities were similar at an IC50 of 0.25 μM (Fig. 6D). The cap-binding site of PB2 is essential for cap-dependent transcription by vRNPs in vitro and in vivo (49). BPR3P0128 may thus target the PB2 subunit or its functionally associated proteins.
DISCUSSION
The polymerase complex of the influenza virus is a potential target for the development of antiviral agents because it is responsible for an essential and highly conserved catalytic function of the virus and because the atomic and quasiatomic structures of its subunit have become available for antiviral drug design (9, 28, 49). The viral polymerase of influenza virus catalyzes the reactions responsible for both capped RNA-primed mRNA transcription (vRNA → mRNA) and unprimed replication of vRNAs in the steps vRNA → cRNA → vRNA. How the polymerase complex carries out these two distinct processes—transcription and replication—is poorly understood. Here, we identified a small compound, BPR3P0128, that plays a role in the regulation of transcription and replication of the influenza virus RNA genome. In particular, incubation with BPR3P0128 markedly decreased the ratio between mRNA and cRNA or vRNA in RNP reconstitution and viral infection assays (Fig. 5). Our data are consistent with those from previous studies of the temporal changes in the amount of RNA (24, 31). Our subsequent results suggested that this compound targets a PB2-associated cap-snatching function (Fig. 6).
BPR3P0128 possesses an essential backbone structure of quinoline (Fig. 1) and might directly target cap-binding-associated functions but not the endoribonuclease activity because this compound is not structurally akin to any known endoribonuclease inhibitors such as flutimide (54). The PB2 protein carries minimal homology to the cap-binding proteins of eukaryotes, and this enzymatic activity has no known cellular counterpart (13). However, BPR3P0128 at a concentration up to 10 μM did not inhibit recombinant PB2 subunit binding to cap-Sepharose, suggesting that BPR3P0128 is not a cap competitor (data not shown). Quinoline and its derivatives have been developed as anti-human immunodeficiency virus compounds, but they do not target cap-related functions (1, 41).
Our results suggest that BPR3P0128 might regulate the balance between viral replication and transcription. Various models of switching from transcription to replication in influenza A viral infections have been proposed (46, 51). NP, which is involved in encapsulation of the vRNA, was identified as a key player because of its RNA binding, which might modify the panhandle structure for the switch (17). The interaction between NP and the polymerase complex was also proposed to potentiate unprimed RNA replication via inhibiting cap-snatching activity (2, 36, 40). The NS2/NEP and PA subunits of the polymerase complex are also associated with the regulation of viral replication and transcription (29, 48). A new model was recently proposed in which virus-generated small RNAs shift the transition from transcription to replication through interactions with the viral polymerase machinery (44). However, some studies have focused on the role of PB2 in tuning transcription and replication because of its cap-binding activity (14, 20), and we speculate that our compound may target PB2.
Two conserved aromatic amino acids, Phe363 and Phe404, which sandwich the N7-methyl guanine of the cap structure in influenza A and B viruses, have been shown to be important for cap binding, but only Phe404 was shown by mutational analysis to play a role in the switch between transcription and replication (14). This crucial residue Phe404 is also conserved in the recent pandemic swine-origin influenza virus (Fig. 7). His357, on the other side of the sandwich, is unique to influenza A virus and is replaced by the more conventional cap-stacking residue tryptophan in influenza B virus (Fig. 7). Mutation of both Phe404 and His357 reduces the accumulation of transcription products and increases the accumulation of replication products (22), and these functions closely resemble those of BPR3P0128. The mode of action of BPR3P0128 might thus target these imperative sites of PB2 to inhibit cap snatching and subsequent transcription. We cannot exclude the possibility that inhibiting transcription provides more template vRNA, which would then be available for replication.
Fig 7.

Sequence alignment of the PB2 cap-binding domain from human and swine influenza A viruses. The secondary structure of A/WSN/1933 is shown over the sequence alignment of strains sensitive to BPR3P0128 (Table 1). Residues His357 and Phe404 of influenza A virus indicated by underlining are principal residues in contact with the cap analogue m7GTP. Phe404 is conserved in influenza A and B viruses, whereas His357 is exclusive to influenza A virus and is replaced by a tryptophan in influenza B virus (22). The full-length PB2 sequences of the influenza B virus tested (Table 1) were not available in the public domain. Four representative sequences from 2005 and 2007 from NCBI are shown here.
Interestingly, BPR3P0128 showed broad-spectrum activity against other RNA viruses, similar to the action of other replication inhibitors such as T-705 and compound A3 (19, 27). Two lines of evidence indicate that the host factor(s) associated with PB2 cap-snatching activity might also be the target of BPR3P0128 in other RNA viruses. First, BPR3P0128 is also very potent in inhibiting EV71 (EC50 = 29 nM), CVB3 (EC50 = 62 nM), and HRV2 (EC50 = 79 nM), although BPR3P0128 at a concentration of 20 μM has no activity against HSV or adenoviruses. Second, no drug-resistant influenza virus could be selected after more than 20 passages of viral propagation under the selection pressure of BPR3P0128 (data not shown). In addition, and most importantly, identifying the target genes of BPR3P0128 could help solve the long-studied problem of the nature of the host factor(s) regulating the switch between transcription and replication by the influenza virus (51). This candidate host factor(s) must fulfill the following criteria. First, it must be essential for replication of the RNA viruses but not the DNA viruses listed in Table 1. Second, it must have both nuclear and cytosolic distributions because influenza virus and the other viruses replicate in the nucleus and cytoplasm, respectively. Third, this factor(s) might be involved in PB2-associated functions through direct or indirect interactions. One candidate molecule is heat shock protein 90 (Hsp90), which interacts with the influenza virus PB2 protein and can stimulate vRNA synthesis (37, 39). Interestingly, inhibiting Hsp90 functions by geldanamycin, a specific Hsp90 inhibitor, reduces the replication activity of influenza virus and other RNA viruses, including poliovirus, CVB3, and rhinoviruses (5, 21); this inhibition is similar to the broad range of inhibitory activity of our compound (Table 1). Another candidate protein is Hsp70, which is functionally related to Hsp90 and is also involved in influenza virus and poliovirus replication (7, 26). Hsp70 interacts with the PB2 subunit and vRNP of influenza virus, and it plays a role in the nuclear export of vRNP (15, 26). Both Hsp70 and Hsp90 have nuclear and cytoplasmic distributions (25, 26, 39). BPR3P0128 might target such cellular proteins to inhibit RNA viruses.
Although our compound is not structurally similar to any known natural or synthetic inhibitors (47), the role of the Hsp family as the target of BPR3P0128 is under investigation. In addition, a number of host factors have been shown to be needed for replication of both influenza virus and poliovirus in host cells through a direct or indirect interaction (8, 57). These communal host factors involved in the virus replication cycle include AKT1, ARAF1, cyclin-dependent kinase 10 (CDK10), CEL, DYRK1B, MAP2K5, mitogen-activated protein kinase 1 (MAPK1), RAB17, and SCN8A. Thus, it is likely that BPR3P0128 can interfere with these crucial host functions, which are needed for influenza virus replication. Inhibition of influenza virus could be achieved through interfering RNA knockdown or pharmacological inhibition of the essential host factors. To our knowledge, BPR3P0128 is the first quinoline-based anti-influenza virus compound identified and characterized to show effective antiviral activity. This compound offers an opportunity for the development of a new anti-influenza virus agent.
ACKNOWLEDGMENTS
This work was supported by the National Science Council (NSC) in Taiwan and Chang Gung Memorial Hospital (grants NSC97-2321-B-400-002, CMRPD160323, and CMRPD160123).
We thank Rei-Lin Kuo for his critical reading of the manuscript and Sung-Nain Tseng for technical assistance.
The experiments were conceived and designed by J.T.-A.H., H.-P.H., and J.-T.H. The experiments were performed by J.-Y.Y., T.-J.L., M.L., Y.C.C., M.-S.W., C.-F.H., W.-F.T., K.S.L., H.-C.H., M.-Y.F., and S.K. J.T.-A.H., H.-P.H., and J.-T.H. analyzed the data. J.T.-A.H. and J.-T.H. wrote the paper.
Footnotes
Published ahead of print 19 September 2011
REFERENCES
- 1. Baba M, et al. 1998. Inhibition of human immunodeficiency virus type 1 replication and cytokine production by fluoroquinoline derivatives. Mol. Pharmacol. 53: 1097–1103 [PubMed] [Google Scholar]
- 2. Biswas SK, Boutz PL, Nayak DP. 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol. 72: 5493–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blaas D, Patzelt E, Kuechler E. 1982. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 10: 4803–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boulo S, Akarsu H, Ruigrok RW, Baudin F. 2007. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 124: 12–21 [DOI] [PubMed] [Google Scholar]
- 5. Chase G, et al. 2008. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377: 431–439 [DOI] [PubMed] [Google Scholar]
- 6. Cianci C, Tiley L, Krystal M. 1995. Differential activation of the influenza virus polymerase via template RNA binding. J. Virol. 69: 3995–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conti C, et al. 1996. Inhibition of poliovirus replication by prostaglandins A and J. in human cells. Antimicrob. Agents Chemother. 40: 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coyne CB, et al. 2011. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 9: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Das K, Aramini JM, Ma LC, Krug RM, Arnold E. 2010. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 17: 530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dawood FS, et al. 2009. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 360: 2605–2615 [DOI] [PubMed] [Google Scholar]
- 11. Dias A, et al. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458: 914–918 [DOI] [PubMed] [Google Scholar]
- 12. Ehrhardt C, Ludwig S. 2009. A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway. Cell. Microbiol. 11: 863–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fechter P, Brownlee GG. 2005. Recognition of mRNA cap structures by viral and cellular proteins. J. Gen. Virol. 86: 1239–1249 [DOI] [PubMed] [Google Scholar]
- 14. Fechter P, et al. 2003. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J. Biol. Chem. 278: 20381–20388 [DOI] [PubMed] [Google Scholar]
- 15. Fislova T, Thomas B, Graef KM, Fodor E. 2010. Association of the influenza virus RNA polymerase subunit PB2 with the host chaperonin CCT. J. Virol. 84: 8691–8699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fodor E, et al. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76: 8989–9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fodor E, Pritlove DC, Brownlee GG. 1994. The influenza virus panhandle is involved in the initiation of transcription. J. Virol. 68: 4092–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Francki RI, Jackson AO. 1972. Immunochemical detection of double-stranded ribonucleic acid in leaves of sugar cane infected with Fiji disease virus. Virology 48: 275–277 [DOI] [PubMed] [Google Scholar]
- 19. Furuta Y, et al. 2009. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 82: 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gastaminza P, Perales B, Falcon AM, Ortin J. 2003. Mutations in the N-terminal region of influenza virus PB2 protein affect virus RNA replication but not transcription. J. Virol. 77: 5098–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 21: 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guilligay D, et al. 2008. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 15: 500–5066 [DOI] [PubMed] [Google Scholar]
- 23. Hara K, Schmidt FI, Crow M, Brownlee GG. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J. Virol. 80: 7789–7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatada E, Hasegawa M, Mukaigawa J, Shimizu K, Fukuda R. 1989. Control of influenza virus gene expression: quantitative analysis of each viral RNA species in infected cells. J. Biochem. 105: 537–546 [DOI] [PubMed] [Google Scholar]
- 25. Henderson B. 2010. Integrating the cell stress response: a new view of molecular chaperones as immunological and physiological homeostatic regulators. Cell Biochem. Funct. 28: 1–14 [DOI] [PubMed] [Google Scholar]
- 26. Hirayama E, Atagi H, Hiraki A, Kim J. 2004. Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J. Virol. 78: 1263–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. 2011. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 108: 5777–5782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsieh HP, Hsu JT. 2007. Strategies of development of antiviral agents directed against influenza virus replication. Curr. Pharm. Des. 13: 3531–3542 [DOI] [PubMed] [Google Scholar]
- 29. Huarte M, et al. 2003. Threonine 157 of influenza virus PA polymerase subunit modulates RNA replication in infectious viruses. J. Virol. 77: 6007–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kao RY, et al. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 28: 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawakami E, et al. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 173: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Layne SP, Monto AS, Taubenberger JK. 2009. Pandemic influenza: an inconvenient mutation. Science 323: 1560–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li ML, Ramirez BC, Krug RM. 1998. RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J. 17: 5844–5852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li ML, Rao P, Krug RM. 2001. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 20: 2078–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Medcalf L, Poole E, Elton D, Digard P. 1999. Temperature-sensitive lesions in two influenza A viruses defective for replicative transcription disrupt RNA binding by the nucleoprotein. J. Virol. 73: 7349–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mena I, et al. 1999. Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J. Virol. 73: 1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Momose F, et al. 2002. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J. Biol. Chem. 277: 45306–45314 [DOI] [PubMed] [Google Scholar]
- 38. Mullin AE, Dalton RM, Amorim MJ, Elton D, Digard P. 2004. Increased amounts of the influenza virus nucleoprotein do not promote higher levels of viral genome replication. J. Gen. Virol. 85: 3689–3698 [DOI] [PubMed] [Google Scholar]
- 39. Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 81: 1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newcomb LL, et al. 2009. Interaction of the influenza A virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J. Virol. 83: 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okamoto H, et al. 2000. Inhibition of the RNA-dependent transactivation and replication of human immunodeficiency virus type 1 by a fluoroquinoline derivative K-37. Virology 272: 402–408 [DOI] [PubMed] [Google Scholar]
- 42. Ozawa M, et al. 2007. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 81: 30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1689 In Knipe D. M., Howley P. M. (ed), Fields virology, 5th ed, vol. 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 44. Perez JT, et al. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc. Natl. Acad. Sci. U. S. A. 107: 11525–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23: 847–858 [DOI] [PubMed] [Google Scholar]
- 46. Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 83: 723–734 [DOI] [PubMed] [Google Scholar]
- 47. Porter JR, Fritz CC, Depew KM. 2010. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr. Opin. Chem. Biol. 14: 412–420 [DOI] [PubMed] [Google Scholar]
- 48. Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 90: 1398–1407 [DOI] [PubMed] [Google Scholar]
- 49. Ruigrok RW, Crepin T, Hart DJ, Cusack S. 2010. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr. Opin. Struct. Biol. 20: 104–113 [DOI] [PubMed] [Google Scholar]
- 50. Schonborn J, et al. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19: 2993–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scull MA, Rice CM. 2010. A big role for small RNAs in influenza virus replication. Proc. Natl. Acad. Sci. U. S. A. 107: 11153–11154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shih SR, et al. 2010. Pyrazole compound BPR1P0034 with potent and selective anti-influenza virus activity. J. Biomed. Sci. 17: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shih SR, et al. 2010. BPR2-D2 targeting viral ribonucleoprotein complex-associated function inhibits oseltamivir-resistant influenza viruses. J. Antimicrob. Chemother. 65: 63–71 [DOI] [PubMed] [Google Scholar]
- 54. Tomassini JE, et al. 1996. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 40: 1189–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ulmanen I, Broni BA, Krug RM. 1981. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 78: 7355–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vreede FT, Jung TE, Brownlee GG. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol. 78: 9568–9572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7: 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80: 5059–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Webster RG, Govorkova EA. 2006. H5N1 influenza—continuing evolution and spread. N. Engl. J. Med. 355: 2174–2177 [DOI] [PubMed] [Google Scholar]
- 60. Wise HM, et al. 2009. A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 83: 8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wong WR, Chen YY, Yang SM, Chen YL, Horng JT. 2005. Phosphorylation of PI3K/Akt and MAPK/ERK in an early entry step of enterovirus 71. Life Sci. 78: 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yeh JY, et al. 2010. Anti-influenza drug discovery: structure-activity relationship and mechanistic insight into novel angelicin derivatives. J. Med. Chem. 53: 1519–1533 [DOI] [PubMed] [Google Scholar]
- 63. Yuan P, et al. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458: 909–913 [DOI] [PubMed] [Google Scholar]