

Abstract
The sulfamethoxazole (SMX)-trimethoprim drug combination is routinely used as prophylaxis against Pneumocystis pneumonia during the first 3 to 6 months after renal transplantation. The objective of this study was to examine the impact of N-acetyltransferase 2 (NAT2) and CYP2C9 polymorphisms on the pharmacokinetics of SMX in 118 renal transplant recipients. Starting on day 14 after renal transplantation, patients were administered 400 mg/day-80 mg/day of SMX-trimethoprim orally once daily. On day 14 after the beginning of SMX therapy, plasma SMX concentrations were determined by a high-performance liquid chromatography method. The SMX area under the concentration-time curve from 0 to 24 h (AUC0-24) for 15 recipients with the NAT2 slow acetylator genotype (NAT2*5/*6, -*6/*6, -*6/*7, and -*7/*7) was significantly greater than that for 56 recipients with the NAT2 rapid acetylator genotype (homozygous for NAT2*4) (766.4 ± 432.3 versus 537.2 ± 257.5 μg-h/ml, respectively; P = 0.0430), whereas there were no significant differences in the SMX AUC0-24 between the CYP2C9*1/*1 and -*1/*3 groups. In a multiple regression analysis, the SMX AUC0-24 was associated with NAT2 slow acetylator polymorphisms (P = 0.0095) and with creatinine clearance (P = 0.0499). Hepatic dysfunction in NAT2 slow acetylator recipient patients during the 6-month period after SMX administration was not observed. SMX plasma concentrations were affected by NAT2 polymorphisms and renal dysfunction. Although standard SMX administration to patients with NAT2 slow acetylator polymorphisms should be accompanied by monitoring for side effects and drug interaction effects from the inhibition of CYP2C9, SMX administration at a low dose (400 mg) as prophylaxis may not provide drug concentrations that reach the level necessary for the expression of side effects. Further studies with a larger sample size should be able to clarify the relationship between SMX plasma concentration and side effects.
INTRODUCTION
The sulfamethoxazole (SMX)-trimethoprim drug combination is routinely used as prophylaxis against Pneumocystis pneumonia during the initial 3- to 6-month period after renal transplantation. The major metabolic pathway for SMX is cytochrome P450 (CYP) 2C9-mediated bioactivation to SMX-hydroxylamine (Fig. 1), which is subsequently converted to N-acetoxy-SMX (Fig. 1) by N-acetylation catalyzed by N-acetyltransferase 2 (NAT2) (5, 15).
Fig 1.
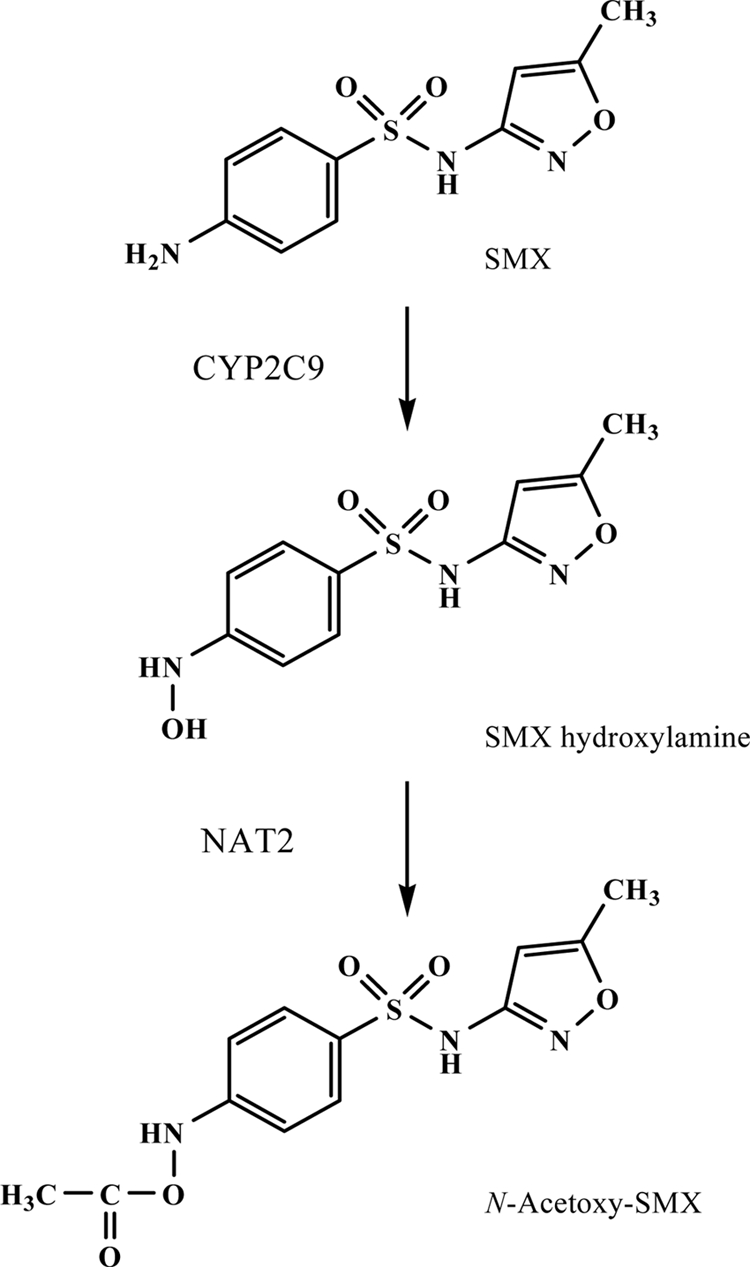
Metabolic pathway for SMX.
NAT2 is a phase II metabolic enzyme involved in the elimination pathways of many therapeutic drugs (10, 19). The alteration of NAT2 acetylator status caused by polymorphisms in the NAT2 gene may affect SMX pharmacokinetics in humans. In the Japanese population, there are 3 point mutations, 341T → C, 590G → A, and 857G → A, which give rise to the variant alleles NAT2*5, -*6, and -*7, respectively, while the wild-type genotype, NAT2*4 (22), is associated with rapid acetylation. Therefore, the Japanese population can be classified into the following 3 groups by NAT2 genotype: rapid acetylators (RA; homozygotes for NAT2*4), intermediate acetylators (IA; carry the NAT2*4/*5, -*4/*6, or -*4/*7 alleles), and slow acetylators (SA; do not have the NAT2*4 allele). Although the contributions of NAT2 polymorphisms to pharmacokinetic variations of SMX are presently poorly understood, a lack of the NAT2*4 allele was reported to be associated with adverse events, such as liver dysfunction, in patients taking SMX-trimethoprim for systemic lupus erythematosus (20). Such adverse events with SMX-trimethoprim in patients with NAT2 SA polymorphisms might account for delayed SMX clearance, resulting in an increase in the SMX plasma concentration; however, in the previous study (20), the SMX plasma concentration was not monitored.
Transplant patients with the CYP2C9*3 (1075A → C) allele are associated with having an impaired metabolic capacity and might be susceptible to higher SMX plasma concentrations than patients with the CYP2C9*1 allele. However, it was reported in an in vitro study that there was no difference in the kinetic parameters for SMX N-hydroxylation between liver microsomes from CYP2C9*1/*1 and CYP2C9*1/*3 patients (6). To our knowledge, such findings have not been reported from in vivo studies. Although the frequencies of the CYP2C9*1/*3 and -*3/*3 genotypes in the Japanese population are approximately 5% and <1% (7, 12, 16), respectively, patients with both NAT2 SA and CYP2C9*3 alleles could have significantly elevated SMX plasma concentrations.
In our present study of renal transplant patients treated with SMX-trimethoprim, we hypothesized that NAT2 polymorphisms might be associated with SMX pharmacokinetics and with adverse reactions to SMX-trimethoprim. The aim of this investigation was to examine the impact of NAT2 and CYP2C9 polymorphisms on the pharmacokinetics of SMX in renal transplant recipients.
MATERIALS AND METHODS
Patients and protocols.
One hundred eighteen Japanese renal transplant recipients were enrolled in this study. Criteria for patient participation were as follows. First, the patient had to be a recipient of his/her first transplant from a living donor. Second, the patient had to be on the standard immunosuppressive regimen, which included tacrolimus (Prograf; Astellas Co. Ltd., Tokyo, Japan), mycophenolate mofetil (MMF) (Cellcept; Chugai Pharmaceutical Co. Ltd., Tokyo, Japan), and prednisolone (Takeda Pharmaceutical Co. Ltd., Osaka, Japan). In addition, the patient must have been ABO compatible, a nonsmoker, and free of hepatic impairment, gastric ulcer disease, or a clinical episode of chronic rejection. The Ethics Committee of Akita University Hospital approved the study protocol, and all recipients gave written informed consent.
On day 14 after renal transplantation, a 400 mg/day-80 mg/day oral dose of SMX-trimethoprim (Baktar; Shionogi Co., Ltd., Osaka, Japan) was started once daily at 0800 h (30 min after breakfast). Tacrolimus and MMF were given in equal doses every 12 h (0900 and 2100 h) starting 2 days prior to surgery, with initial oral doses of 1.0 to 2.0 g/day of MMF and 0.30 mg/kg of body weight/day of tacrolimus. The daily tacrolimus dose was adjusted according to the clinical state of the patient, with the whole blood trough target level being 15 to 20 ng/ml for weeks 0 to 2, 10 to 15 ng/ml for weeks 3 to 4, and <10 ng/ml after 4 weeks. Methylprednisolone was given concomitantly: a dose of 500 mg was given intravenously (i.v.) on the day of surgery, and the dose was reduced to 40 mg/day i.v. during the first week, followed by 20 mg/day of oral prednisolone in the second week, 15 mg/day of prednisolone in the third week, and 10 mg/day thereafter for all 118 recipients. Meals were served at 0730, 1230, and 1800 h daily. Meal content (Japanese food) varied daily for each patient, but the energy, fat, protein, and water content were standardized (energy, 1,700 to 2,400 kcal; protein, 70 to 90 g; fat, 40 to 50 g; and water, 1,600 to 2,000 ml) depending on body weight. Twenty-eight days after renal transplantation (14 days after beginning SMX), whole-blood samples (5 ml) were collected by venipuncture 1, 2, 3, 4, 5, 7, 10, 13, and 22 h after oral SMX-trimethoprim administration.
Genotyping.
DNA was extracted from a peripheral blood sample by using a QIAamp blood kit (Qiagen, Hilden, Germany) and was stored at −80°C until analyzed. Genotyping procedures to identify the CYP2C9*3 (A1075C) allele and the NAT2*5 (T341C), NAT2*6 (G590A), and NAT2*7 (G857A) alleles were performed by the PCR-restriction fragment length polymorphism method described by Moridani et al. and Liu et al. (8, 11).
Analysis of SMX in plasma.
The plasma concentrations of SMX were determined by a high-performance liquid chromatography (HPLC) method. In brief, following the addition of 4,5-diphenylimidazole (100 ng; an internal standard) in methanol (10 μl) to 50-μl plasma samples, 200 μl of acetonitrile was added to the plasma samples, and the solution was vortexed for 30 s. This mixture was centrifuged for 5 min at 13,000 × g. Aliquots of 15 μl of the clear supernatant were filtered through Millipore filters (0.45 μm; Millex-LH, Japan) and then injected directly into the HPLC apparatus. A PU-2080 Plus chromatography pump (Jasco, Tokyo, Japan) equipped with a UV-2075 light source and a UV detector (Jasco) was used. The HPLC column was a Capcell Pak C18 MG Π column (250 mm × 4.6-mm internal diameter; Shiseido, Tokyo, Japan). The mobile phase was 0.5% KH2PO4 (pH 3.5)-acetonitrile (75:25 [vol/vol]) which had been degassed in an ultrasonic bath prior to use. The flow rate was 0.7 ml/min at ambient temperature, and sample detection was carried out at 240 nm. The lower limit of quantification for this assay was 10 ng/ml. The between-day coefficient of variation for SMX was <12.1%. The mean extraction recovery for SMX was 97.2% in the concentration range of 10 to 2,000 ng/ml.
Pharmacokinetic analysis.
Pharmacokinetic analysis of SMX was carried out by a standard noncompartmental method, using WinNonlin (version 5.2; Pharsight Co., Mountain View, CA). The total area under the observed plasma concentration-time curve from 0 to 24 h (AUC0-24) was calculated using the linear trapezoidal rule. The maximum plasma concentration (Cmax) and the time required to reach the peak (Tmax) were obtained directly from the profile.
An estimated creatinine clearance for each patient was calculated using Cockcroft and Gault formulas and the initially analyzed serum creatinine concentration.
Statistical analyses.
The clinical characteristics of patients taking SMX are expressed as numbers or as mean values ± standard deviations (SD) (ranges). The normal distribution of each data type was assessed based on a histogram. Pharmacokinetic parameters of SMX, including Cmax, AUC0-24, and Tmax for each group, are expressed as mean values ± standard deviations or as median values (quartile 1 to quartile 3). The parameters Cmax, AUC0-24, and concentration at each time point were compared between the three groups by using one-way analysis of variance (ANOVA) followed by Bonferroni's correction, and Tmax was compared using the Kruskal-Wallis test. Pearson's correlation coefficient was applied to assess the correlation between the AUC0-24 for SMX and clinical variables (age, weight, and biochemical data), and the results are expressed as a correlation coefficient of determination (r). Stepwise backward-forward selection multiple linear regression analysis was performed to determine the effects of factors examined in the univariate analysis. For each subject, NAT2 genotypes (RA, IA, and SA) were replaced with dummy variables (1 and 0, 0 and 1, and 0 and 0, respectively). Variables with P values of <0.05 for multivariable analysis were entered into the final model. In all of the statistical analyses, a P value of <0.05 was considered statistically significant. Statistical analyses were performed with the statistics software SPSS 14.0J for Windows (SPSS Japan Inc., Tokyo, Japan).
RESULTS
Patient characteristics are listed in Table 1. The mean (±SD) age was 46.3 ± 12.3 years, and the mean (±SD) body weight was 55.6 ± 10.3 kg. There were no patients with serious hepatic dysfunction, but the interpatient creatinine clearance variability was large (55.5 ± 16.5 [17.4 to 95.0] ml/min). The plasma concentration-time profile of SMX for each NAT2 genotype on day 14 after beginning SMX administration is shown in Fig. 2. SMX plasma concentrations 7 and 10 h after SMX administration were higher in the NAT2 SA group than in the NAT2 RA or IA group (P < 0.05 for each). There were no significant differences in the SMX pharmacokinetic parameters, except for AUC0–24, among the three NAT2 genotype groups (Table 2). There was a significant difference in the SMX AUC0–24 between the NAT2 RA and SA groups (537.2 ± 257.5 versus 766.4 ± 432.3 μg-h/ml, respectively; P = 0.0430) (Table 3). On the other hand, there were no significant differences in the SMX AUC0–24 between the two CYP2C9 genotypes (Table 3). There were no significant correlations between the SMX AUC0–24 and clinical characteristics of patients (Table 3). From a stepwise backward-forward selection multiple regression analysis (Table 4), the SMX AUC0–24 was associated with the NAT2 SA genotype (P = 0.0095) and with creatinine clearance (P = 0.0499).
Table 1.
Clinical characteristics of patients taking sulfamethoxazole
| Characteristic | Value |
|---|---|
| No. of patients | 118 |
| Males | 68 |
| Females | 50 |
| Age (yr) (mean ± SD [range]) | 46.3 ± 12.3 (21–70) |
| Body wt (kg) (mean ± SD [range]) | 55.6 ± 10.3 (36.8–81.9) |
| Aspartate transaminase concn (IU/liter) (mean ± SD [range]) | 15.0 ± 6.5 (6–45) |
| Alanine transaminase concn (IU/liter) (mean ± SD [range]) | 18.5 ± 13.8 (2–99) |
| Serum albumin concn (g/dl) (mean ± SD [range]) | 4.1 ± 0.4 (3–4.8) |
| Total bilirubin concn (mg/dl) (mean ± SD [range]) | 0.4 ± 0.2 (0.1–1.1) |
| Serum creatinine concn (mg/dl) (mean ± SD [range]) | 1.4 ± 0.6 (0.5–4.4) |
| Creatinine clearance (ml/min) (mean ± SD [range]) | 55.5 ± 16.5 (17.4–95.0) |
| Platelet count (104/mm3) (mean ± SD [range]) | 23.8 ± 9.3 (10.1–69.4) |
| No. of patients in CYP2C9 genotype group | |
| *1/*1 | 110 |
| *1/*3 | 8 |
| No. of patients in NAT2 genotype group | |
| RA (*4/*4) | 56 |
| IA (*4/*5, *4/*6, *4/*7) | 47 (1, 30, 16) |
| SA (*5/*6, *6/*6, *6/*7, *7/*7) | 15 (2, 3, 8, 2) |
Fig 2.

Mean (± SD) plasma concentration-time profiles of sulfamethoxazole in renal transplant recipients with the NAT2 rapid acetylator (RA; white circles) (n = 56), NAT2 intermediate acetylator (IA; gray circles) (n = 47), and NAT2 slow acetylator (SA; black circles) (n = 15) genotypes on day 14 after once-a-day administration of 400 mg/day-80 mg/day of sulfamethoxazole-trimethoprim.
Table 2.
Pharmacokinetic parameters of sulfamethoxazole in NAT2 genotype groups
| Parameter | Value for genotype groupa |
P value | ||
|---|---|---|---|---|
| RA | IA | SA | ||
| No. of patients | 56 | 47 | 15 | |
| Cmax (μg/ml) | 40.1 ± 18.7 | 39.8 ± 22.3 | 52.0 ± 24.7 | 0.1183 |
| Tmax (h) | 4.0 (4.4–6.7) | 4.0 (4.0–6.6) | 7.0 (3.7–9.0) | 0.3465 |
| AUC0-24 (μg-h/ml) | 537.2 ± 257.5 | 561.8 ± 332.1 | 766.4 ± 432.3 | 0.0431 |
Data for Cmax and AUC0-24 are means ± SD, and data for Tmax are medians (quartile 1 to quartile 3).
Table 3.
Comparison and correlation of sulfamethoxazole AUC0-24 and clinical characteristics
| Characteristic | Mean AUC0-24 (μg-h/ml) ± SD | Correlation coefficient (r) | P value |
|---|---|---|---|
| Sex | 0.9464 | ||
| Male | 574.4 ± 314.4 | ||
| Female | 578.5 ± 330.4 | ||
| CYP2C9 genotype group | 0.7019 | ||
| *1/*1 | 579.2 ± 323.1 | ||
| *1/*3 | 534.1 ± 287.0 | ||
| NAT2 genotype group | 0.0431 | ||
| RA | 537.2 ± 257.5 | 1.0000 (RA vs IA) | |
| IA | 561.8 ± 332.1 | 0.0901 (IA vs SA) | |
| SA | 766.4 ± 432.3 | 0.0430 (RA vs SA) | |
| Age | 0.0626 | 0.5003 | |
| Body wt | 0.0423 | 0.6489 | |
| Aspartate transaminase | −0.1407 | 0.1286 | |
| Alanine transaminase | −0.1183 | 0.2020 | |
| Serum albumin | −0.0602 | 0.5172 | |
| Total bilirubin | 0.0317 | 0.7334 | |
| Serum creatinine | 0.0853 | 0.3561 | |
| Creatinine clearance | −0.1667 | 0.0712 | |
| Platelet | −0.0671 | 0.4705 |
Table 4.
Stepwise backward-forward selection multiple regression analysis of explanatory variables for AUC0-24 of sulfamethoxazole
| Variable | Estimatea | SE | SRCb | P value |
|---|---|---|---|---|
| NAT2 SA | 225.4 | 85.46 | 0.236 | 0.0095 |
| Creatinine clearance | −3.436 | 1.733 | −0.177 | 0.0499 |
| Intercept | 738.1 | 100.4 | < 0.0001 |
Fitted constant associated with each independent variable or intercept.
SRC, standardized regression coefficient.
DISCUSSION
This is the first report on the influence of NAT2 and CYP2C9 genetic polymorphisms on the in vivo pharmacokinetics of SMX. In the present study, we quantified the steady-state SMX plasma concentration on day 14 after repetitive administration of SMX to 118 renal transplant recipients. Although SMX is a substrate of CYP2C9 and NAT2, no significant differences were found in SMX pharmacokinetics between patients with the CYP2C9*1/*1 and -*1/*3 alleles, similar to findings from a previous in vitro study (5). On the other hand, the SMX AUC0–24 was significantly higher in recipients with the NAT2 SA genotype than in those with the NAT2 RA or IA genotype; however, there were no significant differences of other measured biochemical parameters among the NAT2 genotype groups.
Soejima et al. reported that serum alanine aminotransferase (ALT) levels on day 14 after initiating SMX administration were significantly higher in NAT2 SA patients than in NAT2 RA or IA patients (20). However, during the same treatment period in our study, the mean (range) ALT levels in NAT2 SA and RA/IA patients were 18.1 (7 to 47) IU/liter and 18.5 (2 to 99) IU/liter, respectively. Although all recipients in the present study were hospitalized and monitored for asymptomatic liver dysfunction by using routine biochemical tests, for the 15 recipients with the NAT2 SA genotype, side effects caused by an increase of SMX plasma concentration were not confirmed until 6 months after SMX administration (data not shown). In addition, approximately 6 months after renal transplantation, administration of SMX-trimethoprim was stopped. Although the biochemical data were examined with respect to SMX withdrawal, myelosuppression and decreased renal function from SMX treatment were not observed (data not shown). In the present study, recipients took 400 mg/day-80 mg/day of SMX-trimethoprim as prophylaxis against Pneumocystis pneumonia during the 6-month period after renal transplantation. This low dose (400 mg) of SMX might not have been sufficient to reach the plasma concentration necessary to observe side effects. The AUC0–24 range of SMX in the 15 recipients with the NAT2 SA genotype on day 14 after SMX administration was 379 to 1,732 μg-h/ml, while a SMX AUC0–24 greater than this range would be expected to be required for expression of side effects.
The SMX AUC0–24 for two patients with both NAT2 SA and CYP2C9*1/*3 alleles were 400 and 879 μg-h/ml, whereas the mean (± SD) AUC0–24 of SMX for 13 patients with both the NAT2 SA and CYP2C9*1/*1 alleles was 786 ± 453 μg-h/ml. The contribution of CYP2C9 to SMX pharmacokinetics appears to be small, and the metabolic pathway via NAT2 appears to be more important. Cribb et al. have reported that the metabolism of SMX to SMX-hydroxylamine is also catalyzed by neutrophilic myeloperoxidase (3). Thus, there may be a small influence of CYP2C9 polymorphisms, since enzymes other than CYP2C9 could contribute to the metabolism of SMX to SMX-hydroxylamine. In the present study, because the plasma concentration of SMX-hydroxylamine was not monitored, we were not able to compare differences in the pharmacokinetics of SMX-hydroxylamine between patients in the NAT2 SA group and the RA or IA group. A higher plasma concentration of SMX-hydroxylamine, which can undergo oxidation to nitroso-sulfamethoxazole (4, 13, 14), is a possible source of SMX-derived toxicity (2); however, further examination of this proposal is necessary.
In the multivariate analysis, the SMX AUC0–24 was significantly associated with creatinine clearance (P = 0.0499). These data confirmed that SMX is eliminated via the urine, and therefore the SMX concentration could be elevated significantly in patients with a lower creatinine clearance, a finding in agreement with previous reports (17, 18, 21). The elimination half-life of SMX was reported to be 8 to 14 h for patients with normal renal function and 20 to 50 h for patients with end-stage renal disease (17, 18). Therefore, a reduced SMX dosage has been recommended for patients with a creatinine clearance between 15 and 30 ml/min (21). Generally, the dose of SMX-trimethoprim for the treatment of Pneumocystis jirovecii pneumonia is 1,600 mg/day-320 mg/day, given as 4 tablets daily. Consequently, for renal transplant recipients, a SMX-trimethoprim dosage of one tablet with a dose of 400 mg/day-80 mg/day would seem to be safe.
On the other hand, an in vitro study of SMX at a very high concentration (500 μM) showed a 30 to 40% inhibition of the activity of CYP3A4, a member of the CYP3A isozyme family; however, it has been reported that clinical doses of SMX do not affect the pharmacokinetics of cyclosporine and sirolimus, which are CYP3A4 substrates, in renal transplant patients (1, 9). In the present study, the mean (± SD) dose-adjusted AUC0-12 of tacrolimus, a substrate for CYP3A4, in recipients with the NAT2 SA and RA or IA genotype were 43.0 ± 21.9 ng-h/ml/mg and 41.6 ± 18.9 ng-h/ml/mg, respectively (P = 0.787), and an influence of NAT2 polymorphisms on tacrolimus pharmacokinetics was not observed. Thus, the administration of 400 mg/kg SMX does not appear to affect the pharmacokinetics of tacrolimus in renal transplant patients.
In conclusion, SMX plasma concentrations were affected by renal dysfunction and NAT2 polymorphisms, whereas an influence of CYP2C9 polymorphisms on SMX pharmacokinetics was not observed. Patients with NAT2 SA polymorphisms who receive SMX should be monitored for side effects and drug interactions via an inhibitory effect on CYP2C9. However, SMX administration at a low dose (400 mg) as prophylaxis against Pneumocystis pneumonia might not reach the necessary drug level for expression of side effects. By carrying out further studies with a larger sample size, the relationship of SMX plasma concentration and side effects should become clearer.
ACKNOWLEDGMENTS
This work was supported by grants from the Japan Research Foundation for Clinical Pharmacology, Tokyo, Japan, and the Research Foundation for Pharmaceutical Sciences, Tokyo, Japan.
Footnotes
Published ahead of print 21 November 2011
REFERENCES
- 1. Böttiger Y, Brattström C, Bäckman L, Claesson K, Burke JT. 2005. Trimethoprim-sulphamethoxazole does not affect the pharmacokinetics of sirolimus in renal transplant recipients. Br. J. Clin. Pharmacol. 60:566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carr A, Tindall B, Penny R, Cooper DA. 1993. In vitro cytotoxicity as a marker of hypersensitivity to sulphamethoxazole in patients with HIV. Clin. Exp. Immunol. 94:21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cribb AE, Miller M, Tesoro A, Spielberg SP. 1990. Peroxidase-dependent oxidation of sulfonamides by monocytes and neutrophils from humans and dogs. Mol. Pharmacol. 38:744–751 [PubMed] [Google Scholar]
- 4. Cribb AE, Miller M, Leeder JS, Hill J, Spielberg SP. 1991. Reactions of the nitroso and hydroxylamine metabolites of sulfamethoxazole with reduced glutathione. Implications for idiosyncratic toxicity. Drug Metab. Dispos. 19:900–906 [PubMed] [Google Scholar]
- 5. Cribb AE, Spielberg SP, Griffin GP. 1995. N4-hydroxylation of sulfamethoxazole by cytochrome P450 of the cytochrome P4502C subfamily and reduction of sulfamethoxazole hydroxylamine in human and rat hepatic microsomes. Drug Metab. Dispos. 23:406–414 [PubMed] [Google Scholar]
- 6. Gill HJ, et al. 1999. The effect of genetic polymorphisms in CYP2C9 on sulphamethoxazole N-hydroxylation. Pharmacogenetics 9:43–53 [DOI] [PubMed] [Google Scholar]
- 7. Kimura R, et al. 2007. Genotypes of vitamin K epoxide reductase, gamma-glutamyl carboxylase, and cytochrome P450 2C9 as determinants of daily warfarin dose in Japanese patients. Thromb. Res. 120:181–186 [DOI] [PubMed] [Google Scholar]
- 8. Liu HE, Hsiao PY, Lee CC, Lee JA, Chen HY. 2008. NAT2*7 allele is a potential risk factor for adult brain tumors in Taiwanese population. Cancer Epidemiol. Biomarkers Prev. 17:661–665 [DOI] [PubMed] [Google Scholar]
- 9. Maki DG, Fox BC, Kuntz J, Sollinger HW, Belzer FO. 1992. A prospective, randomized, double-blind study of trimethoprim-sulfamethoxazole for prophylaxis of infection in renal transplantation. Side effects of trimethoprim-sulfamethoxazole, interaction with cyclosporine. J. Lab. Clin. Med. 119:11–24 [PubMed] [Google Scholar]
- 10. Meyer UA, Zanger UW. 1997. Molecular mechanisms of genetic polymorphisms of drug metabolism. Annu. Rev. Pharmacol. Toxicol. 37:269–296 [DOI] [PubMed] [Google Scholar]
- 11. Moridani M, et al. 2006. Frequency of CYP2C9 polymorphisms affecting warfarin metabolism in a large anticoagulant clinic cohort. Clin. Biochem. 39:606–612 [DOI] [PubMed] [Google Scholar]
- 12. Mushiroda T, et al. 2006. Association of VKORC1 and CYP2C9 polymorphisms with warfarin dose requirements in Japanese patients. J. Hum. Genet. 51:249–253 [DOI] [PubMed] [Google Scholar]
- 13. Naisbitt DJ, et al. 2002. Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex-restricted antigen presentation. Mol. Pharmacol. 62:628–637 [DOI] [PubMed] [Google Scholar]
- 14. Naisbitt DJ, O'Neill PM, Pirmohamed M, Park BK. 1996. Synthesis and reactions of nitroso sulfamethoxazole with biological nucleophiles: implications for immune-mediated toxicity. Bioorg. Med. Chem. Lett. 6:1511–1516 [Google Scholar]
- 15. Nakamura H, et al. 1995. In vitro formation, disposition and toxicity of N-acetoxy-sulfamethoxazole, a potential mediator of sulfamethoxazole toxicity. J. Pharmacol. Exp. Ther. 274:1099–1104 [PubMed] [Google Scholar]
- 16. Obayashi K, et al. 2006. VKORC1 gene variations are the major contributors of variation in warfarin dose in Japanese patients. Clin. Pharmacol. Ther. 80:169–178 [DOI] [PubMed] [Google Scholar]
- 17. Paap CM, Nahata MC. 1989. Clinical use of trimethoprim/sulfamethoxazole during renal dysfunction. DICP 23:646–654 [DOI] [PubMed] [Google Scholar]
- 18. Rivey MP, Taylor JW, Mullenix TA. 1989. TMP/SMX in renally impaired patients with P. carinii pneumonia. DICP 23:687–689 [PubMed] [Google Scholar]
- 19. Smith CA, Smith G, Wolf CR. 1994. Genetic polymorphisms in xenobiotic metabolism. Eur. J. Cancer 30A:1921–1935 [DOI] [PubMed] [Google Scholar]
- 20. Soejima M, et al. 2007. Association of the diplotype configuration at the N-acetyltransferase 2 gene with adverse events with co-trimoxazole in Japanese patients with systemic lupus erythematosus. Arthritis Res. Ther. 9:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strevel EL, Kuper A, Gold WL. 2006. Severe and protracted hypoglycaemia associated with co-trimoxazole use. Lancet Infect. Dis. 6:178–182 [DOI] [PubMed] [Google Scholar]
- 22. Zielińska E, et al. 1998. Genotyping of the arylamine N-acetyltransferase polymorphism in the prediction of idiosyncratic reactions to trimethoprim-sulfamethoxazole in infants. Pharm. World Sci. 20:123–130 [DOI] [PubMed] [Google Scholar]