

Abstract
Objective
In the US, second non-ocular malignancies are the primary cause of death in retinoblastoma survivors with the germline RB1 mutation. Soft tissue sarcomas are one of the most likely malignancies to pose a risk to these patients, with leiomyosarcoma (LMS) being the most common subtype. As our cohort is followed for a longer period, we discover new second malignancy risks for these patients.
Methods
We estimated the risk for uterine leiomyosarcoma (ULMS) in a cohort of 1854 patients with retinoblastoma who were diagnosed at two US institutions from 1914 through 1996. The standardized incidence ratio and excess absolute risk were calculated by comparison with population data from the Connecticut Tumor Registry or from National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) database. The cumulative risk at 50 years of age was also calculated.
Results
Seven of 525 female hereditary retinoblastoma patients developed ULMS. Five of these patients were used in the risk analysis, resulting in an excess risk of 3.87 per 10,000 women. Among hereditary patients who developed ULMS the excess risk increases dramatically with age: to 20/10,000 for female hereditary retinoblastoma patients aged between 30–39 years, and to 27/10,000 for patients aged 40+ years.
Conclusion
There is a substantial excess risk of ULMS in female hereditary retinoblastoma patients. As more patients survive into their thirties, this number is likely to increase. These findings raise the question of early childbearing, screening and prophylactic measures in hereditary retinoblastoma patients: all issues that would benefit from confirmation from other retinoblastoma cohorts, to allow for better guided counsel of these patients.
Keywords: Retinoblastoma, Uterine Leiomyosarcoma, Secondary Cancers
Introduction
Retinoblastoma is a cancer of the retina in very young children. It is the most common intraocular tumor in children, representing 3% of malignant neoplasms in children younger than 15 months [1]. In patients with the germinal form of retinoblastoma, their germline carries an inactivated RB1 allele, which is then passed to the somatic cells of their progeny. With the loss of a functional RB1-encoded protein, -a cell cycle regulatory retinoblastoma protein (pRb), the patient is at risk for developing a second non-ocular malignancy. In the United States, up to 97% of retinoblastoma patients will survive 5 years [2]. However, they maintain a life long risk for second non-ocular malignancies, which are the foremost cause of death in retinoblastoma survivors with the germline mutation [3]. For example, in our cohort, cumulative incidence for developing a new cancer at 50 years after diagnosis of hereditary Rb is 36 % [4].
The excess risk of second malignancies derives predominantly from soft tissue sarcoma and osteosarcoma [4, 5], and we have reported a strong radiation dose-response for sarcomas in the field of radiation [6]. Leiomyosarcoma (LMS) is the most common subtype of soft tissue sarcoma for both genders with an excess absolute risk (EAR) of 9.1/10,000, in and out of the radiation field; and the majority of LMS are diagnosed 30 years after retinoblastoma diagnosis [5].
As our cohort ages and our follow up period longer, we discover new second malignancy risks for these patients; causing us to question what screening recommendations could be offered. Herein, we report on the increased risk of uterine leiomyosarcoma (ULMS) in hereditary retinoblastoma patients and provide a perspective for patient management relative to these findings.
Methods
Ethics statement: The study was approved by The Institutional Review Board of the National Institutes of Health. Informed written consent was obtained from all participants in this study.
Our cohort has been previously described [4, 6] and consists of 1854 one-year survivors of retinoblastoma who were diagnosed at two US institutions from 1914 through 1996. It includes 1092 (59%) hereditary retinoblastoma patients (defined as having bilateral retinoblastoma or unilateral disease with a pertinent family history of retinoblastoma) and 762 (41%) nonhereditary retinoblastoma patients (defined as unilateral disease with no family history). This present study focused on the 525 female hereditary retinoblastoma patients. Since 1984, this cohort has been investigated for subsequent cancers. Study methods included review of hospital and radiotherapy records, searches of the National Death Index and several follow-up telephone interviews with subjects or family members through 2000. The leiomyosarcomas were classified by both topography and morphology as confirmed by pathology report, autopsy, other medical records or death certificate [5].
Statistical methods have been described previously [5]. The expected number of leiomyosarcoma was based on population data from the Connecticut Tumor Registry for tumors diagnosed before 1970 or from National Cancer Institute Surveillance, Epidemiology, and End Results database for tumors diagnosed after 1970. Rates were generated for ULMS based on International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) morphology classification. Accumulation of person-years of follow-up began 1 year after diagnosis of retinoblastoma and ended at the date of ULMS, date last known alive, the date of death, or December 31, 2000; whichever was earliest. The standardized incidence ratio (SIR) for LMS was calculated as the ratio of observed ULMS to the expected number. The EAR was calculated as the observed minus the expected number of ULMS divided by person-years at risk multiplied by 10,000. All 95 % confidence intervals were determined based on Poisson distribution. The cumulative risk at 50 years of age of ULMS was calculated using the Gooley method [7], which takes into account the competing risk of death. Patient 7 and 4 were diagnosed with ULMS in 2003 and 2010, respectively; however, these patients were not included in the risk analysis, because their diagnosis was ascertained after the study closing date.
Results
The median duration of follow-up of the retinoblastoma cohort was 28.5 years (range 1–69 years) for hereditary patients and 29.6 years (range 1–77 years) for non-hereditary patient. In our cohort, 8 patients developed leiomyosarcoma of the uterus with a mean follow up of 46.9 (+/− 11.1) years (range 34–63 years) and median follow up of 47 years since retinoblastoma diagnosis. Seven (85.7%) patients had hereditary retinoblastoma (Table 1). Of these hereditary retinoblastoma patients, 4 (57.1%) received chemotherapy, 6 (85.7 %) were treated with external beam radiation (EBRT) and 3 (42.9%) were irradiated at an age of less than 1 year (Table 1a). The average age of retinoblastoma diagnosis was 14.0 (+/−10.2) months and of uterine leiomyosarcoma diagnosis was 41.4 (+/− 7.9) years. The average age of death was 44.2 (+/−12.6) years. After excluding one patient who died from oat cell lung cancer 16 yrs after uterine leiomyosarcoma diagnosis, the average interval between ULMS diagnosis and death was 2.4 (+/− 2.9) years. Two patients remain alive after ULMS diagnosis: one having been diagnosed a year ago and a second who is living almost 7 years after uterine leiomyosarcoma diagnosis.
Table 1a.
Selected Characteristics on the 8 Retinoblastoma Patients diagnosed with Uterine Leiomyosarcoma
| Patient | Age at dx (yrs) | Yr of ULMS Dx | Age at Rb Dx (mos) | Hereditary Rb | Dose to Uterus (Gy) | Vital Status | Age at Death (yrs) | Interval until Death* (mos) |
|---|---|---|---|---|---|---|---|---|
| 1 | 32 | 1992 | 23 | Yes | 0.14 | Dead | 39 | 76 |
| 2 | 33 | 1984 | 1 | Yes | 0.08 | Dead | 34 | 3 |
| 3 | 35 | 1996 | 5 | Yes | 0.18 | Dead | 35 | 1 |
| 4 | 44 | 2010 | 18 | Yes | NR | Alive | - | - |
| 5 | 47 | 1991 | 15 | Yes | 0.27 | Dead | 49 | 36 |
| 6 | 49 | 1979 | 29 | Yes | No Rad | Dead | 64 | 192 |
| 7 | 51 | 2003 | 7 | Yes | NR | Alive | - | - |
| 8 | 52 | 1996 | 28 | No | No Rad | Dead | 55 | 36 |
Dx, diagnosis; ULMS, uterine leiomyosarcoma; Rb retinoblastoma; NR, not recorded; No Rad, no radiation. Interval until death defined as months from ULMS diagnosis until death.
Five of 525 female hereditary retinoblastoma patients who developed ULMS, resulted in an excess risk of 3.9 per 10,000 women (excluding the patients diagnosed with ULMS in 2003 and 2010) (Table 2). Among hereditary patients who developed ULMS, the excess risk increased dramatically with age: to 20/10,000 for female hereditary retinoblastoma patients aged between 30–39 years, and to 27/10,000 for patients aged 40+ years (Table 3, Figure 1 & 2). Cumulative risk at 50 years of age for ULMS in hereditary retinoblastoma patients was 3.2% (95% CI 1.0%–8.0%). The excess risk of ULMS in hereditary Rb patients treated with radiation for retinoblastoma was 3.7/10,000 and without radiation was 5.2/10,000; those receiving radiation before 12 months of age was 2.8/10,000 and 5.2/10,000 in those patients receiving radiation after 12 months of age (Table 4). The excess risk of LMS in hereditary patients receiving chemotherapy for retinoblastoma was 4.1/10,000 and 3.9/10,000 in those patients who were not treated with chemotherapy (Table 4).
Table 1b.
Selected Characteristics of 3 Hereditary Retinoblastoma Patients diagnosed with other pelvic malignancies
| Patient | Age at Ca dx (yrs) | Age at Rb Dx (mos) | Dose to Uterus (Gy) | Vital Status | Age at Death (yrs) | Interval until Death (mos) | Diagnosis |
|---|---|---|---|---|---|---|---|
| 9 | 38 | 6 | NR | Dead | 40 | 2 | LMS retroperitoneum |
| 10 | 44 | 17 | 0.17 | Alive | – | – | Uterine adenocarcinoma |
| 11 | 49 | 19 | No Rad | Dead | 49 | 0.03 | Uterine Carcinoma, NOS |
Ca, cancer; Dx, diagnosis; Rb retinoblastoma; LMS, leiomyosarcoma; NOS, not otherwise specified; NR, not recorded; No Rad, no radiation. Interval until death defined as months from ULMS diagnosis until death.
Table 2.
Risk Analysis of Hereditary Retinoblastoma Patients Diagnosed with Uterine Leiomyosarcoma
| No. female subjects | Obs | Exp | SIR | 95% CI | EAR/10E4 | |
|---|---|---|---|---|---|---|
| All Rb patients | 900 | 6 | 0.04 | 164 | 60–356 | 2.59 |
| Hereditary | 525 | 5 | 0.02 | 277 | 90–646 | 3.87 |
| Non-Hereditary | 375 | 1 | 0.02 | 54 | 1.4–299 | 0.96 |
Obs, observed; Exp, expected; SIR, standardized incidence ratio; 95% CI, 95% confidence interval; EAR, excess absolute risk.
Figure 1.
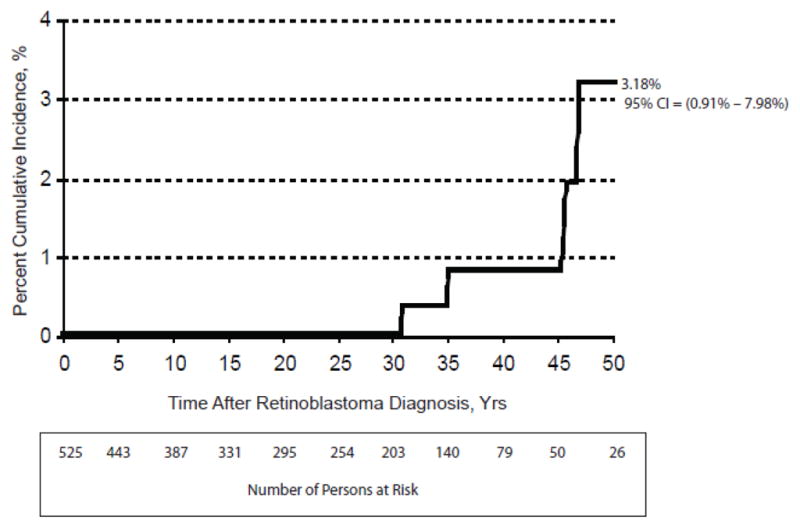
Cumulative risk of ULMS by time since diagnosis of hereditary retinoblastoma.
Figure 2.

Excess risk of ULMS in hereditary retinoblastoma survivors.
Table 3.
Risk Analysis by Time Since Diagnosis of Hereditary Retinoblastoma in Patients with Uterine Leiomyosarcoma
| Years since RB dx | No. entering interval | Obs | Exp | SIR | 95% CI | EAR/10E4 |
|---|---|---|---|---|---|---|
| 0–1 | 525 | 0 | 0 | 0 | ||
| 1–9 yrs | 525 | 0 | 0 | 0 | ||
| 10–19 yrs | 397 | 0 | 0 | 0 | ||
| 20–29 yrs | 308 | 0 | 0 | 0 | ||
| 30–39 yrs | 215 | 3 | 0.01 | 519 | 107–1516 | 20 |
| 40+yrs | 87 | 2 | 0.01 | 199 | 24–717 | 27 |
| All yrs | 525 | 5 | 0.02 | 277 | 90–646 | 3.87 |
Obs, observed; Exp, expected; SIR, standardized incidence ratio; 95% CI, 95% confidence interval; EAR, excess absolute risk.
Two patients developed additional nonocular malignancies. Both of these patients had malignant melanoma, and one also developed basal cell carcinoma of the skin and oat cell lung cancer. Three patients had metastatic ULMS. Patients 2, 6 and 9 had a family history of retinoblastoma in their parents.
Three additional patients were diagnosed with other uterine or retroperitoneal malignancies (Table 1b). Patients 10 and 11 developed uterine adenocarcinoma and carcinoma, not otherwise specified, respectively. Patient 9 was diagnosed with LMS of the retroperitoneum. The average age of diagnosis of these three patients was 43.7 (+/−5.5) years and the average interval between malignancy diagnosis and death was 1.0 (+/− 1.4) years. Patient 10 is still alive and also suffered from cutaneous LMS of the trunk.
Discussion
We reveal a substantial excess risk of ULMS in female hereditary retinoblastoma patients and herein describe this group. Our cohort has been followed for longer than the majority of other cohorts of childhood cancer survivors, and consists of a large number of long-term retinoblastoma survivors. We have previously described LMS as the most common subtype of soft tissue sarcoma in hereditary retinoblastoma patients [5]. LMS are predominantly diagnosed 30 years after retinoblastoma diagnosis and in our cohort occurred most frequently in the head and face of male patients, but are most frequently of pelvic origin in female patients [5]. As we show, the excess risk of ULMS is 3.9 per 10,000 women with hereditary retinoblastoma over all ages. However, as patients enter the age when at risk for ULMS, this excess risk increases dramatically: to 20/10,000 for female hereditary retinoblastoma patients aged between 30–39 years, and to 27/10,000 for patients aged 40+ years.
Other groups have reported LMS in hereditary retinoblastoma patients at anatomical sites including the bladder, thigh, head and neck, abdomen, liver and rectum [8–14]. One case describes LMS of the retroperitoneum [15], while Fletcher et al describe a hereditary retinoblastoma patient with adenocarcinoma of the uterus [16]. Our robust long-term follow-up allows for more patients entering the age when ULMS are commonly diagnosed. As other retinoblastoma cohorts match ours in follow up, they too may discover an excess risk of ULMS in female hereditary retinoblastoma survivors. Furthermore, now that external beam radiation is being replaced by other means of treatment, it is predicted that fewer patients will die of radiation-associated malignancies. This would result in a higher population of patients surviving until their thirties and beyond when ULMS becomes prevalent, giving another reason to expect an increase in ULMS in this population.
According to an analysis of the Surveillance, Epidemiology, and End Results (SEER) Program data, 40% of LMS in women are uterine in origin [17] and LMS make up one third of uterine sarcomas [18]. ULMS have an incidence of 0.3–0.4 per 10,000 women per year, and they account for 1.3% of all uterine malignancies [17, 19]. Most ULMS occur in women over 40 years of age and peaks in the 5th and 6th decades [17, 20–22, 26]. ULMS have a five-year survival rate of 50–60% in stage I, and 15% in more advanced stages [23]; with death typically occurring within 2 years of diagnosis. The mean age of ULMS diagnosis in our cohort was 41.4 years, which is younger than the occurrence of ULMS in the general population. However, the interval until death is approximately 2.4 years, which is comparable to the general population.
Non-genetic risk factors for ULMS are controversial but include tamoxifen [18, 24], oral contraceptives, high body mass index, never-married status and menses onset earlier than 13 years of age [25, 26]. In addition, ULMS have been associated with a number of inconsistent chromosomal abnormalities, including TP53 and PTEN [27, 28]. Furthermore, Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC) is a reported genetic risk factor for ULMS. HLRCC syndrome is a rare disease that involves a predisposition to benign leiomyomas of the skin and uterus and early onset renal cell carcinoma caused by heterozygous germline mutations in fumarate hydratase (FH). While FH loss accounts for a very small proportion of ULMS, its pathogenesis bears some resemblance to retinoblastoma and thus may serve as a useful model. FH appears to act as a tumor suppressor and biallelic inactivation of FH is apparent in most HLRCC-associated tumors [29]. Interestingly, biallelic inactivation FH has also been reported in a single-non-syndromic ULMS [29]. In the Finnish population, there are 5 HRLCC patients with ULMS, with the average age of diagnosis at 32 years [29, 30] and a calculated SIR of 71 in one report [30]. Similar to our findings for RB1, this suggests FH is implicated in the genesis of early onset ULMS [29–31]. Some have suggested that hysterectomy be recommended to HLRCC patients and their families to reduce the risk of uterine malignancy [29]. No HLRCC patients in the United States have been reported to have ULMS. This may be a result of genetic variation or the tendency for young US women to elect for hysterectomy to relieve symptomatic leiomyoma, which acts prophylactically against the development of ULMS [29]. Given our findings of early onset ULMS in hereditary retinoblastoma patients, is it now possible to add hereditary retinoblastoma as a genetic risk factor for ULMS?
Like other tumor suppressor genes, RB1 gene instability may serve as an explanation for leiomyosarcoma formation. The gene product, p105Rb, functions in multiple cellular processes including DNA repair and cell-cycle checkpoint control [16, 32]. It has properties of a tumor suppressor gene so that loss of both copies of the gene favors malignant transformation [33]. Mutations in RB1 or altered expression of p105 Rb have been found in many sarcomas, and significantly associated with proliferative activity and poor survival [34, 35]. For instance, one study demonstrated pRb abnormalities in approximately 70% of the soft tissue sarcomas studied [36]. Investigations into leiomyosarcomas demonstrate the frequent deletions of chromosome 13, particularly within the 13q14–q15 region and support the notion that loss of RB1 is a critial step in LMS tumorigenesis [37]. Additionally, knockout mouse models have demonstrated that concomitant loss of p53 and Rb result in malignant leiomyosarcoma of the ovary [38]. In confirmation of previous studies, this report also suggests that concomitant loss of Rb and p53 may confer better cellular tolerance to insults, including chemotherapeutic agents [38]. As retinoblastoma patients age, they may be more prone to acquiring gene mutations such as those in tumor suppressor genes like p53. If this were to occur in association with an Rb mutation, it may be speculated that any resulting malignant cell (such as ULMS) may have better tolerance or resistance to chemotherapy. Furthermore, specific to the uterus, alterations in the RB1 pathway have been found in ULMS and loss of heterozygosity of RB1 had the second highest incidence of allelic changes in one study of twenty ULMS [39]. Therefore, it is plausible for a germline mutation in one allele of RB1 to confer an increased risk for ULMS.
The contribution of radiation-induced abnormalities to the RB1 gene has been suggested in human sarcoma tumorgenesis [40]. High-dose radiotherapy for cervical cancer has been associated with uterine sarcoma of the mixed mullerian subtype [41]. Low radiation doses, like those received by atomic bomb survivors, suggested a radiation effect on the risk for uterine malignancy in women who were exposed to the bombs prior to age 20 [42]. Furthermore, this childhood radiation effect on secondary ULMS is not limited to patients with retinoblastoma: for instance, a patient with childhood neuroblastoma treated with radiation therapy at 2 months of age was diagnosed with leiomyosarcoma of the uterus at age 33.6 years (and also had a history of melanoma) [43]. The uterus is out of the field of radiation for retinoblastoma and therefore typically receives a very low radiation dose. Likewise, the average radiation dose to the uterus in our cohort ranged from 0.08 to 0.20 Gy [4]; making it less likely for this to impact the risk of ULMS genesis. As confirmation of this, in our cohort, there was not an excess risk of ULMS in those patients who received radiotherapy compared to those that did not; nor even in patients who received radiotherapy before 12 months of age compared to those that received it later in life.
Now that we have established an excess risk for ULMS in hereditary retinoblastoma survivors, the next step involves counseling our patients. The recommended screening techniques for gynecological cancers include annual pelvic exam, transvaginal ultrasonography, endometrial biopsy and CA-125 levels beginning at age 25–35 years. However, there is a lack of sufficient support for these screening techniques in premenopausal and postmenopausal women. Even MRI appearance proves unhelpful in distinguishing benign from malignant uterine cancers [44]; and other imaging techniques such as ultrasound, computed tomography and positron emission tomography are similarly unreliable [45]. Furthermore, a retrospective study of preoperative endometrial sampling in uterine sarcomas predicted the correct histological diagnosis in only 64% of samples [46]. Therefore, preoperative diagnosis remains difficult in the management of all uterine sarcomas including ULMS.
The non-genetic risk factors for ULMS in the general population may interact with the risk of ULMS in hereditary retinoblastoma patients. Therefore it may be prudent to advise hereditary retinoblastoma patients to maintain a lean body mass; particularly in those women with menses onset earlier than 13 years of age. Furthermore, there are suggestions of an underlying genetic susceptibility to both melanoma and leiomyosarcoma in patients without retinoblastoma [47, 48]. As testament to this, two of our patients with ULMS also developed cutaneous melanomas. Patients should be reminded to protect their skin, although it is not known if the cutaneous melanoma associated with hereditary retinoblastoma is affected by excessive ultraviolet radiation exposure.
What is the role of prophylactic surgery for ULMS in hereditary retinoblastoma patients; for example, prophylactic hysterectomy? One may deem hysterectomy as extreme management for a uterine cancer with a cumulative risk of 3.18% at 50 years of age, as in our cohort. But to give perspective to this argument, hysterectomies are the second most common operation performed in the United States: by age 60 years, approximately 1 in 3 American women will have had a hysterectomy [49, 50]. In fact, 10% of hysterectomies are performed to treat cancer, while most are elected to mitigate symptomatic leiomyoma. From 2001–2004, an estimated 3.1 million women in the United States had received a hysterectomy; however, the rate decreased slightly over this time period and given advances in medicine has likely continued to do so [51]. Given the seemingly low threshold to perform hysterectomy, using it for the purpose of reducing the risk of cancer to almost zero is not so far-fetched. We are not asserting prophylactic hysterectomy as the standard of care, but raise the notion as a point of discussion for future studies.
Some presume ULMS are the malignant counterpart to leiomyoma; however, it is estimated that less that 0.1% of leiomyomas progress to LMS [52]. Nevertheless, more recently, investigations have demonstrated similar transcriptional profiles [53], immunohistochemical and genetic aberrations between leiomyoma and LMS [54, 55]. This may suggest both tumors have similar genetics playing a role in their pathogenesis. In our telephone interview of 416 female survivors in 2000, there were a reported 18 hereditary and 7 non-hereditary patients with uterine leiomyoma; this included patient 1 who then developed ULMS and patient 10 who was later diagnosed with uterine adenocarcinoma (of note, the daughter of patient 6 also had leiomyoma). Patient 4 was not included in the telephone interview but she also had a history of leiomyoma and was diagnosed with ULMS. 14 of the 18 hereditary retinoblastoma patients and all of the non-hereditary patients had hysterectomies and thus were not at risk for ULMS. It is unclear whether the presence of leiomyoma in hereditary retinoblastoma patients predisposes them to developing ULMS. This phenomenon will continue to be monitored in our cohort and collaboration by other groups would be useful.
Perhaps the most important information gleaned from our study, is the relatively young age for ULMS diagnosis in hereditary retinoblastoma patients compared to the general population. The average age of ULMS diagnosis in our cohort was 41.4 years, but ranged from 32 to 52 years. This highlights the clear need for early childbearing in hereditary retinoblastoma patients; and efforts should be made to counsel patients to this effect.
As other cohorts of retinoblastoma patients continue to be followed longer, it is advisable to evaluate for ULMS incidence and its risk patterns. It would be helpful for our patients to elucidate additional risk factors like smoking, endogenous and exogenous estrogen, childbearing demographics, body mass indices and treatment or age characteristics. We have discovered a profound excess risk for ULMS in hereditary retinoblastoma, but it would be ideal to expand this to include additional data from other cohorts. This would create a more robust profile of these women at risk and allow for better–guided counsel of women with hereditary retinoblastoma.
Table 4.
Risk Analysis by Treatment and Age of Hereditary Retinoblastoma Diagnosis in Patients with Uterine Leiomyosarcoma
| No. female subjects | Obs | Exp | SIR | 95% CI | EAR/10E4 | |
|---|---|---|---|---|---|---|
| Any Radiation | ||||||
| Yes | 459 | 4 | 0.01 | 298 | 81–756 | 3.65 |
| No | 63 | 1 | 0 | 215 | 5.4–1196 | 5.22 |
| Any Chemo | ||||||
| Yes | 200 | 3 | 0 | 433 | 52–1562 | 4.06 |
| No | 317 | 2 | 0.01 | 229 | 47–670 | 3.87 |
| Age at Rb Dx | ||||||
| 0–12 mos | 301 | 2 | 0.01 | 291 | 35–1050 | 2.79 |
| >12 mos | 224 | 3 | 0.01 | 269 | 55–786 | 5.21 |
Obs, observed; Exp, expected; SIR, standardized incidence ratio; 95% CI, 95% confidence interval; EAR, excess absolute risk.
Research highlights.
There is a substantial excess risk of ULMS in hereditary retinoblastoma patients.
Among hereditary patients, the excess risk increases dramatically with age.
These findings raise questions of how to best counsel these retinoblastoma patients.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, Division of Cancer Epidemiology and Genetics and by The Fund for Ophthalmic Knowledge.
Footnotes
None of the authors have any relevant financial relationships with commercial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schefler AC, Abramson DH, Dunkel IJ, McCormick B. Neoplasms of the Eye. In: Kufe DW, Frei E, Holland JF, Weichselbaum RR, Pollock RE, Bast RC, Hong WK, Hait WN, editors. Cancer Medicine 8. Shelton, CT: People’s Medical Publishing House-USA; 2010. [Google Scholar]
- 2.Ries LAG, Harkins D, Krapcho M, Mariotto A, Miller BA, Feuer EJ, et al., editors. SEER Cancer Statistics Review, 1975–2003. Bethesda (MD): National Cancer Institute; 2006. [Google Scholar]
- 3.Yu Cl, Tucker MA, Abramson DH, Furukawa K, Seddon JM, Stovall M, et al. Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst. 2009;101:581–591. doi: 10.1093/jnci/djp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005;23:2272–9. doi: 10.1200/JCO.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 5.Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF., Jr Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24–31. doi: 10.1093/jnci/djk002. [DOI] [PubMed] [Google Scholar]
- 6.Wong FL, Boice JD, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, et al. Cancer Incidence after retinoblastoma: radiation dose and sarcoma risk. JAMA. 1997;278:1262–7. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- 7.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18:695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 8.Liang SX, Lakshmanan Y, Woda BA, Jiang Z. A high-grade primary leiomyosarcoma of the bladder in a survivor of retinoblastoma. Arch Pathol Lab Med. 2001;125:1231–4. doi: 10.5858/2001-125-1231-AHGPLO. [DOI] [PubMed] [Google Scholar]
- 9.Klippenstein KA, Wesley RE, Glick AD. Orbital leiomyosarcoma after retinoblastoma. Ophthalmic Surg Lasers. 1999;30:579–83. [PubMed] [Google Scholar]
- 10.Minagawa T, Okaneya T, Kamigaito M, Nishizawa S, Ogawa T, Kawakami M, et al. Leiomyosarcoma of the urinary bladder in a patient with bilateral retinoblastoma. Int J Urol. 2008;15:548–50. doi: 10.1111/j.1442-2042.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 11.Pauser U, Grimm H. Intramucosal leiomyosarcoma of the stomach following hereditary retinoblastoma in childhood - a case report and review of the literature. World J Surg Oncol. 2008;6:131. doi: 10.1186/1477-7819-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelli N, Thiefin G, Diebold MD, Bouche O, Aucouturier JP, Zeitoun P. Primary leiomyosarcoma of the liver 37 years after successful treatment of hereditary retinoblastoma. Gastroenterol Clin Biol. 1996;20:502–505. [PubMed] [Google Scholar]
- 13.Marees T, van Leeuwen FE, de Boer MR, Imhof SM, Ringens PJ, Moll AC. Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009;45:3245–53. doi: 10.1016/j.ejca.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771–1779. doi: 10.1093/jnci/djn394. [DOI] [PubMed] [Google Scholar]
- 15.Venkatraman L, Goepel JR, Steele K, Dobbs SP, Lyness RW, McCluggage WG. Soft tissue, pelvic, and urinary bladder leiomyosarcoma as second neoplasm following hereditary retinoblastoma. J Clin Pathol. 2003;56:233–6. doi: 10.1136/jcp.56.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime Risks of Common Cancers Among Retinoblastoma Survivors. J Natl Cancer Inst. 2004;96:357–63. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 17.Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the Surveillance, Epidemiology and End Results program, 1978–2001. Int J Cancer. 2006;119:2922–2930. doi: 10.1002/ijc.22239. [DOI] [PubMed] [Google Scholar]
- 18.Wickerham DL, Fisher B, Wolmark N, Bryant J, Costantino J, Bernstein L, et al. Association of tamoxifen and uterine sarcoma. J Clin Oncol. 2002;20:2758–2760. doi: 10.1200/JCO.2002.20.11.2758. [DOI] [PubMed] [Google Scholar]
- 19.Leibsohn D, d’Ablaing G, Mishell DR, Jr, Schlaerth JB. Leiomyosarcoma in a series of hysterectomies performed for presumed uterine leiomyosarcomas. Am J Obstet Gynecol. 1990;162:968–74. doi: 10.1016/0002-9378(90)91298-q. [DOI] [PubMed] [Google Scholar]
- 20.Gadducci A, Landoni F, Sartori E, Zola A, Maggino T, Lissoni A, et al. Uterine leiomyosarcoma: Analysis of treatment failures and survival. Gynecol Oncol. 1996;62:25–32. doi: 10.1006/gyno.1996.0185. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz LB, Diamond MP, Schwartz PE. Leiomyosarcomas: clinical presentation. Am J Obstet Gynecol. 1993;168:180–3. doi: 10.1016/s0002-9378(12)90910-2. [DOI] [PubMed] [Google Scholar]
- 22.Giuntoli RL, Metzinger DS, DiMarco CS, Cha SS, Sloan JA, Keeney GL, et al. Retrospective review of 208 patient with leiomyosarcoma of the uterus: prognostic indicators, surgical management, and adjuvant therapy. Gynecol Oncol. 2003;89:460–469. doi: 10.1016/s0090-8258(03)00137-9. [DOI] [PubMed] [Google Scholar]
- 23.Major FJ, Blessing JA, Silverberg SG, Morrow CP, Creasman WT, Currie JL, et al. Prognostic factors in early-stage uterine sarcoma. A Gynecologic Oncology Group study. Cancer. 1993;71:1702–9. doi: 10.1002/cncr.2820710440. [DOI] [PubMed] [Google Scholar]
- 24.Mourits MJ, De Vries EG, Willemse PH, Ten Hoor KA, Hollema H, Van der Zee AG. Tamoxifen treatment and gynecologic side effects: a review. Obstet Gynecol. 2001;97:855. doi: 10.1016/s0029-7844(00)01196-0. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz SM, Weiss NS. Marital status and the incidence of sarcomas of the uterus. Cancer Res. 1990;50:1886–9. [PubMed] [Google Scholar]
- 26.Schwartz SM, Weiss NS, Daling JR, Gammon MD, Liff JM, Watt J, et al. Exogenous sex hormone use, correlates of endogenous hormone levels, and the incidence of histologic types of sarcoma of the uterus. Cancer. 1996;77:717–24. doi: 10.1002/(sici)1097-0142(19960215)77:4<717::aid-cncr18>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 27.de Vos S, Wilczynski SP, Fleischhacker M, Koeffler P. p53 alterations in uterine leiomyosarcomas versus leiomyomas. Gynecol Oncol. 1994;54:205–8. doi: 10.1006/gyno.1994.1194. [DOI] [PubMed] [Google Scholar]
- 28.Amant F, de la Rey M, Dorfling CM, van der Walt L, Dreyer G, Dreyer L, et al. PTEN mutations in uterine sarcomas. Gynecol Oncol. 2002;85:165–9. doi: 10.1006/gyno.2002.6601. [DOI] [PubMed] [Google Scholar]
- 29.Kiuru K, Launonen V. Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) Curr Mol Med. 2004;4:869–875. doi: 10.2174/1566524043359638. [DOI] [PubMed] [Google Scholar]
- 30.Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, Salovaara R, Herva R, Kolvisto PA, et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet. 2006;43:523–6. doi: 10.1136/jmg.2005.036400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ylisaukko-Oja SK, Kiuru M, Lehtonen HJ, Lehtonen R, Pukkala E, Arola J, et al. Analysis of fumarate hydratase mutations in a population-based series of early onset uterine leiomyosarcoma patients. Int J Cancer. 2006;119:283–7. doi: 10.1002/ijc.21798. [DOI] [PubMed] [Google Scholar]
- 32.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 33.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polsky D, Mastorides S, Kim D, Dudas M, Leon L, Leung D, et al. Altered patterns of RB expression define groups of soft tissue sarcoma patients with distinct biological and clinical behavior. Histol Histopathol. 2006;21:743–52. doi: 10.14670/HH-21.743. [DOI] [PubMed] [Google Scholar]
- 35.Dei Tos AP, Maestro R, Doglioni C, Piccinin S, Libera DD, Bolocchi M, et al. Tumor suppressor genes and related molecules in leiomyosarcoma. Am J Pathol. 1996;148(4):1037–1045. [PMC free article] [PubMed] [Google Scholar]
- 36.Karpeh MS, Brennan MF, Cance WG, Woodruff JM, Pollack D, Casper ES, et al. Altered patterns of retinoblastoma gene product expression in adult soft tissue sarcomas. Br J Cancer. 1995;72:986–991. doi: 10.1038/bjc.1995.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otaño-Joos M, Mechtersheimer G, Ohl S, Lehnert T, Willeke F, Moller P, et al. Analysis of chromosome copy number changes in leiomyosarcoma through molecular cytogenetic methods. Verh Dtsch Ges Pathol. 1999;82:207–9. [PubMed] [Google Scholar]
- 38.Clark-Knowles KV, Senterman MK, Collins O, Vanderhyden BC. Conditional inactivation of Brca1, p53 and Rb in mouse ovaries results in the development of leiomyosarcomas. PLoS One. 2009;4(12):e8534. doi: 10.1371/journal.pone.0008534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhai YL, Nikaido T, Orii A, Horiuchi A, Toki T, Fujii S. Frequent occurrence of loss of heterozygosity among tumor suppressor genes in uterine leiomyosarcoma. Gynecol Oncol. 1999;75:453–9. doi: 10.1006/gyno.1999.5629. [DOI] [PubMed] [Google Scholar]
- 40.Brachman DG, Hallahan DE, Beckett MA, Yandell DW, Weichselbaum RR. p53 gene mutations and abnormal retinoblastoma protein in radiation-induced human sarcomas. Cancer Res. 1991;51:6393–6. [PubMed] [Google Scholar]
- 41.Boice JD, Jr, Engholm G, Kleinerman RA, Blettner M, Stovall M, Lisco H, et al. Radiation dose and second cancer risk in patients treated for cancer of the cervix. Radiat Res. 1988;116:3–55. [PubMed] [Google Scholar]
- 42.Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, et al. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res. 2007;168:1–64. doi: 10.1667/RR0763.1. [DOI] [PubMed] [Google Scholar]
- 43.Paulino AC, Fowler BZ. Secondary neoplasms after radiotherapy for childhood solid tumor. Pediatr Hematol Oncol. 2005;22:89–101. doi: 10.1080/08880010590896459. [DOI] [PubMed] [Google Scholar]
- 44.Cornfeld D, Israel G, Martel M, Weinreb J, Schwartz P, McCarthy S. MRI appearance of mesenchymal tumors of the uterus. Eur J Radiol. 2010;74:241–9. doi: 10.1016/j.ejrad.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Kitajima K, Murakima K, Kaji Y, Sugimura K. Spectrum of PDG PET/CT findings of uterine tumors. Am J Roentgenol. 2010;195:737. doi: 10.2214/AJR.09.4074. [DOI] [PubMed] [Google Scholar]
- 46.Bansal N, Herzog TJ, Burke W, Cohen CJ, Wright JD. The utility of preoperative endometrial sampling for the detection of uterine sarcomas. Gynecol Oncol. 2008;110:43. doi: 10.1016/j.ygyno.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 47.Berking C, Brady MS. Cutaneous melanoma in patients with sarcoma. Cancer. 1997;79:843–8. doi: 10.1002/(sici)1097-0142(19970215)79:4<843::aid-cncr22>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 48.Cohen RJ, Curtis RE, Inskip PD, Fraumeni JF., Jr The risk of developing second cancers among survivors of childhood soft tissue sarcoma. Cancer. 2005;103:2391–6. doi: 10.1002/cncr.21040. [DOI] [PubMed] [Google Scholar]
- 49.Carlson KJ, Eisenstat SA, Ziporyn T. The Harvard Guide to Women’s Health. Cambridge, MA: Harvard University Press; 1996. Hysterectomy; pp. 308–313. [Google Scholar]
- 50.Griffith H Winter. The Complete Guide to Symptoms, Illness and Surgery. 3. New York: Berkeley Publishing; 1995. Hysterectomy; pp. 818–825. [Google Scholar]
- 51.Whiteman MK, Hillis SD, Jamieson DJ, Morrow B, Podgornik MN, Brett KM, Marchbanks PA. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol. 2008;198(1):34.e1–7. doi: 10.1016/j.ajog.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 52.Buttram VC, Jr, Reiter RC. Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril. 1981;36:433–445. doi: 10.1016/s0015-0282(16)45789-4. [DOI] [PubMed] [Google Scholar]
- 53.Christacos NC, Quade BJ, Dal Cin P, Morton CC. Uterine leiomyomata with deletions of Ip represent a distinct cytogenetic subgroup associated with unusual histologic features. Genes Chromosomes Cancer. 2006;45(3):304–12. doi: 10.1002/gcc.20291. [DOI] [PubMed] [Google Scholar]
- 54.Mittal KR, Chen F, Wei JJ, Rijhvani K, Kurvathi R, Streck D, et al. Molecular and immunohistochemical evidence for the origin of uterine leiomyosarcomas from associated leiomyoma and symplastic leiomyoma-like areas. Mod Pathol. 2009;22(10):1303–11. doi: 10.1038/modpathol.2009.96. [DOI] [PubMed] [Google Scholar]
- 55.Levy B, Mukherjee T, Hirschhorn Molecular Cytogenetic Analysis of Uterine Leiomyoma and Leiomyosarcoma by Comparative Genomic Hybridization. Cancer Genet Cytogenet. 2000;121:1–8. doi: 10.1016/s0165-4608(00)00225-9. [DOI] [PubMed] [Google Scholar]