

Abstract
Rhesus TRIM5α (TRIM5αrh) is a cytosolic protein that potently restricts HIV-1 at an early postentry stage, prior to reverse transcription. The ability of TRIM5αrh to block HIV-1 infection has been correlated with a decrease of pelletable HIV-1 capsid during infection. To genetically dissect the ability of TRIM5α to block reverse transcription, we studied a set of TRIM5αrh RING domain mutants that potently restrict HIV-1 but allow the occurrence of reverse transcription. These TRIM5αrh RING variants blocked HIV-1 infection after reverse transcription but prior to integration, as suggested by the routing of nuclear viral DNA to circularization in the form of 2-long terminal repeat (2-LTR) circles. The folding of RING domain variants was similar to that of the wild type, as evaluated by nuclear magnetic resonance. RING domain changes that allowed the occurrence of reverse transcription were impaired in their ability to decrease the amount of pelletable capsid compared with wild-type TRIM5α. Similar effects of this particular group of mutations were observed with human TRIM5α inhibition of N-tropic murine leukemia virus (N-MLV). Interestingly, TRIM5αrh RING domain variants also prevented the degradation of TRIM5αrh that occurs following cell entry of HIV-1. These data correlated the block of reverse transcription with the ability of TRIM5α to accelerate uncoating. Collectively, these results suggest that TRIM5αrh blocks HIV-1 reverse transcription by inducing premature viral uncoating in target cells.
INTRODUCTION
Several newly discovered proteins endogenously expressed in primates show the ability to dominantly block retroviral infection and cross-species transmission by interfering with the early phase of viral replication (27, 46, 51). Of particular interest are members of the tripartite motif (TRIM) family of proteins (43). The splicing variant alpha of TRIM5 from rhesus macaque (TRIM5αrh) is an ∼53-kDa cytosolic protein that potently restricts HIV-1 (24, 49). TRIM5αrh blocks HIV-1 and certain other retroviruses soon after viral entry but prior to reverse transcription (24, 51). The retroviral capsid protein (CA) is the viral determinant for susceptibility to restriction by TRIM5α (38). Studies on the fate of the HIV-1 capsid in the cytosol of infected cells have correlated restriction with a decreased amount of cytosolic particulate capsid (10, 13, 41, 52), suggesting that TRIM5αrh acts by inducing premature uncoating in target cells.
TRIM5αrh is composed of four distinct domains: RING, B-box 2, coiled-coil, and B30.2(SPRY) (43). The RING domain of TRIM5αrh is an E3 ubiquitin ligase (12, 23, 26, 28, 31, 32, 57). The E3 ligase activity of TRIM5αrh correlates with the ability of TRIM5αrh to block HIV-1 (31). The B-box 2 domain of TRIM5αrh and other TRIM proteins, such as TRIM63, self-associates into dimeric complexes that are important for TRIM5α higher-order self-association (HOSA) and capsid binding avidity; these B-box 2 domain functions are essential for full and potent restriction of HIV-1 (11, 14, 20, 22, 34, 39). The coiled-coil domain enables TRIM5αrh dimerization (23, 28), which is critical for interaction of the B30.2(SPRY) domain with the HIV-1 capsid (15, 47, 52). The B30.2(SPRY) domain provides the capsid recognition motif that dictates the specificity of restriction (35, 45, 50, 53, 59).
TRIM5αrh exhibits an intrinsic intracellular turnover of 50 to 60 min that is dependent on an intact RING domain, but this property is apparently not important for restriction (10, 12, 57). However, TRIM5αrh degrades at a faster rate than its normal turnover when in the presence of HIV-1 capsid (44). Interestingly, TRIM5αrh capsid-dependent degradation is prevented by chemical inhibition of proteasome activity. Substantial evidence has linked the ubiquitin-proteasome system to HIV-1 restriction by TRIM5αrh. Retroviral restriction by TRIM5α results in abortive reverse transcription in target cells. However, inhibition of proteasome activity by drugs allows the completion of reverse transcription in the presence of TRIM5αrh without affecting the inhibition of HIV-1 (1, 13, 40, 52, 55). These results suggest that TRIM5α might act at multiple steps in the retroviral life cycle.
To genetically dissect the ability of TRIM5α to block reverse transcription, we screened a large set of TRIM5αrh RING domain mutants in order to find variants that would potently restrict HIV-1 while still allowing the occurrence of reverse transcription. Interestingly, we found that TRIM5αrh RING domain residue Y63 is important for the ability of the protein to block reverse transcription. Changes in position Y63 impaired the ability of TRIM5αrh to block HIV-1 reverse transcription without affecting retroviral restriction. Corresponding changes in the TRIM5αhu orthologue Y62 generated variants that allowed the occurrence of N-tropic murine leukemia virus (N-MLV) reverse transcription but potently restricted infection. TRIM5αrh Y63 variants blocked HIV-1 after reverse transcription but prior to integration, as circularization of nuclear viral DNA in the form of 2-long terminal repeat (2-LTR) was observed. Next, we tested the ability of the mutants to decrease the amount of pelletable capsid or accelerate uncoating during infection. RING domain changes that allowed the occurrence of reverse transcription were impaired in their ability to accelerate uncoating. These results correlated the occurrence of reverse transcription with the inability of TRIM5α to decrease the amount of pelletable capsid (or acceleration of uncoating) during infection, suggesting that reverse transcription occurs prior to or during uncoating. Altogether, these results suggested that the ability of TRIM5α to block reverse transcription is linked to the acceleration of uncoating.
MATERIALS AND METHODS
Creation of cells stably expressing TRIM5α variants.
Retroviral vectors encoding wild-type (WT) or mutant rhesus monkey TRIM5αrh proteins were created using the pLPCX vector, as previously described (31). Cf2Th canine thymocytes were transduced with the different constructs and selected in 5 μg/ml of puromycin (Sigma).
Infection with viruses expressing GFP.
Recombinant HIV-1 and N-MLV expressing green fluorescent protein (GFP) were prepared as described previously (13). All recombinant viruses were pseudotyped with the vesicular stomatitis virus (VSV)-G glycoprotein. For infections, 3 × 104 Cf2Th cells seeded in 24-well plates were incubated at 37°C with virus for 24 h. Cells were washed and returned to culture for 48 h and then subjected to fluorescence-activated cell sorter (FACS) analysis with a FACScan (Becton Dickinson). HIV-1 and N-MLV viral stocks were titrated by serial dilution on Cf2Th cells to determine the concentration of infectious viruses.
Protein analysis.
Cellular proteins were extracted with radioimmunoprecipitation assay (RIPA) buffer, as previously described (31). Detection of protein by Western blotting utilized monoclonal antibodies directed against the hemagglutinin (HA) epitope tags (Roche), FLAG epitope tags (Sigma), and monoclonal antibodies against β-actin (Sigma) conjugated to Alexa Fluor 680. Bands were detected by scanning blots with the LI-COR Odyssey imaging system using both 700 and 800 channels, and integrated intensities were determined using the LI-COR Odyssey band quantitation software with the median top-bottom background subtraction method.
TRIM5α self-ubiquitylation.
Proteins encoding FLAG-tagged mutant and wild-type TRIM5αrh proteins were prepared as previously described (31). Next, similar amounts of mutant and wild-type TRIM5α proteins were treated with 5 μM ubiquitin aldehyde, a potent inhibitor of all ubiquitin C-terminal hydrolases, ubiquitin-specific proteases, and deubiquitylating enzymes (BostonBiochem). The inhibitor-treated fractions containing mutant and wild-type TRIM5αrh were incubated in a final reaction mixture containing 200 nM E1 (human recombinant UBE1) (BostonBiochem), 100 nM E2 (human recombinant UbcH5b) (BostonBiochem), 200 μM ubiquitin tagged with a myc epitope (human recombinant ubiquitin), and ATP (energy regeneration solution containing MgCl2, ATP, and ATP-regenerating enzymes to recycle hydrolyzed ATP) (BostonBiochem). The reaction was incubated at 37°C for 1 h, and collected fractions were analyzed by Western blotting using horseradish peroxidase (HRP)-conjugated antibodies against FLAG and myc. Similar reactions were performed in the absence of recombinant E1 and E2 enzymes to determine the contribution of endogenous E1 and E2 enzymes to TRIM5αrh ubiquitylation.
HOSA of TRIM5α.
Human 293T cells were independently transfected with plasmids encoding FLAG-tagged and HA-tagged mutant or wild-type TRIM5αrh proteins. Forty-eight hours later, the cells expressing each TRIM5αrh variant were lysed in 1 ml of whole-cell extract buffer (50 mM Tris [pH 8.0], 280 mM NaCl, 0.5% Igepal-10% glycerol, 1 mM dithiothreitol [DTT], protease inhibitor cocktail [Roche]). Lysates were centrifuged at 14,000 rpm for 1 h at 4°C. Postspin lysates were then precleared using Protein-A-agarose (Sigma) for 1 h at 4°C; a small aliquot of each lysates was stored as an input sample. Precleared lysates containing the differently tagged proteins were mixed in a 1/1 ratio and incubated with anti-FLAG-agarose beads (Sigma) for 2 h at 4°C to precipitate the FLAG-tagged proteins. Beads containing the immunoprecipitate were washed four times in whole-cell extract buffer. Next, immune complexes were eluted with 200 μg/ml of FLAG tripeptide in whole-cell extract buffer. The eluted samples were separated by SDS-PAGE and analyzed by Western blotting using anti-HA or anti-FLAG antibodies conjugated to Alexa Fluor 680.
Binding of TRIM5αrh variants to HIV-1 capsid complexes.
The HIV-1 CA-NC protein was expressed, purified, and assembled as previously described (19, 21). HIV-1 CA-NC particles were assembled in vitro by diluting the CA-NC protein to a concentration of 0.3 mM in 50 mM Tris-HCl (pH 8.0), 0.5 M NaCl and 2 mg/ml DNA oligo(TG)50. The mixture was incubated at 4°C overnight and centrifuged at 8,600 × g for 5 min. These CA-NC complexes were used for the binding assay as follows. 293T cells were transfected with plasmids expressing wild-type or mutant TRIM5αrh proteins. Forty-eight hours after transfection, cell lysates were prepared as follows: washed cells were resuspended in hypotonic lysis buffer (10 mM Tris [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT). The cell suspension was frozen and thawed and then incubated on ice for 10 min. Next, the lysate was centrifuged at full speed in a refrigerated Eppendorf microcentrifuge (∼14,000 × g) for 5 min. The supernatant was supplemented with 1/10 volume of 10× phosphate-buffered saline (PBS) and then used in the binding assay. In some cases, samples containing the TRIM5αrh variants were diluted with extracts prepared in parallel from untransfected cells. To test binding, 5 μl of CA-NC complexes assembled in vitro were incubated with 200 μl of cell lysate at room temperature for 1 h. A fraction of this mixture was stored (input). The mixture was spun through a 70% sucrose cushion (70% sucrose, 1× PBS, and 0.5 mM DTT) at 100,000 × g in an SW55 rotor (Beckman) for 1 h at 4°C. After centrifugation, the supernatant was carefully removed and the pellet was resuspended in 1× SDS-PAGE loading buffer (pellet). The level of TRIM5αrh proteins was determined by Western blotting with an anti-HA antibody as described above. The level of HIV-1 CA-NC protein in the pellet was assessed by Western blotting with an anti-p24 capsid antibody.
Assays of the fate of the HIV-1 and N-MLV capsids.
HIV-1 virus-like particles (VLPs) were produced by calcium phosphate cotransfection of plasmids containing the following genes: HIV-1 gag-pol, VSV-G envelope, and rev protein at a weight ratio of 15:3:1. N-MLV VLPs were produced by calcium phosphate cotransfection of 293T cells with pCIG-N (6), along with pVPack-VSV-G at a weight ratio of 15:4. Stably transduced Cf2Th cells (1.5 × 106) expressing the different TRIM5α variants were seeded in 80-cm2 flasks. The following day, the cells were incubated with 5 to 10 ml (approximately 2.5 × 105 to 5.0 × 105 reverse transcriptase units) of HIV-1 or N-MLV VLPs at 4°C for 30 min to allow viral attachment to the cells. The cells were then shifted to 37°C until they were harvested at 4 to 12 h postinfection. Cells were washed three times using ice-cold PBS and detached by treatment with 1.0 ml of pronase (7.0 mg/ml in Dulbecco's modified Eagle's medium [DMEM]) for 5 min at 25°C. The cells were then washed three times with PBS. The cells were resuspended in 2.5 ml of hypotonic lysis buffer (10 mM Tris-HCl [pH 8.0], 10 mM KCl, 1 mM EDTA, and one Complete protease inhibitor tablet) and incubated on ice for 15 min. The cells were lysed using 15 strokes in a 7.0-ml Dounce homogenizer with pestle B. Cellular debris was cleared by centrifugation for 3 min at 3,000 rpm. To allow assessment of the input for HIV-1 p24 and MLV p30 capsid proteins, 100 μl of the cleared lysate was collected, made 1× in SDS sample buffer, and analyzed by SDS-PAGE and Western blotting. Then, 2.0 ml of the cleared lysate was layered onto a 50% sucrose (weight:volume) cushion in 1× PBS and centrifuged at 125,000 × g for 2 h at 4°C in a Beckman SW41 rotor. Following centrifugation, 100 μl of the topmost portion of the supernatant was collected and made 1× in SDS sample buffer; this sample is referred to as the soluble p24 (HIV-1 CA) or p30 (N-MLV CA) fraction. The pellet was resuspended in 50 μl 1× SDS sample buffer and is referred to as the particulate p24 or p30 fraction. All samples were then subjected to SDS-PAGE and Western blotting. The HIV-1 p24 and MLV p30 proteins were detected using a mouse anti-p24 antibody (Immunodiagnostics) and a rat monoclonal antibody purified from the R187 hybridoma cell line (American Type Culture Collection), respectively. The HIV-1 samples were also quantified by p24 enzyme-linked immunosorbent assay (ELISA).
Quantitative real-time PCR for the detection of HIV-1 and N-MLV reverse transcription.
Canine Cf2Th cells expressing wild-type and mutant TRIM5αrh proteins were challenged with HIV-1–GFP at a multiplicity of infection (MOI) of 0.2. Viruses were pretreated with DNase to prevent contamination from carryover plasmid DNA. An infection using heat-inactivated virus (60°C for 30 min) was performed as a control for carryover plasmid DNA in the PCR. After 6 h, the cells were lysed and DNA was extracted using a Qiagen blood tissue DNA extraction kit. PCRs were prepared using the QuantiTect probe PCR kit. Each sample contained 100 ng of total cellular DNA. PCR was carried out using two primers and a probe that amplify a 263-bp fragment of GFP: GFP-fwd, 5′-GAC GTA AAC GGC CAC AAG-3′; GFP-rev, 5′-GGT CTT GTA GTT GCC GTC GT-3′; and GFP-Probe, 5′-56-FAM-CCT ACG GCA AGC TGA CCC TGA-36-TAMRA-3′. The calibration curve was prepared using an HIV-1–GFP plasmid.
Quantitative real-time PCR for detection of HIV-1 2-LTR circles.
Canine Cf2Th cells expressing wild-type and mutant TRIM5αrh proteins were seeded at a density of 0.2 million cells per well in 12-well plates 1 day prior to inoculation. VSV-G-pseudotyped Env-defective HIV-1 particles (R9 clone) were treated with DNase I (20 μg/ml with 10 μM MgCl2) for 1 h at 37°C to remove contaminating plasmid DNA. Cultures were inoculated with the virus (10 ng p24 in 1 ml) in the presence of 20 μg/ml DEAE-dextran. Twenty-four hours after inoculation, the medium was aspirated, cell monolayers were rinsed in PBS, and the cells were subsequently detached with trypsin. Cells were pelleted and the total cellular DNA was purified using DNeasy columns (Qiagen). 2-LTR circles were quantified by the method of Dismuke and Aiken (16) using plasmid p2-LTR as a standard (7). As a control to determine the level of contaminating HIV-1 plasmid DNA, parallel analyses were performed with cultures inoculated in the presence of the reverse transcriptase inhibitor efavirenz (1 μg/ml). Because the level of 2-LTR circles is affected by the efficiency both of nuclear entry and of integration, HIV-1 2-LTR circles were analyzed in cells cultured in the presence of the integrase inhibitor raltegravir to eliminate the consequences of any effects of the TRIM5α mutations on the efficiency of integration.
Assays of HIV-1-induced destabilization of TRIM5α.
TRIM5αrh-expressing Cf2Th cells were precultured in medium containing cycloheximide for 1 h and then inoculated with VSV-G-pseudotyped HIV-1 and cultured for 5 h in cycloheximide-containing medium. Cells were harvested and lysates prepared and analyzed by immunoblotting using monoclonal antibody specific for the HA tag on TRIM5α, as previously described (44). Blots were reprobed with actin-specific monoclonal antibody for normalization, and images obtained and bands quantified using the Odyssey imaging software. The level of TRIM5α was expressed as a percentage of the levels in corresponding control samples that were cultured in the absence of the virus.
NMR measurements of TRIM5α RING domains.
The DNA fragments encoding the wild-type or mutant RING domain of TRIM5αrh (1 to 79) and TRIM5αhu (1 to 78) were amplified via PCR as a fusion with an N-terminal His tag and tobacco etch virus (TEV) protease cleavage site (56). The 15N-labeled proteins were produced by cell-free protein synthesis with optimization for zinc-binding proteins (25, 33) and were purified by immobilized metal affinity chromatography using an automated system (2). The eluted protein from the cell-free reaction mixture was cleaved with TEV protease to remove the His tag and was used for nuclear magnetic resonance (NMR) measurements. The final concentrations of samples were 0.24 mM TRIM5αrh RING (wild-type), 0.23 mM TRIM5αrh RING (Y63E), 0.32 mM TRIM5αhu RING (wild-type), and 0.26 mM TRIM5αhu RING (Y62E). Proteins were eluted in 20 mM Tris-HCl buffer at pH 7.5, 300 mM NaCl, 1 mM dithiothreitol, 34 mM nitrilotriacetic acid (NTA)-Zn, with the addition of D2O to 10% (vol/vol). The two-dimensional 1H-,15N-HSQC spectra were recorded at 25°C on a Bruker Avance 600 spectrometer equipped with pulse-field gradient triple-resonance CryoProbe.
RESULTS
Identification of TRIM5α RING domain variants that potently restrict HIV-1 after reverse transcription.
By correlating the E3 ligase activity with HIV-1 restriction, we previously demonstrated that the E3 ubiquitin ligase activity of the RING domain is important for potent restriction of HIV-1 by TRIM5αrh (31). Because deletions in the RING domain of TRIM5αrh did not relieve restriction (data not shown) yet allowed the occurrence of reverse transcription, we sought to identify mutations in the RING domain of TRIM5α that potently block HIV-1 while compromising the ability of the protein to block reverse transcription. For this purpose, we stably expressed a large set of TRIM5αrh RING domain variants in canine Cf2Th cells, as part of our previously described screen (31), and tested them for the ability to block infection and reverse transcription. Interestingly, we found mutations in the RING domain of TRIM5αrh that potently restricted HIV-1 after reverse transcription (Fig. 1A, B, and E and Table 1). TRIM5αrh RING domain Y63D, Y63E, and Y63K variants potently blocked HIV-1 (Fig. 1A) but completely restored the reverse transcription process (Fig. 1B). By contrast, Y63A, Y63F, and Y62S variants blocked HIV-1 and prevented the occurrence of reverse transcription to a similar extent as the wild-type TRIM5αrh. In order to corroborate these findings, we generated similar changes on the RING domain of human TRIM5α (TRIM5αhu). In agreement, TRIM5αhu RING domain Y62F, Y62D, Y62E, and Y62K variants potently blocked N-MLV infection but allowed the occurrence of reverse transcription (Fig. 1C, D, and E and Table 2). TRIM5αhu RING domain Y62A and Y62S variants blocked N-MLV infection and prevented the occurrence of reverse transcription to a lesser extent than the wild-type TRIM5αhu. These results provide genetic evidence that TRIM5α can act at more than one step in the HIV-1 retroviral life cycle, as it has been previously suggested by experiments using proteasome inhibitors and TRIMCyp fusion constructs (44, 55, 58).
Fig 1.

Restriction of HIV-1 and N-MLV infection after reverse transcription. Cf2Th cells were transduced with the LPCX vector expressing HA-tagged wild-type and mutant TRIM5α proteins. Stable cell lines were selected with 5 μg/ml of puromycin. CF2Th cell lines expressing the indicated wild-type and TRIM5α RING domain variants or containing the empty LPCX vector were challenged with different amounts of HIV-1–GFP (A) or N-MLV–GFP (C). Infection was determined by measuring the number of GFP-positive cells 48 h postinfection by flow cytometry. Similar results were obtained in three independent experiments, and the result of one experiment is shown. Similarly, CF2Th cell lines expressing the indicated wild-type and mutant TRIM5α proteins or containing the empty LPCX vector were challenged at an MOI of 0.4 with DNase-pretreated HIV-1–GFP (B) or N-MLV–GFP (D) viruses. After 7 h, cells were lysed and total DNA was extracted. The levels of viral DNA were measured by quantitative real-time PCR, using a probe against GFP, as described in Materials and Methods. Similar results were obtained in three independent experiments, and the standard deviation is shown. (E) The expression level of mutant and wild-type TRIM5α proteins was assayed by Western blotting using HRP-conjugated antibodies against HA. (F) CF2Th cell lines expressing the indicated wild-type and mutant TRIM5αrh proteins or containing the empty LPCX vector were challenged at an MOI of 0.4 with DNase-pretreated VSV-G-pseudotyped Env-defective HIV-1 viruses (R9 clone). After 24 h, cells were lysed and total DNA was extracted. The levels of HIV-1 2-LTR circles were measured by quantitative real-time PCR, using specific primers to detect the junction of 2-LTR circles, as described in Materials and Methods. Similar results were obtained in three independent experiments, and the standard deviation is shown. In panels A and C, black arrows point out the viral doses of HIV-1 and N-MLV used to measure reverse transcription.
Table 1.
Phenotypes of TRIM5α RING Y63 variants
| TRIM5αrh variant | Restriction potency against HIV-1a | HIV-1 reverse transcriptionb | HIV-1 2-LTR circle formationc | % TRIM5 self-ubiquitylation (± SD)d | Particulate HIV-1 cytoplasmic capsidse | Mean binding to HIV-1 CA-NC complexes (± SD)f | % HOSA (± SD)g |
|---|---|---|---|---|---|---|---|
| WT | + | − | − | 100.0 | − | 1 | 100.0 |
| Y63A | + | − | − | 50.0 (8.3)*** | − | 0.9 (0.2) NS | 18.6 (4.3) |
| Y63F | + | − | − | 80.9 (11.5)* | ND | 0.9 (0.1) NS | 79.3 (7.3) |
| Y63D | + | + | + | 31.0 (4.5)*** | + | 1.2 (0.1) NS | 8.7 (5.7) |
| Y63E | + | + | + | 40.6 (5.6)*** | + | 0.8 (0) NS | 19.4 (7.2) |
| Y63K | + | + | + | 30.6 (10.1)*** | + | 0.4 (0.2)*** | 18.6 (5.4) |
| Y63S | + | − | − | 50.6 (9.7)*** | − | 1.0 (0.1) NS | 15.7 (4.3) |
Restriction was measured by infecting cells expressing the indicated TRIM5αrh variants with HIV-1 expressing the GFP protein. After 48 h, the percentage of GFP-positive cells (infected cells) was determined by flow cytometry. +, Restriction comparable to that of the wild-type protein. Experiments were performed at least three times.
HIV-1 reverse transcription was measured by real-time PCR 7 h after infection, as described in Materials and Methods. −, Absence of reverse transcription. +, Occurrence of reverse transcription at levels similar to those observed in control cells transduced with the empty LPCX vector when infected with HIV-1 (or N-MLV, as shown in Table 2). Experiments were performed at least three times.
Formation of HIV-1 2-LTR circles was measured by real-time PCR 24 h after infection, as described in Materials and Methods. The presence (+) or absence (−) of HIV-1 2-LTR circles is indicated. Experiments were performed at least three times.
To investigate the E3 ubiquitin ligase activity of TRIM5αrh RING domain variants, the ability of these variants to undergo self-ubiquitylation was assayed. Semipurified Flag-tagged TRIM5α variants from transfected 293T cells by immunoprecipitation were incubated with E1, E2, myc-tagged ubiquitin, and ATP, for 1 h at 37°C as described in Materials and Methods. Samples were analyzed by Western blotting using antibodies against FLAG and myc for the detection of TRIM5α and ubiquitin, respectively. The amount of TRIM5α ubiquitylated protein was determined by subtracting the amount of nonubiquitylated TRIM5α protein remaining in the reaction mixture incubated with E1 and E2 from the TRIM5α protein in the control reaction mixture, which was not incubated with E1 and E2. The value of nonubiquitylated TRIM5α for the reactions was quantified by using fluorescent Western blotting. TRIM5α self-ubiquitylation was expressed as the percentage of total TRIM5α variant input protein. Experiments were performed at least three times and standard deviations (SD) are shown. Statistical differences when compared with wild-type TRIM5α are given as follows: ***P < 0.001; *P < 0.05 (two-way analysis of variance [ANOVA] followed by the Bonferroni posttest).
The amount of particulate capsids for HIV-1 during infection of cells expressing the indicated TRIM5α RING domain variants was measured as described in Materials and Methods. −, Absence of particulate capsids during infection, similar to what is observed in the presence of the wild-type TRIM5α. +, Presence of particulate capsids in an amount similar to that observed in HIV-1 (or N-MLV, as shown in Table 2) infection of control cells that were transduced with the empty vector LPCX. Experiments were performed at least twice. ND, not determined.
Binding to the HIV-1 capsid complexes was determined for each TRIM5αrh RING domain variant as described in Materials and Methods. Binding is expressed as the amount of the TRIM5αrh variant bound to HIV-1 capsid complexes divided by the amount of bound wild-type TRIM5αrh at a similar input level. Experiments were repeated at least three times; the means and standard deviations (SD) are shown. Note that, because the binding ratios are calculated at input levels at which some binding of the mutant TRIM5αrh protein to the HIV-1 capsid complexes can be detected, these ratios overestimate the relative CA-binding affinities of the mutant proteins. Statistical differences when compared with wild-type TRIM5α binding are given as follows: ***P < 0.001; NS, not significant with P > 0.05; two-way ANOVA followed by the Bonferroni posttest.
Each TRIM5αrh RING domain variant was assayed for HOSA as described in Materials and Methods. The percentage represents the fraction of the TRIM5αrh variant coprecipitated with itself, relative to the coprecipitation of wild-type TRIM5αrh with itself. Experiments were performed at least three times.
Table 2.
Phenotypes of TRIM5α RING Y62 variants
| TRIM5αhu variant | Restriction potency against N-MLVa | N-MLV reverse transcriptionb | Particulate N-MLV cytoplasmic capsidsc |
|---|---|---|---|
| WT | + | − | − |
| Y62A | + | +/− | +/− |
| Y62F | + | + | + |
| Y62D | + | + | + |
| Y62E | + | + | + |
| Y62K | + | + | + |
| Y62S | + | − | − |
Restriction was measured by infecting cells expressing the indicated TRIM5αrh variants with N-MLV expressing the GFP protein. See Table 1 footnote a for further details.
N-MLV reverse transcription. See Table 1 footnote b for further details.
Amount of particulate capsids for N-MLV. See Table 1 footnote e for further details.
TRIM5αrh RING domain variants block HIV-1 infection after reverse transcription but prior to integration.
Because TRIM5αrh RING domain variants blocked HIV-1 infection after reverse transcription, we decided to test the ability of these variants to generate HIV-1 2-LTR circles. Formation of 2-LTR circles would suggest the nuclear transport of the preintegration complex and the consequent routing of nuclear viral DNA to circularization (7). For this purpose, we challenged canine Cf2Th cells stably expressing the different TRIM5αrh RING domain variants with HIV-1 at an MOI of 0.4 and extracted DNA after 24 h for assessment of HIV-1 2-LTR circles by real-time PCR (7). TRIM5αrh RING domain Y63D, Y63E, and Y63K variants allowed more formation of HIV-1 2-LTR circles than did wild-type TRIM5αrh (Fig. 1F and Table 1). In agreement with these results, TRIM5αrh RING domain changes that block HIV-1 reverse transcription (Fig. 1B) did not lead to the formation of HIV-1 2-LTR circles (Fig. 1F). These results indicated that TRIM5αrh RING domain Y63D, Y63E, and Y63K variants restrict HIV-1 after reverse transcription but prior to integration.
Folding of RING domain mutants.
The NMR structure of the RING domain revealed that tyrosine 63 of TRIM5αrh is located in the hydrophobic core of the domain (Fig. 2A) (31). Because changes in the hydrophobic core of the domain might result in protein misfolding, we tested the effect of these changes in the folding of the RING domain. To examine the fold of RING domain mutants, the wild type and the Y63E variant of TRIM5αrh (1 to 79) were expressed in the presence of labeled amino acids, as described in Materials and Methods. Two-dimensional 1H-, 15N-HSQC spectrum analysis of the labeled proteins revealed that the RING domain bearing the Y63E mutation was folded similarly to the wild-type RING domain (Fig. 2B). However, there were at least six important chemical shifts in the Y63E spectrum compared to the wild type that could be responsible for the observed phenotypes. Similar results were obtained for the RING domain of TRIM5αhu (1 to 78) bearing the mutation Y62E (data not shown). These results demonstrated that the RING domain of TRIM5αrh and TRIM5αhu bearing the Y63E and Y62E mutations, respectively, folded similarly to the wild-type domain.
Fig 2.

Protein folding of RING domain mutants. (A) Structure of the RING domain of TRIM5αrh illustrating the position of tyrosine 63 (red). The side chain of tyrosine 63 is part of the hydrophobic core of the RING domain and is surrounded by the hydrophobic side chains of residues L28, I39, I61, P65, and I68. (B) The two-dimensional 1H, 15NHSQC spectra of wild-type (black) and Y63E (red) RING domains are shown. The overlay of the spectral data (black and red) showed that the RING domain Y63E mutation does not disrupt protein folding.
Effects of RING changes in TRIM5α on the fate of the retroviral capsid in infected cells.
Previous studies demonstrated that restriction of HIV-1 and N-MLV infection by TRIM5α proteins is accompanied by an accelerated conversion of the cytosolic retroviral capsid from particulate to soluble forms (5, 10, 11, 13, 37, 41, 48, 52). These results suggested that TRIM5α destabilizes the core in order to restrict retroviral infection. Because stability of the core is important for the occurrence of reverse transcription and productive infection (10, 13, 18, 36), we hypothesized that TRIM5α variants that allow the occurrence of reverse transcription do not decrease the amount of pelletable capsid during infection. To test this hypothesis, we examined the ability of different RING domain variants to decrease the amount of pelletable capsid during infection using the fate of the capsid assay, as previously described (5, 10, 37, 41, 52, 53). TRIM5αrh RING domain Y63E, Y63D, and Y63K variants were not able to decrease the amount of HIV-1 pelletable capsid compared to the wild-type protein (Fig. 3A and Table 1). Interestingly, the same changes fully allowed the occurrence of reverse transcription compared to the wild-type protein (Fig. 1B). By contrast, TRIM5αrh RING domain Y63S and Y63A variants decreased the amount of HIV-1 pelletable capsid during HIV-1 infection and prevented the occurrence of reverse transcription (Fig. 3A). Similarly, we studied the fate of the N-MLV capsid in canine Cf2Th cells stably expressing the various TRIM5αhu RING domain variants. In agreement with our previous results, TRIM5αhu RING variants that did not inhibit reverse transcription, including the Y62E, Y62F, Y62K, and Y62D variants, lost the ability to decrease the amount of pelletable capsid during N-MLV infection (Fig. 3B and Table 2). By contrast, variants that prevented reverse transcription, such as the Y62S and Y62A variants, decreased the amount of pelletable N-MLV capsid during infection to an extent similar to that of the wild-type TRIM5αhu (Fig. 3B). These results showed that TRIM5α mutants that had lost their ability to decrease the amount of pelletable capsid also lost their capacity to prevent reverse transcription.
Fig 3.

Fate of the retroviral capsid in cells expressing TRIM5α variants. (A) CF2Th cell lines expressing the indicated wild-type and mutant TRIM5αrh proteins or containing the empty LPCX vector were incubated with equivalent amounts of HIV-1–GFP at 4°C for 30 min. The cells were washed and returned to 37°C, and infection was allowed to proceed for 12 h. Cell extracts were fractionated on sucrose gradients as described in Materials and Methods. Input, soluble, and pellet fractions were analyzed by Western blotting using antibodies against the HIV-1 p24 capsid protein. (B) CF2Th cell lines expressing the indicated wild-type and mutant TRIM5αhu proteins or containing the empty LPCX vector were incubated with similar amounts of N-MLV–GFP at 4°C for 30 min. The cells were washed and returned to 37°C, and infection was allowed to proceed for 4 h. Cell extracts were fractionated on sucrose gradients as described in Materials and Methods. Input, soluble, and pellet fractions were analyzed by Western blotting using antibodies against the N-MLV p30 capsid protein. The lower bar graphs represent the fluorescent quantification of pelletable capsid shown as a percentage. Similar results were obtained in three independent experiments and the standard deviation is shown.
Assaying the E3 ubiquitin ligase activity of TRIM5αrh RING domain variants.
The RING domain of TRIM5αrh exhibits self-ubiquitylation activity and can use UbcH5 as an E2-conjugating enzyme (12, 23, 28, 31, 32, 57). To measure the ability of the TRIM5αrh RING domain variants to undergo RING domain-dependent self-ubiquitylation, we used our previously described ubiquitylation assay (31). Purified RING domain variants and wild-type TRIM5αrh proteins from human cell extracts were incubated with recombinant UBE-1 (E1), recombinant UbcH5a(E2), an energy regeneration system, and myc-tagged ubiquitin. TRIM5αrh self-ubiquitylation is observed only when E2 enzymes were added to the reaction (Fig. 4). The amount of ubiquitylated TRIM5α protein was determined by subtracting the amount of nonubiquitylated TRIM5α protein remaining in the reaction mixture that was incubated with E2 enzymes from the TRIM5α protein in the control reaction, which was not incubated with E2 enzyme. The value of nonubiquitylated TRIM5α was quantified by using a fluorescent Western blotting assay, as described in Materials and Methods. TRIM5α self-ubiquitylation was expressed as the percentage of the total TRIM5α variant input protein (Tables 1 and 2). Interestingly, TRIM5αrh RING domain Y63A, Y63D, Y63E, and Y63K variants were impaired in their ability to undergo polyubiquitylation (Fig. 4). To the contrary, TRIM5αrh RING domain Y63F and Y63S variants were polyubiquitylated to a lesser extent than the wild-type protein (Fig. 4). Notably, all RING domain variants in these studies accumulated monoubiquitylated forms compared to wild-type TRIM5αrh (Fig. 4). As controls, we used TRIM5αrh R60A, a RING domain variant that does not undergo mono- or poly ubiquitylation (31). These results showed that mutations at residue Y63 accumulate monoubiquitylated forms of the protein.
Fig 4.

E3 ubiquitin ligase activity of TRIM5αrh RING domain variants. Human 293T cells were transfected with plasmids encoding FLAG-tagged mutant and wild-type TRIM5αrh proteins. Forty-eight hours later, the cells expressing each TRIM5αrh variant were lysed in whole-cell extract and immunoprecipitated using anti-FLAG-agarose beads as described in Materials and Methods. Beads containing the immunoprecipitated TRIM5αrh variants were washed and eluted with 200 μg/ml of FLAG tripeptide in whole-cell extract buffer as described in Materials and Methods. Samples were supplemented with 5 μM ubiquitin aldehyde, a potent inhibitor of all ubiquitin C-terminal hydrolases, ubiquitin-specific proteases, and deubiquitinating enzymes. Similar amounts of inhibitor-treated samples containing mutant and wild-type TRIM5αrh were incubated with 200 nM enzyme E1 (human recombinant UBE1), 200 μM ubiquitin tagged with a myc epitope (human recombinant ubiquitin), and an energy regeneration solution containing MgCl2, ATP, and ATP-regenerating enzymes to recycle hydrolyzed ATP. The final reaction was supplemented or not with 100 nM enzyme E2 (human recombinant UbcH5a) as indicated. The reaction mixture was incubated at 37°C for 1 h, and collected fractions were analyzed by Western blotting using HRP-conjugated antibodies against FLAG to detect the levels of TRIM5αrh variants (anti-FLAG). To detect TRIM5αrh ubiquitylated forms, membranes were blotted using HRP-conjugated antibodies against myc (anti-myc). The results of three independent experiments were similar; the result of a single experiment is shown.
TRIM5αrh RING domain variants binding to HIV-1 capsid.
We analyzed the ability of the TRIM5αrh RING domain variants to bind HIV-1 CA, since this is required for restriction of HIV-1 by TRIM5αrh (15, 47, 52). To measure the binding of the different TRIM5αrh RING domain variants to in vitro-assembled HIV-1 CA-NC complexes, we used our previously described quantitative binding assay, which adjusts input levels of TRIM5α variants to more accurately compare capsid-binding abilities (14). Most TRIM5αrh RING domain variants retained wild-type binding to HIV-1 capsid-nucleocapsid complexes, with the exception of the Y63K variant, which was bound to capsid-nucleocapsid complexes with lower affinity (Fig. 5 and Tables 1 and 2). These results suggested that these mutants interact with the HIV-1 capsid in a manner similar to the wild-type protein.
Fig 5.
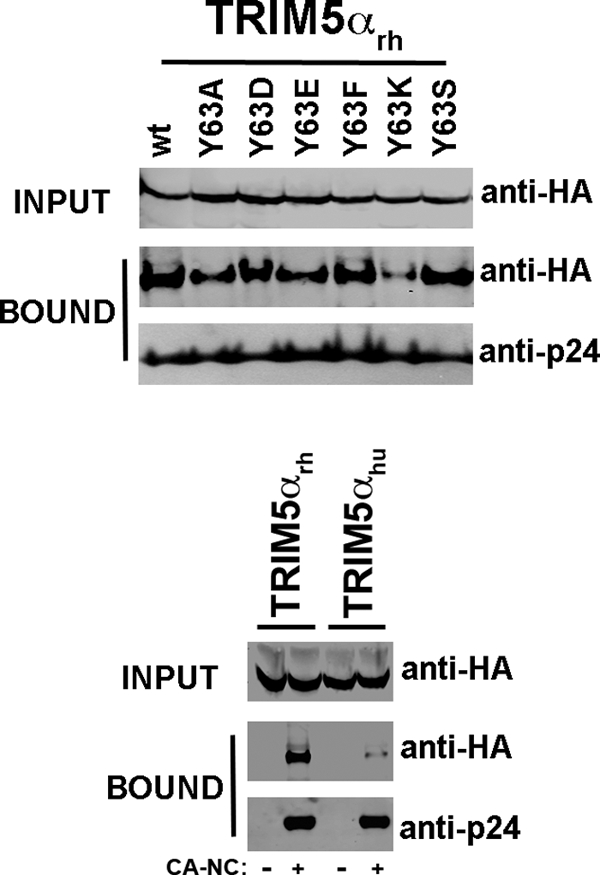
Binding of TRIM5αrh RING domain variants to the HIV-1 CA. 293T cells were transfected with plasmids expressing the indicated wild-type and mutant TRIM5αrh proteins tagged with HA epitopes. Thirty-six hours after transfection, cells were lysed. The lysates were incubated at room temperature for 1 h with HIV-1 CA-NC complexes that had been assembled in vitro. The mixtures were applied onto a 70% sucrose cushion and centrifuged. INPUT represents the lysates analyzed by Western blotting before being applied to the 70% cushion. The input mixtures were Western blotted using anti-HA antibodies. The pellet from the 70% cushion (BOUND) was analyzed by Western blotting using antibodies against the HA tag and HIV-1 CA-NC protein. The blots were quantitated as described in Materials and Methods, and values for binding are shown in Tables 1 and 2. The results of three independent experiments were similar; the result of a single experiment is shown.
HOSA of TRIM5αrh RING domain variants.
Higher-order self-association is important for the ability of TRIM5αrh to restrict HIV-1 (11, 14, 20, 29). The B-box 2 domain is critical in the formation of higher-order complexes, which are known to increase the avidity of TRIM5αrh for the HIV-1 capsid (14, 29); residue R121 in the B-box 2 domain of TRIM5αrh is essential for the ability of TRIM5αrh to form hexagonal structures on the surface of the HIV-1 capsid (11, 14, 20). Because changes in the RING domain of TRIM5αrh can affect higher-order self-association (30, 31), we tested the ability of these variants to form HOSA complexes. Interestingly, most of the mutants were affected to different degrees, with the exception of the Y63F mutant (Fig. 6 and Tables 1 and 2). These results suggested that the RING domain is involved in the ability of TRIM5αrh to form HOSA complexes.
Fig 6.

TRIM5αrh RING domain variant higher-order self-association (HOSA). 293T cells were transfected with plasmids expressing the indicated wild-type or mutant TRIM5α proteins with a FLAG or HA epitope tag. Cells expressing wild-type and mutant TRIM5αrh proteins were lysed 48 h after transfection. The cell lysates containing similar inputs were mixed (INPUT), and the indicated mixtures were used for immunoprecipitation by an antibody directed against the FLAG epitope, as described in Materials and Methods. Elution of the immunocomplexes was performed with a FLAG tripeptide and analyzed by Western blotting using anti-HA and anti-FLAG antibodies (higher-order self-associates). The results of three independent experiments were similar; the result of a single experiment is shown.
RING mutations prevent the HIV-1-induced degradation of TRIM5αrh.
In a previous study, Rold and Aiken (44) reported that cellular TRIM5αrh is destabilized upon encountering a restriction-sensitive retroviral capsid. The virus-induced TRIM5αrh degradation was prevented by compounds that inhibit the activity of the 26S proteasome, indicating that proteasome activity is required and suggesting that TRIM5α engagement of the capsid recruits and/or activates proteasomal degradation of the protein. However, it is unknown whether the RING domain and its associated ubiquitin ligase activity play a role in virus-induced TRIM5α degradation. To test this, we analyzed the levels of TRIM5αrh in cells exposed to VSV-G-pseudotyped HIV-1 particles. Cells were pretreated with cycloheximide to halt protein synthesis, and the inhibitor was present during the 5-h assay period. In addition, the VSV-G-pseudotyped HIV-1 stock was pretitered in the experiment to ensure that the dose was sufficient to induce maximal degradation of the wild-type TRIM5αrh protein. The levels of TRIM5αrh were determined by immunoblotting using an antibody specific for the C-terminal HA tag, and the blots were reprobed with an actin-specific antibody to normalize the quantitation. To quantify the basal degradation of the protein, we analyzed samples collected from cells not exposed to VSV-G-pseudotyped HIV-1 at the beginning of the experiment and after the 5-h culture period.
As previously reported (44), exposure of cells to VSV-G-pseudotyped HIV-1 particles resulted in degradation of wild-type TRIM5αrh during the 5-h time course (Fig. 7A). The cellular levels of the protein were reduced by an average of 77% relative to the control cells not exposed to the virus. By contrast, all of the Y63 mutations rendered TRIM5α less sensitive to HIV-induced degradation, albeit to different extents. Y63K, Y63E, and Y63D TRIM5α RING mutants, which failed to inhibit HIV-1 reverse transcription, were the least affected by exposure to the virus, exhibiting no significant loss relative to the respective controls. Of the mutants, that with Y63F was the most sensitive, exhibiting 63% degradation (Fig. 7B). The Y63S and Y63A mutants were intermediate, exhibiting 30% and 18% degradation, respectively (Fig. 7B). It is possible that some of the effects of the Y63S, Y63K, Y63E, Y63D, and Y63A mutations result from the higher steady-state cellular levels of these TRIM5α proteins, which may limit the ability of the incoming HIV-1 particles to saturate the restriction factor. Overall, the results suggest that mutations in the RING domain can render TRIM5α resistant to destabilization induced by incoming HIV-1 cores.
Fig 7.

HIV-1-induced degradation of TRIM5αrh RING domain variants. (A) TRIM5αrh-expressing Cf2Th cell lines were cultured in cycloheximide-containing media in the presence or absence of HIV-1 particles for the indicated time periods, and cytoplasmic lysates were prepared and analyzed by immunoblotting for TRIM5αrh. Due to the lower steady-state levels of the wild-type and Y63F mutant TRIM5αrh proteins, the panels for these samples were obtained by scanning the immunoblot with the LI-COR Odyssey at a higher laser intensity than for the other mutants. (B) Quantification of the effect of HIV-1 on TRIM5αrh protein levels after 5 h of culture. Values shown are the averages of duplicate parallel assays. Paired values depict the results from two independent experiments.
DISCUSSION
In this study, we identified TRIM5αrh RING domain mutants that potently blocked retrovirus infection without inhibiting reverse transcription. These mutants are strong genetic evidence that TRIM5αrh can act at more than one step on the retroviral life cycle. Our results are consistent with the observation that proteasomal inhibitors allowed the occurrence of reverse transcription while minimally affecting the ability of TRIM5αrh to block HIV-1 infection (52, 55). This evidence suggested that TRIM5αrh can target the HIV-1 viral life cycle at more than one step. To further understand the nature of the post-reverse transcription block, we measured the generation of HIV-1 2-LTR circles during the infection of cells expressing different TRIM5αrh RING domain mutants. HIV-1 infection of cells expressing these variants allowed formation of HIV-1 2-LTR circles but did so to a lesser extent than cells containing the empty vector LPCX (Fig. 1F). After reverse transcription, the routing of viral DNA to the formation of 2-LTR circles is an indication that the preintegration complex has been transported to the nucleus (54). This suggests that TRIM5αrh may directly inhibit the integration of HIV-1 preintegration complexes that have entered the nucleus, consistent with our recent finding that TRIM5αrh shuttles between the nucleus and the cytoplasm (9). A second possibility is that mutations on position Y63 result in a TRIM5αrh protein that might damage the uncoating process, leaving a viral DNA substrate that could be end ligated and circularized but not integrated. However, further investigation is required to understand the mechanism used by mutant TRIM5αrh proteins to block HIV-1 after reverse transcription.
To rule out the possibility that the phenotypes described in the present work were the result of mutations that simply unfold the RING domain, we demonstrated that the RING domain bearing the mutations Y63E and Y62E in TRIM5αrh and TRIM5αhu, respectively, are folded similarly to the wild-type protein (Fig. 2). Proper folding implies that these mutations lost a specific function of the RING domain, which causes the observed phenotypes. The fact that the RING Y63E folding is slightly different from the wild-type domain might explain why all the mutants, with the exception of the Y63F mutant, lost higher-order self-association capabilities while maintaining the capacity to bind HIV-1 CA-NC complexes. The chemically shifted residues in the RING Y63E structure might be a “hot spot” for the self-association of TRIM5αrh. The future assignment of the chemically shifted residues will help on the identification of this potential hot spot.
We also measured the fate of the capsid for HIV-1 and N-MLV during infection of cells expressing different RING domain variants of TRIM5αrh and TRIM5αhu, respectively. Remarkably, in this group of mutants, the acceleration of uncoating was correlated with inhibition of reverse transcription, suggesting that a decrease of pelletable capsid or an acceleration of uncoating by TRIM5α disrupts reverse transcription. These data add to our understanding of early steps on HIV-1 infection that reverse transcription is dependent on proper uncoating and occurs before or during uncoating but not afterwards. These results were in agreement with several observations suggesting that stability of the viral core is important for reverse transcription and infection. (i)There are changes in the stability of the incoming retroviral core in the host cell achieved by mutagenesis of the HIV-1 capsid that result in a loss of infectivity. Mutations in the capsid protein of HIV-1 that diminish (or increase) the stability of the HIV-1 core interfere with reverse transcription (18). (ii) HIV-1 cores isolated from particles produced in the absence of the accessory protein Vif exhibit low stability and are poorly infectious. Careful studies of these viruses revealed a defect in reverse transcription (36). (iii) The use of proteasome inhibitors during HIV-1 infection increases the stability of the incoming cores and at the same time augments reverse transcription (10, 13). Taken together, these results indicate that the acceleration of uncoating by TRIM5α precludes efficient reverse transcription in target cells. One possibility is that the reverse transcription complex is tightly associated with the core and that disruption of the core ultimately prevents reverse transcription. HIV-1 uncoating in vitro is accompanied by dissociation of the RT enzyme from the viral ribonucleoprotein complex within the core, suggesting that premature uncoating may lead to impaired reverse transcription processivity (17). Finally, these findings are consistent with the idea that reverse transcription occurs before or during uncoating but not afterwards, as has been previously suggested (3, 4).
TRIM5αrh undergoes self-ubiquitylation activity, and this ability correlates with the ability of TRIM5αrh to block HIV-1 (31). In the present work, we measured the ability of TRIM5αrh RING domain variants to undergo self-ubiquitylation. Interestingly, all of the mutants formed preferentially monoubiquitylated forms. For example, the Y63E protein had a very long half-life relative to the wild-type protein (data not shown), suggesting that monoubiquitylation might stabilize the protein. One possibility is that when TRIM5αrh binds to the HIV-1 capsid by forming hexagonal structures on the surface of the core (20), it recruits an E2 enzyme to allow TRIM5αrh polyubiquitylation. Removal and degradation of polyubiquitylated TRIM5αrh from the surface of the core will trigger acceleration of uncoating (8, 44). By contrast, a TRIM5αrh mutant protein that preferentially undergoes monoubiquitylation will not accelerate uncoating and will allow the completion of reverse transcription, as shown here. However, additional experiments need to be done in order to understand the role of TRIM5αrh polyubiquitylation in the acceleration of uncoating. Because TRIM5αrh polyubiquitylation is also involved in the signaling ability of TRIM5αrh (42), further investigation is also required to test whether TRIM5αrh-mediated uncoating is related to signaling.
Analysis of the intracellular stability of the TRIM5αrh mutants following exposure to HIV-1 particles revealed that several of the mutations confer resistance to HIV-1-induced degradation. Three of the most protective mutations (Y63K, Y63E, and Y63D) also abolished the ability of TRIM5αrh to block reverse transcription; however, the correlation between reverse transcription inhibition and virus-induced TRIM5αrh degradation was not strictly quantitative. While the degradation of the three charged mutants may have been limited by the higher steady-state levels of these proteins (probably reflecting enhanced stability), other TRIM5αrh mutants (the Y63S and Y63A mutants) also exhibited higher steady-state levels than the wild-type protein but appeared to be somewhat more sensitive to virus-induced degradation than the charged amino acid substitution mutants. Based on these results, we suggest that the E3 ligase activity of TRIM5αrh may play a role in the virus-induced degradation of TRIM5αrh. It will be of interest to identify substrates of the TRIM5αrh ubiquitin ligase activity that may contribute to TRIM5αrh degradation.
Because potent restriction of HIV-1 by TRIM5αrh requires capsid binding and the formation of HOSA complexes, we tested the ability of TRIM5αrh RING domain variants in terms of these properties. As expected, the variants retained wild-type ability to bind capsid. However, the ability to form HOSA complexes was affected, in agreement with previous observations suggesting that the RING domain is involved in HOSA (30, 31). Even though the variants lost the ability to form HOSA complexes, we did not find a correlation between HOSA and either blockage of reverse transcription or restriction in this particular set of mutants.
Overall, this work provides genetic evidence that TRIM5α can act at more than one step of retroviral replication and that TRIM5α-mediated acceleration of uncoating contributes to the block in retroviral reverse transcription. Future investigations will attempt to understand the possible block against HIV-1 imposed by TRIM5α within the nucleus.
ACKNOWLEDGMENTS
We thank Andre Rosowsky for critical reading of the manuscript. We also thank Eiko Seki, Kazuharu Hanada, Masaomi Ikari, and Manami Sato for sample preparation of the TRIM5α RING proteins used for NMR studies. Efavirenz and raltegravir were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
This work has been supported by a K99/R00 Pathway to Independence Award to F.D.-G. from the National Institutes of Health, 4R00MH086162-02, and an American Foundation for AIDS Research Mathilde Krim Fellowship Phase II in Basic Biomedical Research (amfAR Research Grant #107787-47-RKHF). This work was also funded by NIH R01 AI076121 to C.A. and NIH R01 AI087390 to F.D.-G.
Footnotes
Published ahead of print 23 November 2011
REFERENCES
- 1. Anderson JL, et al. 2006. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 80: 9754–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aoki M, et al. 2009. Automated system for high-throughput protein production using the dialysis cell-free method. Protein Expr. Purif. 68: 128–136 [DOI] [PubMed] [Google Scholar]
- 3. Arfi V, et al. 2009. Characterization of the behavior of functional viral genomes during the early steps of human immunodeficiency virus type 1 infection. J. Virol. 83: 7524–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arfi V, et al. 2008. Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J. Virol. 82: 6557–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berube J, Bouchard A, Berthoux L. 2007. Both TRIM5alpha and TRIMCyp have only weak antiviral activity in canine D17 cells. Retrovirology 4: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bock M, Bishop KN, Towers G, Stoye JP. 2000. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J. Virol. 74:7422–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7: 631–634 [DOI] [PubMed] [Google Scholar]
- 8. Diaz-Griffero F. 2011. Caging the beast: TRIM5α binding to the HIV-1 core. Viruses 3: 423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diaz-Griffero F, Gallo DE, Hope TJ, Sodroski J. 2011. Trafficking of some old world primate TRIM5alpha proteins through the nucleus. Retrovirology 8: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz-Griffero F, et al. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology 369:400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz-Griffero F, et al. 2007. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5alpha B-box 2 domain. J. Virol. 81: 10362–10378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz-Griffero F, et al. 2006. Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 349: 300–315 [DOI] [PubMed] [Google Scholar]
- 13. Diaz-Griffero F, et al. 2008. A human TRIM5alpha B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology 378: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diaz-Griffero F, et al. 2009. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 83: 10737–10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diaz-Griffero F, et al. 2006. Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351: 404–419 [DOI] [PubMed] [Google Scholar]
- 16. Dismuke DJ, Aiken C. 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J. Virol. 80: 3712–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forshey BM, Aiken C. 2003. Disassembly of human immunodeficiency virus type 1 cores in vitro reveals association of Nef with the subviral ribonucleoprotein complex. J. Virol. 77: 4409–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76: 5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283: 80–83 [DOI] [PubMed] [Google Scholar]
- 20. Ganser-Pornillos BK, et al. 2011. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. U. S. A. 108: 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78: 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Javanbakht H, Diaz-Griffero F, Stremlau M, Si Z, Sodroski J. 2005. The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5alpha. J. Biol. Chem. 280: 26933–26940 [DOI] [PubMed] [Google Scholar]
- 23. Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J. 2008. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J. Virol. 82: 11669–11681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keckesova Z, Ylinen LM, Towers GJ. 2004. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. U. S. A. 101: 10780–10785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kigawa T, et al. 1999. Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett. 442: 15–19 [DOI] [PubMed] [Google Scholar]
- 26. Kim J, Tipper C, Sodroski J. 2011. Role of TRIM5{alpha} RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade, and restriction of simian immunodeficiency virus. J. Virol. 85: 8116–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kirmaier A, et al. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8: e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langelier CR, et al. 2008. Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 82:11682–11694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Sodroski J. 2008. The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 82: 11495–11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X, Yeung DF, Fiegen AM, Sodroski J. 2011. Determinants of the higher-order association of the restriction factor TRIM5{alpha} and other tripartite motif (TRIM) proteins. J. Biol. Chem. 286: 27959–27970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lienlaf M, et al. 2011. Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5{alpha}rh: structure of the RING domain of TRIM5{alpha}. J. Virol. 85: 8725–8737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maegawa H, Miyamoto T, Sakuragi J, Shioda T, Nakayama EE. 2010. Contribution of RING domain to retrovirus restriction by TRIM5alpha depends on combination of host and virus. Virology 399: 212–220 [DOI] [PubMed] [Google Scholar]
- 33. Matsuda T, et al. 2006. Cell-free synthesis of zinc-binding proteins. J. Struct. Funct. Genomics 7: 93–100 [DOI] [PubMed] [Google Scholar]
- 34. Mrosek M, et al. 2008. Structural analysis of B-Box 2 from MuRF1: identification of a novel self-association pattern in a RING-like fold. Biochemistry 47: 10722–10730 [DOI] [PubMed] [Google Scholar]
- 35. Nakayama EE, Miyoshi H, Nagai Y, Shioda T. 2005. A specific region of 37 amino acid residues in the SPRY (B30.2) domain of African green monkey TRIM5alpha determines species-specific restriction of simian immunodeficiency virus SIVmac infection. J. Virol. 79: 8870–8877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohagen A, Gabuzda D. 2000. Role of Vif in stability of the human immunodeficiency virus type 1 core. J. Virol. 74: 11055–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohkura S, et al. 2011. Novel escape mutants suggest an extensive TRIM5alpha binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 7: e1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Owens CM, Yang PC, Gottlinger H, Sodroski J. 2003. Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J. Virol. 77:726–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD. 2005. Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J. Virol. 79: 8969–8978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 79: 15567–15572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perron MJ, et al. 2007. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 81: 2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pertel T, et al. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472: 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reymond A, et al. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20: 2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rold CJ, Aiken C. 2008. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 4: e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. U. S. A. 102:2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430: 569–573 [DOI] [PubMed] [Google Scholar]
- 47. Sebastian S, Luban J. 2005. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song B, et al. 2005. TRIM5alpha association with cytoplasmic bodies is not required for antiretroviral activity. Virology. [DOI] [PubMed] [Google Scholar]
- 50. Song B, et al. 2005. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J. Virol. 79: 6111–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stremlau M, et al. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427: 848–853 [DOI] [PubMed] [Google Scholar]
- 52. Stremlau M, et al. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103: 5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stremlau M, Perron M, Welikala S, Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J. Virol. 79: 3139–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suzuki Y, Craigie R. 2007. The road to chromatin - nuclear entry of retroviruses. Nat. Rev. Microbiol. 5: 187–196 [DOI] [PubMed] [Google Scholar]
- 55. Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 103: 7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yabuki T, et al. 2007. A robust two-step PCR method of template DNA production for high-throughput cell-free protein synthesis. J. Struct. Funct. Genomics 8: 173–191 [DOI] [PubMed] [Google Scholar]
- 57. Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T. 2008. Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 275: 1540–1555 [DOI] [PubMed] [Google Scholar]
- 58. Yap MW, Dodding MP, Stoye JP. 2006. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 80: 4061–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yap MW, Nisole S, Stoye JP. 2005. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 15: 73–78 [DOI] [PubMed] [Google Scholar]