

Abstract
Fibrochondrogenesis is a severe, recessively inherited skeletal dysplasia shown to result from mutations in the gene encoding the proα1(XI) chain of type XI collagen, COL11A1. The first of two cases reported here was the affected offspring of first cousins and sequence analysis excluded mutations in COL11A1. Consequently, whole-genome SNP genotyping was performed to identify blocks of homozygosity, identical-by-descent, wherein the disease locus would reside. COL11A1 was not within a region of homozygosity, further excluding it as the disease locus, but the gene encoding the proα2(XI) chain of type XI collagen, COL11A2, was located within a large region of homozygosity. Sequence analysis identified homozygosity for a splice donor mutation in intron 18. Exon trapping demonstrated that the mutation resulted in skipping of exon 18 and predicted deletion of 18 amino acids from the triple helical domain of the protein. In the second case, heterozygosity for a de novo 9 bp deletion in exon 40 of COL11A2 was identified, indicating that there are autosomal dominant forms of fibrochondrogenesis. These findings thus demonstrate that fibrochondrogenesis can result from either recessively- or dominantly-inherited mutations in COL11A2.
Keywords: Fibrochondrogenesis, skeletal dysplasia, COL11A2, collagen, cartilage
INTRODUCTION
Fibrochondrogenesis (OMIM 228520) is a severe, recessively inherited skeletal dysplasia characterized by short long bones and short ribs with broad metaphyses, a flat midface, and vertebral bodies which show distinctive hypoplastic posterior ends and rounded anterior ends, giving the vertebral bodies a pinched appearance on lateral radiographic views. The chest is small and leads to perinatal respiratory problems which usually, but not always, result in lethality [Eteson et al., 1984; Whitley et al., 1984; Bankier et al., 1991; Martinez-Frias et al., 1996; Al-Gazali et al., 1999; Randrianaivo et al., 2002; Leeners et al., 2004; Tompson et al., 2010; Akawi et al., 2011]. Affected individuals who have survived the neonatal period have high myopia, mild-to-moderate hearing loss and a severe skeletal dysplasia [Tompson et al., 2010; Akawi et al., 2011].
Fibrochondrogenesis has been shown to result from mutations in the gene encoding the proα1(XI) chain of type XI procollagen (COL11A1; MIM 120280) [Tompson et al., 2010; Akawi et al., 2011]. The functional type XI collagen molecule is a heterotrimeric protein assembled from three different polypeptides, the proα1(XI), proα2(XI) and proα3(XI) chains [Morris et al., 1987]. The proα1(XI) and proα2(XI) chains are encoded by COL11A1 and COL11A2 (MIM 120290), respectively, whereas the proα3(XI) chain is a posttranslational variant encoded by COL2A1 (MIM 120140) [Wu et al., 1995]. We report on the identification of COL11A2 mutations in two cases of fibrochondrogenesis, one in which there was recessive inheritance and a second in which the phenotype was produced by a new dominant mutation. These data establish a second locus for the phenotype and reveal a distinct mode of inheritance in some cases.
MATERIALS AND METHODS
Patients
Informed consent was obtained from all individuals under an IRB-approved protocol. DNA was extracted from peripheral blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA).
Whole-genome SNP Genotyping
For the SNP array genotyping, DNA was processed and hybridized to GeneChip Human Mapping 250K arrays (NspI) per the manufacturer's recommendations (Affymetrix, Santa Clara, CA). Genotypes were called with GeneChip DNA analysis software (Affymetrix) and then analyzed using IBD Finder (http://dna.leeds.ac.uk/ibdfinder/) with a maximum error rate of one in six.
Mutation Analysis
All exons and flanking splice acceptor / donor sequences of COL11A1 (MIM 120280; NM_001854.3), COL11A2 (OMIM 120290; NM_080680.2) and COL2A1 (MIM 120140; NM_001844.4) were amplified by PCR and sequenced by Sanger dideoxynucleotide methodology using ABI 3730 sequencers.
Exon Trapping Splicing Assay
Phusion Hot Start High-Fidelity DNA polymerase (Thermo Fisher Scientific, Lafayette, CO) was used to amplify a 681 bp genomic region containing COL11A2 exons 17-19 from a control subject and from case R09-068A. The PCR products were inserted into the multiple cloning site of the exon trapping vector, pSPL3, using XhoI and BamHI (Fig. 2; Life Technologies, Baltimore, MD, USA). Normal and mutant minigenes were transfected into Cos-7 cells using FuGene 6 (Roche Applied Science, Indianapolis, IN), and 24 hours later total RNA was isolated using the RNeasy Mini Kit (QIAGEN). A High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) with random primers was used to generate cDNA, and the splicing vector transcribed cDNA species were then specifically amplified by PCR using primer pairs corresponding to sequences in COL11A2 exons 17 and 19, respectively (Fig. 2). The resulting RT-PCR products from the normal and mutant minigenes were visualized by gel electrophoresis, purified by gel extraction and their sequences determined.
Figure 2. Skipping of exon 18 resulting from the splice donor IVS18+3insG mutation in case R09-068A.
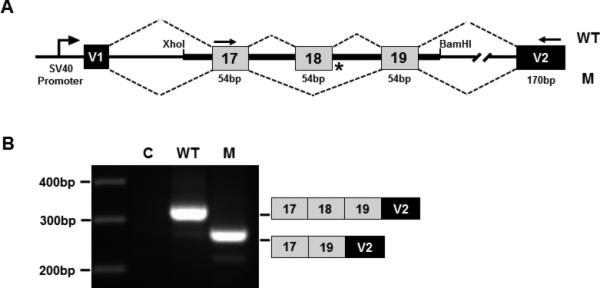
(A) Splicing construct showing the COL11A2 genomic region and vector/collagen exon organization. V1 and V2 are the vector exons. Normal and mutant COL11A2 genomic DNA fragments containing exons 17-19 were cloned into the XhoI and BamHI restriction endonuclease cleavage sites of the vector. (B) Gel electrophoresis image of fragments generated by RT-PCR from RNA derived from transfected cells using vector only (C), normal (WT) and mutant (M) constructs. Diagrammatic representations of the PCR product exon structures, based on sequence analysis, are shown on the right. PCR primers used to generate the fragments are indicated by arrows in (A).
Microsatellite Analysis
Twelve pan-genomic microsatellites were amplified by PCR, using fluorescent FAM, HEX or TET-labeled primers. Amplified products were resolved using an ABI 3730 sequencer and sized by application of the Genemapper software package from Applied Biosystems.
RESULTS
The index case, International Skeletal Dysplasia Registry (ISDR) reference R09-068A, was the offspring of first cousins of Saudi Arabian descent. The couple previously had an affected child and has two unaffected offspring. Clinical and radiographic information was only available for the second affected child, who died at birth. The clinical phenotype consisted of midface hypoplasia with a small nose and anteverted nares, significant shortening of all limb segments with relatively normal hands and feet, and a small thorax with a protuberant abdomen. Radiographs showed characteristic findings of fibrochondrogenesis that included severe shortening of the long bones with very widened metaphyses, moderate platyspondyly, delayed ossification of the cervical vertebral bodies, ischia and pubis, short cupped ribs giving a “bell-shaped” appearance to the thorax, and small ilia with irregular metaphyses (Fig. 1, Table I). A lateral view of the spine was unavailable. Based on previous reports showing that fibrochondrogenesis can result from mutations in COL11A1, sequence analysis of the coding exons and splice junctions of COL11A1 was carried out commercially (Connective Tissue Gene Tests, Allentown, PA), but did not reveal any mutations. As the parents were known to be consanguineous, suggesting an autosomal recessive pattern of inheritance, whole-genome SNP genotyping was performed to identify blocks of homozygosity wherein the disease locus would be located. There were many regions of homozygosity across the genome, an expected consequence of the close relationship between the parents. The COL11A1 gene was not within a region of homozygosity, further supporting locus heterogeneity in fibrochondrogenesis. Interestingly, the gene encoding the proα2(XI) chain of type XI procollagen (COL11A2) was within a 26.7 Mb region of homozygosity, the largest block of homozygosity identified (data not shown). To test the hypothesis that the phenotype was produced by homozygosity for a mutation in COL11A2 (NM_080680.2), mutation analysis was carried out by sequence analysis of all 66 coding exons and splice junction consensus sequences. Sequence analysis revealed homozygosity for a splice site change, IVS18+3insG (Table II), which was predicted to result in aberrant mRNA processing. The parents were both identified as carriers of the mutation as was the one unaffected sibling studied. To determine the effect of the IVS18+3insG change on splicing, an exon trapping splicing assay was performed, utilizing engineered normal and mutant COL11A2 minigene expression in Cos-7 cells (Fig. 2). These data showed skipping of exon 18 in RNA derived from the mutant construct but not from the control.
Figure 1. Radiograph of autosomal recessive fibrochondrogenesis case R09-068A at full term.
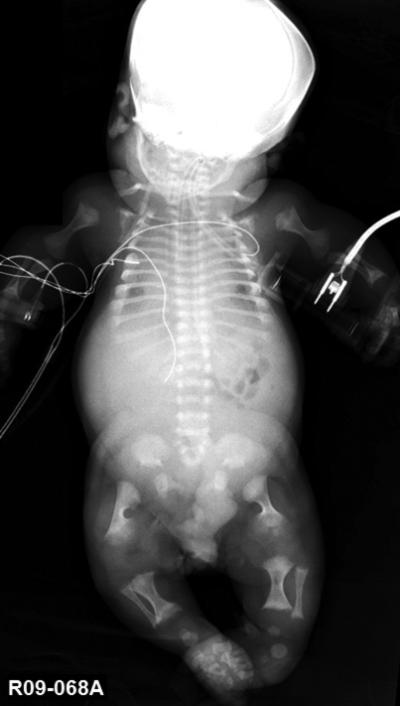
Anterior-posterior view. Note the short long-bones with substantial metaphyseal flaring, short ribs with anterior cupping, platyspondyly, delayed ossification of the cervical vertebral bodies, and small ilia with hypoplastic ischia and pubis.
Table I.
Clinical and radiographic phenotype of fibrochondrogenesis cases R09-068A and R10-691A versus other differential diagnoses in the newborn period.
| R09-068A (AR) | R10-691A (AD) | FIB (AR) | OSMED (AR) | OSMED (AD) | KD (AD) | |
|---|---|---|---|---|---|---|
| Midface hypoplasia | +++ | +++ | +++ | ++ | ++ | + |
| Anteverted nares | +++ | +++ | +++ | ++ | ++ | - |
| Frontal bossing | +++ | +++ | +++ | + | + | - |
| Short, cupped ribs | +++ | +++ | +++ | + | - | ++ |
| Platyspondyly | +++ | +++* | +++* | ++ | + | ++ |
| Widened metaphyses | +++ | ++ | +++ | ++ | + | +++ |
| Shortened long bones | +++ | ++ | +++ | + | + | ++ |
| “Bell-shaped” thorax | +++ | +++ | +++ | - | - | - |
| Small ilia with hypoplastic ischia/pubis | + | + | + | - | - | + |
Hypoplastic posterior ends of the vertebral bodies. FIB, Fibrochondrogenesis; KD, Kniest dysplasia; OSMED, Otospondylomegaepiphyseal dysplasia; AR, Autosomal recessive; AD, Autosomal dominant; Absent (-); Mild (+); Moderate (++); Severe (+++).
Table II. COL11A2 mutations in fibrochondrogenesis cases.
The COL11A2 nucleotide changes are shown with respect to mRNA sequence NM_080680.2. The corresponding predicted amino acid changes are numbered from the initiating methionine residue with respect to protein sequence NP_542411.2. Exons are numbered sequentially 1-66.
| Patient ID | Inheritance | Exon | Nucleotide change | Predicted amino acid change |
|---|---|---|---|---|
| R09-068A | Recessive | IVS18 | IVS18+3insG | p.556_573del18 |
| R10-691A | Dominant | 40 | c.2899_2907del9 | p.967_969del3 |
Mutation analysis was carried out on an additional fibrochondrogenesis case, R10-691A, the offspring of a non-consanguineous phenotypically normal couple. The child, who died at birth, had the typical facial features of fibrochondrogenesis presenting with a relatively large skull with a wide anterior fontanelle, midface hypoplasia with a small nose and anteverted nares, micrognathia, significant shortening of all limb segments with relatively normal hands and feet, and a small thorax with a protuberant abdomen. Radiographs displayed features characteristic of fibrochondrogenesis, including platyspondyly, shortening of the long bones with widened metaphyses, a “bell-shaped” thorax and short ribs with metaphyseal cupping, and hypoplastic ischia, pubis and ilia (Fig. 3, Table I). A lateral view of the spine revealed posteriorly narrowed vertebral bodies consistent with fibrochondrogenesis. Sequence analysis of the coding exons, splice junctions and branch sites of COL11A1 and COL2A1 did not identify any mutations. Sequence analysis of COL11A2 revealed heterozygosity for a 9 bp deletion in exon 40 (c.2899_2907del9; Table II) which predicted a 3 amino acid deletion (p.967_969del3) within the triple helical domain of proα2(XI). Analysis of parental DNA showed that neither carried the c.2899_2907del9 sequence change (Fig. 4), and genotypes at 12 informative pan-genomic microsatellite markers were consistent with parentage (data not shown), demonstrating that the mutation reflected a de novo event.
Figure 3. Radiograph of dominant fibrochondrogenesis case R10-691A at full term.
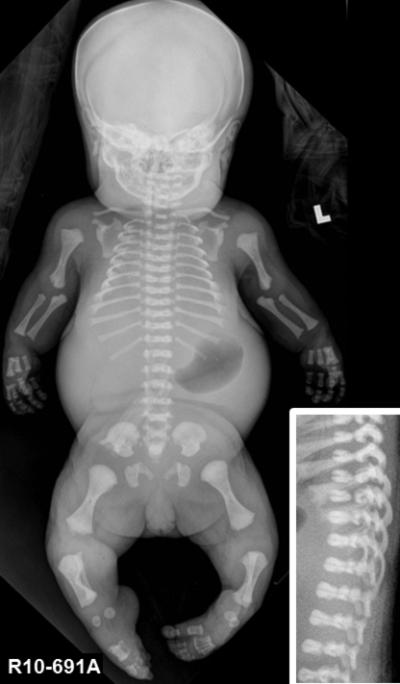
Anterior-posterior view. (Inset) Lateral view. Note the platyspondyly with hypoplastic posterior and rounded anterior ends of the vertebral bodies, giving them a pinched appearance.
Figure 4. New dominant COL11A2 mutation in fibrochondrogenesis.
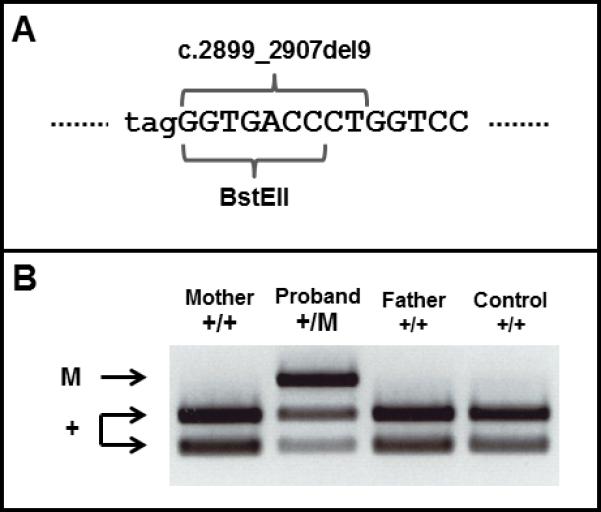
(A) Genomic sequence showing the region of COL11A2 in exon 40 (upper case) harboring the mutation and the adjacent splice acceptor site of intron 39 (lower case). The position of the c.2899_2907del9 mutation and overlapping BstEII restriction endonuclease site are indicated. (B) Agarose gel electrophoresis of BstEII digested PCR products. The PCR product was undigested in the mutant (M) allele as it lacked the BstEII site, whereas all normal alleles (+) generated two digestion fragments.
DISCUSSION
The first fibrochondrogenesis case, R09-068A, resulted from homozygosity for a COL11A2 splice donor mutation that led to skipping of exon 18. Since homozygosity for null mutations in COL11A2 is associated with otospondylomegaepiphyseal dysplasia (OSMED, OMIM 215150), a non-lethal skeletal dysplasia with much less severe skeletal abnormalities [Melkoniemi et al., 2000; Temtamy et al., 2006], we infer that the mutation is not a null allele and that the presence of abnormal chains had a detrimental effect on type XI collagen function. This inference is further supported by the observation that mice deficient for proα2(XI) have skeletal manifestations that markedly resemble OSMED and not fibrochondrogenesis [Li et al., 2001]. For the exon skipping mutation, transcripts lacking the 54 bp exon are predicted to encode proα2(XI) chains harboring an 18 amino acid deletion at position 556-573 (NP_542411.2), toward the amino-terminal end of the triple helical domain. The carrier parents were phenotypically normal, without abnormalities in either hearing or vision, indicating that heterozygosity for the defect is tolerated. This is in contrast to two fibrochondrogenesis carriers heterozygous for substitutions of two different triple helical glycine residues in COL11A1, in which mild short stature and myopia were observed [Tompson et al., 2010].
The second case studied, R10-691A, was heterozygous for a COL11A2 mutation that predicted a more distal 3 amino acid deletion within the triple helical domain of the proα2(XI) chain, corresponding to amino acids 967-969 (NP_542411.2). No other mutations were identified in COL11A2, and mutations were not found in either COL11A1 or COL2A1. The mutation was not present in the parental DNA and parentage was confirmed by microsatellite analysis, proving that the mutation was de novo. These data thus indicate that there is an autosomal dominant form of fibrochondrogenesis. Furthermore, there is precedent for mutations in COL11A2 producing both dominant and recessive forms of the same disorder, with reported cases of dominant and recessive forms of OSMED [Pihlajamaa et al., 1998; Temtamy et al., 2006] as well as nonsyndromic hearing loss [McGuirt et al., 1999; Chen et al., 2005].
We cannot exclude the theoretical possibility of digenic inheritance with the de novo COL11A2 mutation inherited with a second mutation from a carrier parent. However, the possibility of compound COL11A1/COL11A2 or COL2A1/COL11A2 mutant genotypes has been excluded by sequence analysis. Furthermore, both parents were of normal height and had normal vision and hearing, so the possible second mutation would have to be of no phenotypic consequence. While there are rare examples in the literature for new mutations in recessive disorders, where the affected individuals have inherited a point mutation carried by one parent in addition to a new mutation (Cremonesi et al., 1996), these have typically been observed for disorders in which the carrier frequency is high. Fibrochondrogenesis is a very rare disorder, perhaps less than 1 in 1,000,000 births in outbred populations, which would imply a carrier frequency of 1 in 500. Consequently, it is more likely that this case resulted solely from the new mutation in COL11A2, consistent with the conclusion that there are autosomal dominant forms of the disease.
Several dominantly inherited type XI collagenopathies have been reported to result from incorporation of molecules containing abnormal proα2(XI) chains into fibrils. Autosomal dominant forms of OSMED (OMIM 277610) can result from substitution for a triple helical glycine residue [Pihlajamaa et al., 1998]. In-frame triplet-repeat deletions of 18 amino acids in two cases and 9 amino acids in a third have been described for non-ocular Stickler syndrome (MIM 184840) [Vikkula et al., 1995; Sirko-Osadsa et al., 1998; Vuoristo et al., 2004]. Finally, nonsyndromic hearing loss (OMIM 601868) caused by cysteine for arginine or glutamate for glycine amino acid substitutions within the triple helical domain have been identified in two independent families [McGuirt et al., 1999]. Thus mutations that lead to the synthesis of abnormal proα2(XI) chains can result in a spectrum of dominantly inherited phenotypes ranging in severity from hearing loss to perinatal lethal fibrochondrogenesis, likely reflecting the nature and location of the molecular defect.
The newborn radiographic phenotype of fibrochondrogenesis, consisting of shortened long bones with metaphyseal flaring leading to a dumbbell-shaped appearance, along with platyspondyly and epiphyseal abnormalities, bears some similarity to the phenotypes of OSMED and Kniest dysplasia (Table I). This similarity is evident in case R10-691A in which, despite perinatal lethality, there was a milder radiographic appearance. In particular, there was less pronounced metaphyseal widening and irregularity of the long bones, reduced anterior cupping of the ribs and a lack of caudally protruding spurs from the basilar portion of the ilia as compared with some fibrochondrogenesis cases, including the recessive case presented here. However, the dominant case exhibited the characteristic clinical findings of fibrochondrogenesis, especially the typical craniofacial abnormalities, distinguishing it from the other, related disorders.
OSMED can result from homozygosity for null mutations in COL11A2 [Temtamy et al., 2006], heterozygosity for missense mutations in COL11A2 [Pihlajamaa et al., 1998] or heterozygosity for an in-frame deletion in the triple helical domain of the COL2A1 product [Miyamoto et al., 2005]. Kniest dysplasia has been shown to result from heterozygosity for mutations in COL2A1 [Winterpacht et al., 1993]. These genotypic differences distinguish the fibrochondrogenesis cases studied here from the other phenotypes in this spectrum of disease, and provide insight into the effect of discrete mutations and molecular mechanisms that alter the function of the main structural component of cartilage, the type II/XI collagen heterotypic fibril. The phenotypic similarities among these disorders likely arise from the contribution of the gene products affected in all three disorders to the cartilage collagen fibril.
This study has identified COL11A2 as a second locus for fibrochondrogenesis, has indicated that synthesis of structurally abnormal proα2(XI) collagen molecules can produce fibrochondrogenesis, and has extended the spectrum of severity among the type XI collagenopathies resulting from COL11A2 mutations. The data further demonstrate that there are dominant forms of fibrochondrogenesis, and this finding must now be taken into account when counseling families in that recurrence risk will need to consider the possibility of parental germline mosaicism for dominant mutations. Analysis of additional cases will help determine the relative frequencies of mutations in COL11A1 and COL11A2 in fibrochondrogenesis as well as the proportion of cases with dominant and recessive inheritance.
ACKNOWLEDGMENTS
We thank the families who participated in this work. The authors have no conflicts of interest. This work was supported in part by grants from the National Institutes of Health (DE019567, HD22657 and M01-RR00425).
REFERENCES
- Akawi NA, Al-Gazali L, Ali BR. Clinical and molecular analysis of UAE fibrochondrogenesis patients expands the phenotype and reveals two COL11A1 homozygous null mutations. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01734.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Al-Gazali LI, Bakir M, Dawodu A, Haas D. Recurrence of fibrochondrogenesis in a consanguineous family. Clin Dysmorphol. 1999;8:59–61. [PubMed] [Google Scholar]
- Bankier A, Fortune D, Duke J, Sillence DO. Fibrochondrogenesis in male twins at 24 weeks gestation. Am J Med Genet. 1991;38:95–98. doi: 10.1002/ajmg.1320380121. [DOI] [PubMed] [Google Scholar]
- Chen W, Kahrizi K, Meyer NC, Riazalhosseini Y, Van Camp G, Najmabadi H, Smith RJ. Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J Med Genet. 2005;42:e61. doi: 10.1136/jmg.2005.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremonesi L, Cainarca S, Rossi A, Padoan R, Ferrari M. Detection of a de novo R1066H mutation in an Italian patient affected with cystic fibrosis. Hum Genet. 1996;98:119–121. doi: 10.1007/s004390050171. [DOI] [PubMed] [Google Scholar]
- Eteson DJ, Adomian GE, Ornoy A, Koide T, Sugiura Y, Calabro A, Lungarotti S, Mastroiacovo P, Lachman RS, Rimoin DL. Fibrochondrogenesis: Radiologic and histologic studies. Am J Med Genet. 1984;19:277–290. doi: 10.1002/ajmg.1320190210. [DOI] [PubMed] [Google Scholar]
- Leeners B, Funk A, Cotarelo CL, Sauer I. Two sibs with fibrochondrogenesis. Am J Med Genet. 2004;127:318–320. doi: 10.1002/ajmg.a.20620. [DOI] [PubMed] [Google Scholar]
- Li SW, Takanosu M, Arita M, Bao Y, Ren ZX, Maier A, Prockop DJ, Mayne R. Targeted disruption of Col11a2 produces a mild cartilage phenotype in transgenic mice: comparison with the human disorder otospondylomegaepiphyseal dysplasia (OSMED). Dev Dyn. 2001;222:141–152. doi: 10.1002/dvdy.1178. [DOI] [PubMed] [Google Scholar]
- Martinez-Frias ML, Garcia A, Cuevas J, Rodriguez JI, Urioste M. A new case of fibrochondrogenesis from Spain. J Med Genet. 1996;33:429–431. doi: 10.1136/jmg.33.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkoniemi M, Brunner HG, Manouvrier S, Hennekam R, Superti-Furga A, Kääriäinen H, Pauli RM, van Essen T, Warman ML, Bonaventure J, Miny P, Ala-Kokko L. Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am J Hum Genet. 2000;66:368–377. doi: 10.1086/302750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirt W, Prasad S, Griffith A, Kunst H, Green G, Shpargel K, Runge C, Huybrechts C, Mueller R, Lynch E, King MC, Brunner HG, Cremers CW, Takanosu M, Li SW, Arita M, Mayne R, Prockop DJ, Van Camp G, Smith RJ. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat Genet. 1999;23:413–419. doi: 10.1038/70516. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Nakashima E, Hiraoka H, Ohashi H, Ikegawa S. A type II collagen mutation also results in oto-spondylo-megaepiphyseal dysplasia. Hum Genet. 2005;118:175–178. doi: 10.1007/s00439-005-0058-0. [DOI] [PubMed] [Google Scholar]
- Morris N, Bachinger H. Type XI collagen is a heterotrimer with the composition (1 alpha, 2 alpha, 3 alpha) retaining non-triple-helical domains. J Biol Chem. 1987;262:11345–11350. [PubMed] [Google Scholar]
- Pihlajamaa T, Prockop DJ, Faber J, Winterpacht A, Zabel B, Giedion A, Wiesbauer P, Spranger J, Ala-Kokko L. Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymüller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am J Med Genet. 1998;80:115–120. doi: 10.1002/(sici)1096-8628(19981102)80:2<115::aid-ajmg5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Randrianaivo H, Haddad G, Roman H, Delezoide AL, Toutain A, Le Merrer M, Moraine C, Lise A. Fetal fibrochondrogenesis at 26 weeks’ gestation. Prenat Diagn. 2002;22:806–810. doi: 10.1002/pd.423. [DOI] [PubMed] [Google Scholar]
- Sirko-Osadsa DA, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH. Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J Pediatr. 1998;132:368–371. doi: 10.1016/s0022-3476(98)70466-4. [DOI] [PubMed] [Google Scholar]
- Tompson SW, Bacino CA, Safina NP, Bober MB, Proud VK, Funari T, Wangler MF, Nevarez L, Ala-Kokko L, Wilcox WR, Eyre DR, Krakow D, Cohn DH. Fibrochondrogenesis results from mutations in the COL11A1 type XI collagen gene. Am J Hum Genet. 2010;87:708–712. doi: 10.1016/j.ajhg.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temtamy SA, Männikkö M, Abdel-Salam GM, Hassan NA, Ala-Kokko L, Afifi HH. Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical and radiological findings in sibs homozygous for premature stop codon mutation in the COL11A2 gene. Am J Med Genet A. 2006;140:1189–1195. doi: 10.1002/ajmg.a.31205. [DOI] [PubMed] [Google Scholar]
- Vikkula M, Madman ECM, Lui VCH, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SEC, de Waal Malefijt MC, van den Hoogen FHJ, Ropers HH, Mayne R, Cheah KSE, Olsen BR, Warman ML, Brunner HG. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80:431–437. doi: 10.1016/0092-8674(95)90493-x. [DOI] [PubMed] [Google Scholar]
- Vuoristo MM, Pappas JG, Jansen V, Ala-Kokko L. A stop codon mutation in COL11A2 induces exon skipping and leads to non-ocular Stickler syndrome. Am J Med Genet A. 2004;130A:160–164. doi: 10.1002/ajmg.a.30111. [DOI] [PubMed] [Google Scholar]
- Whitley CB, Langer LO, Jr, Ophoven J, Gilbert EF, Gonzalez CH, Mammel M, Coleman M, Rosemberg S, Rodriques CJ, Sibley R, Horton WA, Opitz JM, Gorlin RJ. Fibrochondrogenesis: Lethal, autosomal recessive chondrodysplasia with distinctive cartilage histopathology. Am J Med Genet. 1984;19:265–275. doi: 10.1002/ajmg.1320190209. [DOI] [PubMed] [Google Scholar]
- Winterpacht A, Hilbert M, Schwarze U, Mundlos S, Spranger J, Zabel BU. Kniest and Stickler dysplasia phenotypes caused by collagen type II gene (COL2A1) defect. Nat Genet. 1993;3:323–6. doi: 10.1038/ng0493-323. [DOI] [PubMed] [Google Scholar]
- Wu J, Eyre D. Structural analysis of cross-linking domains in cartilage type XI collagen: insights on polymeric assembly. J Biol Chem. 1995;270:18865–18870. doi: 10.1074/jbc.270.32.18865. [DOI] [PubMed] [Google Scholar]
- Mendelian Inheritance in Man (MIM) http://www.ncbi.nlm.nih.gov/Omim IBD Finder, http://dna.leeds.ac.uk/ibdfinder/