

Abstract
Autophagy is a homeostatic process that functions to balance cellular metabolism and promote cell survival during stressful conditions by delivering cytoplasmic components for lysosomal degradation and subsequent recycling. During viral infection, autophagy can act as a surveillance mechanism that delivers viral antigens to the endosomal/lysosomal compartments that are enriched in immune sensors. Additionally, activated immune sensors can signal to activate autophagy. To evade this antiviral activity, many viruses elaborate functions to block the autophagy pathway at a variety of steps. Alternatively, some viruses actively subvert autophagy for their own benefit. Manipulated autophagy has been proposed to facilitate nearly every stage of the viral lifecycle in direct and indirect ways. In this review, we synthesize the extensive literature on virus–autophagy interactions, emphasizing the role of autophagy in antiviral immunity and the mechanisms by which viruses subvert autophagy for their own benefit.
Keywords: Autophagy, Viruses, Innate immunity, Adaptive immunity, Selective autophagy, Metabolism
1. Introduction
Macroautophagy, hereafter called autophagy, is a process that generates membrane structures that engulf and sequester portions of the cytoplasm into enclosed double-membrane vesicles, autophagosomes, and delivers the contents to the lysosome for degradation (Fig. 1 A). This allows the cell to recycle nutrients and remove unwanted cytosolic components, such as damaged organelles and intracellular pathogens [1]. In addition to performing housekeeping functions, autophagy is induced by numerous stress stimuli to elicit an appropriate metabolic counter-response and prolong cell survival. Its multiple physiological roles are a likely reason that deficiencies in autophagy are associated with cancers, neurodegenerative diseases, and autoimmune disorders [2], [3].
Fig. 1.
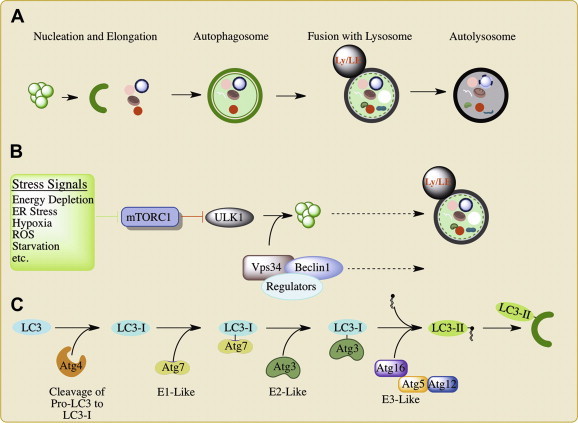
An overview of autophagy. A. Following initial autophagosome nucleation, the nascent autophagosome (phagophore) expands and engulfs cytoplasmic contents then closes to form a completed autophagosome. The autophagosome fuses with the lysosome to deliver its cargo for degradation and recycling of nutrients. B. Induction of autophagy is stimulated through a variety of stress signals that inhibit mTORC1. This leads to the activation of the ULK1/2 kinase complex which licenses autophagosome biogenesis. The phosphatidylinosital-3-kinase (PI3K) Vps34 forms a complex with Beclin-1 to drive the membrane expansion and fusion events essential to autophagy. Interaction with various Beclin-1 interacting proteins (“regulators”) facilitates the coordination of these events. C. LC3-I is conjugated to phosphatidylethanolamine (PE) to generate LC3-II by a ubiquitin-like conjugation system. Once conjugated, LC3-II inserts into the growing autophagosome and serves as a marker of autophagosomes. LE, late endosome; Ly, lysosome; MVBs, multivesicular bodies; E1, E2, and E3 ligases for the ubiquitin-like conjugation systems; ROS, reactive oxygen species.
Autophagy is also an important component and regulator of the host response against viral infections. As a branch of the immune system, autophagy has both surveillance and effector functions that are important for the detection and clearance of viral pathogens [4], [5]. Autophagy can deliver viral components to endosomal compartments to stimulate innate immune signaling and to provide processed antigens for MHC presentation. Furthermore, the intrinsic capacity of the autophagosome to capture and degrade intracellular pathogens (xenophagy) adds to the antiviral capacity of the autophagosome. Consequently, viruses have developed a variety of ways to suppress and subvert the autophagy machinery for their own benefit. While the role of autophagy in immunity is relatively well understood [6], there is less consensus about how viruses co-opt autophagy for their benefit. Recent research has displayed several proviral subversions of autophagy, although the mechanisms are often unclear. In this review, we will highlight recent work on the antiviral role of autophagy, and how viruses overcome or subvert autophagy.
2. Autophagy
Autophagy is a highly conserved process that relies on the same core machinery from yeast to mammals. Although initially observed in mammalian cells, genetic screens in yeast provided a powerful tool to identify many autophagy-related genes (ATGs), over 30 to date [7]. These ATGs play fundamental roles in the biogenesis of the autophagosome, from its nucleation to its maturation (Fig. 1A). In its classical and most conserved form, autophagy is a response to nutrient starvation, and as such is controlled principally by key regulators and sensors of cellular energy and metabolism [8]. However, much research has shown that autophagy can be induced by signs of cellular damage, such as reactive oxygen species (ROS), ceramides, mitochondrial depolarization, or ER stress, as well as signs of infection, including the activation of innate immune sensors by pathogen-associated molecular patterns (PAMPs) and cytokine production [8].
The various stimuli that activate autophagy largely (but not exclusively) converge upon the master regulator mTOR (Fig. 1B) [8]. mTOR is a serine/threonine kinase that forms two separate complexes (mTORC1 and mTORC2) and acts as a global regulator of cellular metabolism. In mammals, mTORC1 negatively regulates autophagy under normal conditions through the binding, phosphorylation and consequent inactivation of the pro-autophagy ULK1/2 kinase complex [9]. Under nutrient deprivation, mTORC1 dissociates from the ULK1/2 complex, leading to its activation. Although many of its direct substrates remain unknown, ULK1/2 kinase activity is necessary for the subsequent recruitment of autophagy-related genes to the site of phagophore nucleation [9].
The Vps34-Beclin-1 complex is a central regulator of autophagosome biogenesis (Fig. 1B) [10], [11]. Vps34 is a class III phosphatidylinositol-3-kinase (PI3K) that generates phosphatidylinostiol-3-phosphate (PI3P), which recruits many ATGs and autophagy regulators to the phagophore. The activity of Vps34 is regulated by its interaction with Beclin-1, normally sequestered away from Vps34 by Bcl-2 (B-cell lymphoma 2), and a variety of Beclin-1 binding partners [10]. The exchange of Beclin-1 binding partners during autophagy has been shown to facilitate the nucleation and expansion of the autophagosome [12], [13], [14]. Furthermore, Vps34-Beclin-1 binding proteins also regulate the fusion of the autophagosome with endocytic compartments and the lysosome [15], [16], [17].
LC3, the mammalian ortholog of Atg8, is a common marker of the autophagosome, and has significant roles in the expansion and closure of the autophagosome, as well as the selective recruitment of cargo to the autophagosomal membrane [18], [19], [20], [21]. LC3 exists most prominently in two forms, LC3-I and LC3-II, the latter of which is conjugated to phosphatidylethanolamine (PE) in one of the two ubiquitin-like conjugation systems essential for autophagy (Fig. 1C) (the other being Atg5-12, reviewed in [22]). LC3 is cleaved at its C-terminus by Atg4, and then conjugated to phosphatidylethanolamine (PE) by Atg7 (E1), Atg3 (E2), and the Atg5-12-16 complex (E3) to generate LC3-II [19]. LC3-II associates exclusively with the autophagosome, defining it as a classical marker of autophagosome formation.
In addition to the non-specific, bulk degradation of cytosol, autophagosomes can be targeted to specific cargo in a process termed “selective autophagy” [18], [23]. While selective and bulk autophagy both depend on the same core machinery to nucleate, elongate, and mature the autophagosome; selective autophagy uses special adaptors to facilitate the uptake of marked cargo (Fig. 2 A). Several selective autophagies have been demonstrated for various organelles, intracellular pathogens, and protein aggregates [18]. In general, the targeting of cargo for selective autophagy depends on the decoration of the target using various autophagy adaptors (p62, NBR1, NDP52, ALFY, Nix etc.) that bind LC3 and the cargo [18], [24]. For several adaptors, such as p62, NBR1, and NDP52, recognition of the cargo is facilitated by the interaction between ubiquitin-binding domains on these adaptors and the specific ubiquitylation of the cargo (Fig. 2A) [25], [26], [27], [28]. However, ubiquitin-independent roles have been suggested for the recognition of some targets by their adaptors [29]. Recognition leads to the specific entrapment of these molecules or organelles in autophagosomes, and the cargo is then delivered to lysosomes for degradation by the canonical pathway.
Fig. 2.
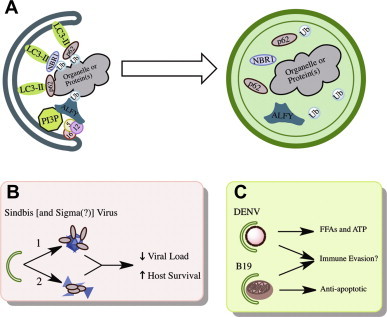
Selective autophagy. A. Autophagosomes can be directed to engulf specific cargo through the action of adaptor molecules that bind to the cargo, often by a post-translational modification such as ubiquitination or acetylation, and LC3. This allows for the culling of organelles, aggregated proteins, and invasive pathogens. B. Autophagy genes and known adaptors are important for the restriction of Sindbis and sigma virus in vivo. C. Dengue and human parvovirus B19 induce selective autophagies (of lipid droplets and mitochondria, respectively) that facilitate viral replication through the remodeling of host lipid metabolism (DENV) and the suppression of apoptosis (B19).
3. Mechanisms of viral induction of autophagy
The large amount of stimuli that can signal downstream to autophagy induction affords the cell the ability to respond to a variety of stimuli in a similar fashion. This also means that viruses can trip many switches that lead to induction of autophagy. Several studies have suggested a variety of events in the viral lifecycle that can trigger downstream autophagy. Broadly these involve signaling downstream of their receptor interactions [30], [31], [32], cellular stress [33], [34], [35], and the triggering of immune sensors (Fig. 3 ).
Fig. 3.
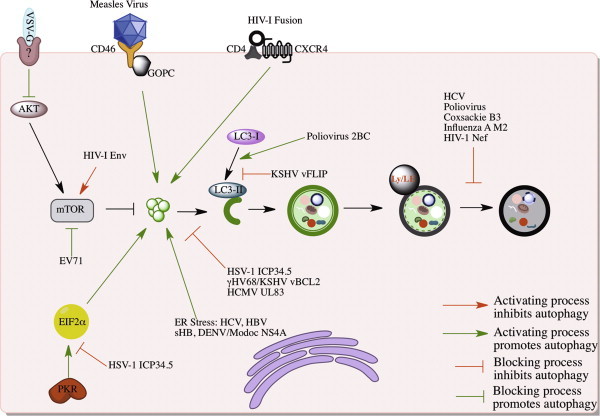
Viral modulation of autophagy. The activation (green lines) and inhibition (red lines) of autophagy can be achieved by the modulation of mTOR signaling, receptor interactions, various stresses and sensors. Alternatively, several viruses have been shown to block various stages of autophagosomal maturation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.1. Virus–receptor interactions
In the case of some viral infections, the initial virus-cell engagement stimulates signaling that induces autophagy. Studies with VSV in Drosophila showed that binding of VSV to the cell is sufficient to induce autophagy [30]. Infection with UV-inactivated VSV or VSV-G virus-like particles induced Atg8-I to Atg8-II conversion and formed GFP-Atg8 puncta in infected flies. VSV infection, or incubation with the virus-like particles, led to the down regulation of mTOR activity by inactivation of Akt signaling [30]. The specific signaling events leading to Akt inactivation remain to be determined. However, the authors suggest that either a Drosophila TLR or engagement of a viral receptor may be the trigger [30].
The cell surface receptor CD46 binds several viral pathogens, including measles virus, human herpesvirus 6, and adenovirus types B and D, in addition to some bacterial pathogens. At least in the case of measles virus infection, CD46 engagement induces autophagy [31]. This requires the cellular CD46-interacting protein, GOPC. Viral binding of CD46 leads to the association of GOPC with the Vps34-Beclin-1 complex and subsequent activation of autophagy [31]. It remains to be tested whether other CD46-interacting viruses stimulate autophagy by a similar mechanism.
HIV-1 engagement of CD4+ T-cells can also induce autophagy via an Env-CXCR4 interaction [32]. Interestingly, this induction is independent of CXCR4 signaling [36]. Rather, autophagy induction requires the C-terminal domain of the fusogenic gp41 subunit of Env, suggesting that the formation and insertion of the six-helix bundle into the cellular membrane is an important trigger [36]. Of note, HIV-1 Env with CXCR4 tropism does not induce autophagy in macrophages, which can also express CXCR4, suggesting a cell type specificity to the triggering of autophagy by HIV-1 [37].
3.2. ER stress
Many viral infections induce cellular stresses, such as ER stress and ROS production, that can also induce autophagy [8]. ER stress is perceived through the unfolded protein response (UPR), a convergence of several cellular pathways that respond to an accumulation of misfolded proteins inside the ER with the concerted goal of expanding the ER to handle the amount of protein inside as well as stopping further proteins from entering the ER [38]. Viral infections often overwhelm the ER with the amount of protein that is being translated and needs to be properly folded. Furthermore, the folding of highly glycosylated (e.g., viral glycoproteins) or hydrophobic proteins places even greater demand on the folding machinery of the ER [38]. Thus, the sheer volume of proteins moving through the ER during viral infections leads to an overload of the ER and subsequent activation of the UPR [39].
The UPR has been suggested to be the autophagy induction signal for several viral pathogens (Fig. 3) [33], [34], [35], [40], [41], [42]. HCV induction of ER stress and the UPR has been suggested to lead to induction of autophagy [33]. Silencing of any of the three UPR signaling molecules decreased LC3-II conversion and HCV replication. However, it has not been shown that HCV-induced ER stress is directly activating autophagy, as opposed to an indirect requirement [33]. Furthermore, transfection of dengue and Modoc virus NS4A, as well as HBV sHB, has been shown to induce autophagy via the UPR. The NS4A protein of Modoc, dengue, and many other flaviviruses, deforms the ER, and while this physical stress on the ER may be sufficient for autophagy induction, it is not clear that this may be the sole (or primary) inducer of autophagy in the context of infection [34]. For instance, in the case of HBV infection, HBV X protein expression was reported to sensitize cells to starvation induced autophagy by up regulating Beclin-1 transcript levels [43]. Subsequent work further reaffirmed a role for HBV-X in autophagy induction, however, the proposed mechanism did not implicate Beclin-1 transcript levels, but rather the interaction with Vps34 and increased PI3K activity [42]. Indeed, neither study has been able to replicate the induction mechanism of previous labs, possibly owing to differences in viral strains and cell types.
4. Antiviral autophagy
Autophagy is an integral part of the immune system that functions both in the sensing of viral infection and as an antiviral effector mechanism (Fig. 4 ) [44]. As a process that converts cytosol into lumen and then fuses with endocytic compartments, the autophagosome has the unique ability to deliver cytosolic pathogen-associated molecular patterns (PAMPs) into the proximity of endosomal pattern recognition receptors (PRRs) and MHC loading compartments (Fig. 4A,C). Furthermore, the autophagosome can directly entrap and degrade virions and virion components (Fig. 4B) (xenophagy). These aspects make autophagy well suited for its role in mediating the antiviral immune response.
Fig. 4.

Antiviral activities of autophagy. Autophagy can restrict viral replication and spread through the direct capture of virions, viral nucleic acid, or viral proteins, and the subsequent delivery of these to different compartments. A) Autophagy delivers viral nucleic acid to endosomal TLRs to stimulate innate immune sensors. In turn, innate immune sensors can also stimulate autophagy. B) Fusion with the lysosome can directly degrade virion components. C) The autophagosome can deliver virions viral antigens to MHC-I and -II complexes to stimulate antigen presentation.
4.1. Autophagy and antiviral PRRs
Detection of viral infection depends on many cytosolic and endosomal immune sensors, which generally recognize varying aspects of viral nucleic acid [45]. Toll-like receptors (TLRs) 3, 7/8, and 9, which recognize dsRNA, ssRNA, CpG DNA, respectively, are located in endosomes while other immune sensors, including DNA sensors, RIG-I like RNA helicase receptors (RLRs), PKR, and NOD-like receptors, are located primarily in the cytosol [45]. Since many RNA viruses deliver their nucleic acid to the cytosol and/or replicate in the cytosol it is apparent how the cytosolic PRRs are activated by these infections. In the case of the endosomal TLRs, it is less obvious, since the nucleic acid is protected from the endosomal environment by its capsid during endocytosis, or alternatively, entry does not occur through an endosomal compartment for viruses that fuse at the plasma membrane. Nevertheless, TLRs are important in mediating the immune response against a number of viral infections, frequently by unclear mechanisms.
Although it is likely that TLRs may encounter nucleic acid via scavenging remnants of infected cells or by a subset of capsids destabilizing in endosomes, it is also clear that mechanisms exist to deliver cytosolic PAMPs to the endosome [46], [47], [48]. For several viruses, autophagy delivers viral PAMPs to endosomal TLRs for their activation (Fig. 4A). In VSV infection, the production of antiviral IFN-α by plasmacytoid dendritic cells (pDCs) was shown to be dependent upon the autophagic delivery of replication intermediates to TLR7 [46]. By using TLR7−/− mouse pDCs as well as UV-inactivated VSV, it was shown that IFN-α production depended exclusively on active viral replication and recognition of viral intermediates by TLR7. Treatment of infected pDCs with 3-methyladenine (3-MA) or Wortmannin to block Vps34 activity attenuated IFN-α production in response to infection, suggesting that autophagy was important for delivery of VSV intermediates to TLR7 endosomes [46]. Atg5−/− mice showed a similar phenotype, and also carried a larger viral load systemically, suggesting that autophagy is important for the regulation of VSV replication throughout the organism.
HIV-1 appears to have evolved mechanisms to block the autophagic delivery of PAMPS to the endosome [48]. HIV-1 blocks autophagy in dendritic cells in an Env-dependent manner, and this inhibition is important for evading the fusion of autophagosomes with endocytic compartments enriched with immune molecules to form “immunoamphisomes” [48]. Infection with an Env-mutant that could not block autophagy increased innate immune signaling and the release of antiviral cytokines [48]. Furthermore, exposure of DCs to HIV-1 reduced the activation of TLRs 4 and 8 upon stimulation with their cognate ligands to a similar extent in DCs treated with irrelevant, ATG5, or LC3 siRNAs suggesting an important role for autophagy in mediating the TLR response [48].
The importance of autophagy in delivering viral PAMPs to endosomal PRRs is preserved in non-immune cells as well, as evidenced by the autophagy-dependent activation of TLR3 in kidney fibroblasts infected with Coxsackie Virus B3 (CVB3) [47]. The authors demonstrated that TLR3+ vesicles regularly colocalized with LC3 puncta, and that poly(I:C) and CVB3-dependent activation of TLR3 was sensitive to inhibitors of autophagy induction, autophagosome acidification, and lysosomal proteases. Moreover, in the absence of virus or poly(I:C), TLR3 could still be found colocalized with LC3 puncta [47]. This would suggest that under basal conditions, the autophagosome may fuse with TLR3+ endosomal compartments allowing these amphisomes to serves as platforms for sensing the presence of viral PAMPs and then subsequently trigger their downstream signaling.
In addition to delivering viral PAMPs to PRRs, autophagy is also induced following the activation of innate immune sensors, including PKR and several TLRs [49], [50], [51], [52]. The stimulation of TLRs 3, 4, and 7 with various purified PAMPs can induce autophagy in a MyD88 and/or TRIF-dependent manner [49], [51]. Both MyD88 and TRIF, the adaptors through which TLRs mediate their signaling, directly interact with Beclin-1 upon TLR activation, and this interaction is required for the induction of autophagy. The interaction of Beclin-1 with MyD88/TRIF displaces Bcl-2 from Beclin-1, resulting in formation of active Vps34-Beclin-1 complexes [51]. The requirement of TLR adaptors for viral induction of autophagy has not yet been reported.
PKR is both an innate immune sensor that binds dsRNA and an effector that phosphorylates eIF2α to inhibit cap-dependent translation. As such, many viruses have evolved mechanisms to antagonize PKR [53]. Herpes simplex virus I (HSV-I) ICP34.5 directs the cellular phosphatase PP1 to dephosphorylate eIF2α so that protein translation can proceed [54], [55], [56]. Infection with HSV-1 deleted in the genes encoding ICP34.5 (γ134.5) results in translational arrest and attenuated neurovirulence [57]. It also produces a PKR-dependent autophagy. This induction is dependent on translational arrest induced by PKR’s phosphorylation of eIF2α [50]. Specifically, expression of an eIF2α mutant that cannot be phosphorylated by PKR does not lead to the induction of autophagy after infection with ∆ICP34.5 HSV-1 mutant [50]. Although, the mechanistic link between eIF2α phosphorylation and autophagy induction is unclear, it is not limited to PKR. The phosphorylation of eIF2α by the ER stress sensor PERK is also important for ER stress induced autophagy [58].
The delivery of PAMPs to PRRs by autophagosomes and the subsequent induction of autophagy by PRRs upon their activation suggest a positive feedback loop. This may serve at least three purposes, all of which are consequent of autophagic engulfment of viral pathogens: i) to sequester virions and fuse with the early and late endosomal compartments, and thus continue to present viral PAMPs to endosomal PRRs; ii) to deliver viral proteins to be processed for presentation on MHC-I and -II molecules to stimulate the adaptive immune response; or iii) to degrade the virion in a cell autonomous manner by fusing with the lysosome (xenophagy). Furthermore, the induction of autophagy downstream of cytosolic immune sensor activation can serve to further facilitate the delivery and degradation of virions and their PAMPs by autophagy.
4.2. Autophagy processes and delivers viral antigens for MHC presentation
Autophagy is also important for the adaptive immune response via the processing and delivery of antigens for presentation on MHC-I and MHC-II molecules. Canonically, MHC-I molecules present endogenous antigens that are degraded in the cytosol by the immuno-proteasome, and then translocated into the ER where these peptides are loaded onto MHC-I molecules and transported to the cell surface [59]. The MHC-I molecules can then stimulate a CD8+ T-cell response. In contrast, MHC-II molecules present peptides derived from exogenous antigens that are processed by lysosomal hydrolases in vacuolar compartments and present them on the surface to stimulate a CD4+ T-Cell response [59]. However, it had been noted that luminal antigens can be presented on MHC-I molecules and stimulate a CD8+ T-Cell response in a process known as “cross-presentation.” Similarly, endogenous antigens are known to be presented on MHC-II molecules [59].
The presentation of viral antigens on MHC-I and MHC-II molecules is influenced by autophagy (Fig. 4C) [60], [61], [62], [63], [64], [65]. MHC-I presentation of HSV-1 antigens on macrophages proceeds in a biphasic pattern. At early time points of infection, the autophagosomal machinery was dispensable for processing of MHC-I peptides [63]. However, later in infection autophagy was necessary for optimal processing and presentation of HSV-1 antigens on MHC-I molecules. This effect was heightened following infection with a γ134.5 mutant HSV-1 unable to inhibit autophagy. Interestingly, in wild type HSV-1 infection, atypical autophagosomes were described wherein four layered membrane structures that derived from the nuclear envelope accumulated [63]. The nature of these structures is unclear, but they enclose HSV-1 virions, degrade and deliver virion antigens for MHC-I loading, and are dependent upon autophagy induction. It remains unclear how degradation by the autophagosome (or any vacuolar compartment) leads to presentation of antigens on the ER-resident MHC-I molecule [63], [66].
The processing of endogenous antigens and their presentation on MHC-II depends on the delivery of cytosolic antigens to the endosome. The EBNA1 protein of Epstein Barr virus is delivered to MHC-II compartments via autophagic uptake and subsequent fusion of the autophagosome with the MHC-II vacuolar compartment [62], [64]. Blocking autophagosomal acidification with chloroquine produced increases in EBNA1 containing autophagosomes, and blocking autophagy by depletion of Atg12 or treatment with 3-MA demonstrated a severe decrease in the ability of these cells to stimulate MHC-II-dependent activation of T-cells [62]. In vivo studies demonstrated the requirement of Atg5 for proper MHC-II antigen presentation of extracellular microbial antigens. DCs from Atg5−/− mice, unlike their wild type counterparts, were unable to prime a protective antiviral response in Th1 immune cells [65]. Furthermore, basal autophagy is important for the continuous delivery of antigen to MHC-II molecules, suggesting a major role for autophagy in the presentation of MHC-II antigens in vivo during infection [60]. Interestingly, fusing the matrix protein of influenza to LC3 was shown to enhance the MHC-II activation of cytotoxic CD4+ T-cells suggesting this may be a potential vaccine strategy [60].
In addition to the delivery of antigen to MHC loading compartments by the autophagosome, the importance of autophagy for MHC-II antigen presentation can also be linked to its ability to promote phagosome–lysosome fusion [60]. DCs from Atg5−/− mice contain fewer phagosomes that acquired lysosomal markers, suggesting an impairment of phagosome maturation. This led to a decreased ability for the phagosome to process antigens for presentation on MHC-II molecules and then activate CD4+ T-cells [60]. Thus in some instances, non-canonical roles of the autophagy machinery are essential for optimal antiviral immune function.
4.3. Xenophagy
In addition to the ability to deliver viral antigens into the arms of the innate and adaptive immune responses, studies have suggested autophagy can directly restrict viral infection in a wide variety of multicellular eukaryotes (Fig. 4B). This was first demonstrated by the ectopic expression of Beclin-1 in murine neurons, which produced a decrease in Sinbdis virus titer in vivo [67]. This was associated with limited pathology including decreased encephalomyelitis and increased survival of infected mice. Xenophagy was also shown to restrict infections of HSV-1 viruses deleted for γ134.5, and to limit the associated neuronal pathology. Later work showed that the restriction of Sinbdis virus was due to xenophagic degradation of Sinbdis virus capsid [68].
Xenophagic degradation of Sinbdis virus requires the targeting of the viral capsid for degradation by the selective autophagy adaptor p62 (Fig. 2B) [68]. The signal for recognition is unclear, as it does not involve binding to an ubiquitinated capsid. In infected neurons of Atg5 deficient or WT mice, no accumulation of ubiquitin was observed in the cells or on the capsid [68]. However, p62 contains several protein–protein interaction domains that may otherwise facilitate ubiquitin-independent interaction [69], [70]. Whether other protein–protein interaction domains within p62 are necessary for the targeting of Sindbis virus capsid remains to be tested. Interestingly, the Drosophila homolog of p62, Ref(2)P, is important for the restriction of sigma virus, and genetic analyses have mapped the relevant domain of Ref(2)P to outside of its ubiquitin-binding domain [71], [72], [73], [74]. While currently unclear, it will be interesting to determine whether the Ref(2)P restriction of sigma virus is autophagy-dependent and the antiviral role of autophagy adaptors is evolutionarily conserved (Fig. 2B). Future work in these systems should provide insight into additional ways that autophagy adaptors recognize cargo.
In organisms that lack an adaptive immune system, autophagy is also an essential part of the antiviral response. During tobacco mosaic virus (TMV) infection, autophagy plays a role in limiting viral dissemination throughout the plant and restricting the plant’s hypersensitive response to infected cells [75]. Deletion of various autophagy genes led to the increased spread of programmed cell death outside the infected cells, and greater pathology to the plant [75]. In addition to the Ref(2)P restriction of sigma virus, autophagy is essential for Drosophila defense against VSV [30]. Deletion of any of several Drosophila ATGs renders Drosophila exquisitely sensitive to VSV infection and increases the lethality of infection [30]. Thus, while autophagy may have evolved additional functions in safeguarding the host from infection, the importance of virion degradation for cellular defense is highly conserved throughout eukaryotes.
5. Viral impairment of autophagy
The significance of autophagy to antiviral defense can also be appreciated by the number of strategies that viruses have evolved to impair autophagy (Fig. 3). Several different viruses antagonize Beclin-1, and this antagonism can result in distinct outcomes depending on the virus. The herpesviruses Kaposi’s Sarcoma herpesvirus (KSHV), gammaherpesvirus 68 (gHV68), and HSV-1, encode viral proteins that competitively bind Beclin-1, inhibit Beclin-1-Vps34 interaction, and block autophagy initiation (Fig. 3) [68], [76], [77]. HIV-1 Nef and Influenza M2, on the other hand, block autophagosome maturation in a manner dependent upon their association with Beclin-1 (Fig. 3). For Nef and M2, it is unclear whether the interaction with Beclin-1 is direct or mediated through interaction with a larger complex [78], [79]. Since autophagosomal initiation and maturation are regulated by changes in components of the Beclin-1/Vps34 complex, it will be interesting to understand the molecular mechanism of how Nef and M2 selectively block autophagosome maturation.
In addition to targeting Beclin-1, viruses enumerate many other mechanisms to block autophagy at a variety of steps (Fig. 3). Upon entry into CD4+ T-cells, HIV-1 Env activates mTOR signaling, shutting down the autophagic response that is initiated upon fusion of HIV-1 into CD4+ T-cells [48]. KHSV also encodes a homolog of the FLICE-like inhibitor protein (v-FLIP) that blocks the interaction of LC3 and Atg3, inhibiting the lipidation of LC3 and subsequent phagophore expansion [80]. Homologous v-FLIPs were found in herpesvirus saimiri and molluscum contagiosum virus that also were capable of blocking autophagy (Fig. 3). In many more viruses, however, the mechanism of blocking autophagy is relatively unclear. For example, HCMV UL83 is capable of blocking mTOR-dependent and mTOR-independent autophagy stimuli through an unknown mechanism [81]. Additionally, both hepatitis B and C viruses have been reported to induce autophagy but then somehow inhibit autophagosome acidification [33], [35], [42]. In the case of SIV-1 infection of glial cells, it has been shown that supernatants from these infected cells are capable of blocking autophagy in surrounding neurons [82]. Such a paracrine suppression of autophagy may have roles in neuronal killing and the associated dementia during SIV-1 and HIV-1 infections [83]. Undoubtedly, as we learn more about the pathways involved in autophagy, we will also find viruses that antagonize these new players.
6. Proviral autophagy
Although autophagy plays a prominent antiviral role in many infections, a number of viruses have evolved ways to manipulate autophagy for their benefit (Fig. 5 ). In these viral infections, experimental inhibition of autophagy decreases infectious virus production, although in many cases, the proviral mechanisms are unclear. Frequently, these viruses induce an incomplete autophagy wherein autophagosomes are formed, but do not mature to fuse with the lysosome. Seminal work in poliovirus and other positive-stranded RNA viruses suggested a role for autophagosomes in replication complex formation. More recently, evidence from various viruses shows that autophagy can play an important role in nearly all steps of the viral lifecycle. Broadly, proviral autophagy can be broken down into two large categories: direct and indirect. A direct proviral role for autophagy suggests that the autophagy machinery physically interacts with a viral component to the benefit of the virus. Alternatively, some viruses can induce an autophagy program that indirectly supports infection by modifying cellular physiology.
Fig. 5.
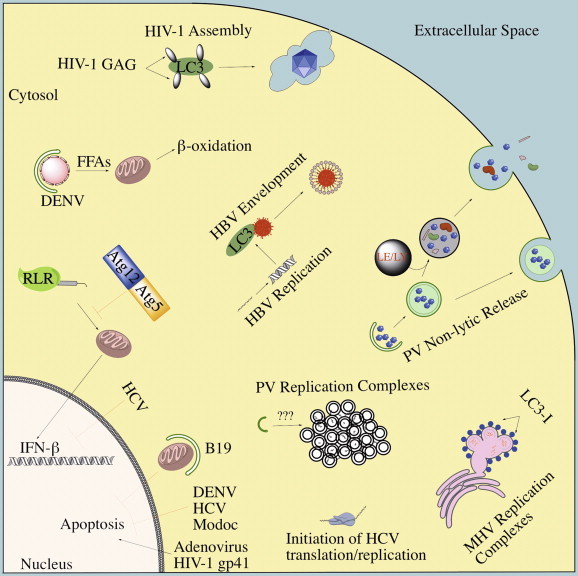
Proviral roles for autophagy. The diverse proviral roles that have been proposed for distinct viruses are shown and discussed in the text.
6.1. Direct roles of proviral autophagy
6.1.1. Genome replication
In large part, the direct roles for proviral autophagy have centered on the role of autophagosomes in generating the membranes that positive-stranded RNA viruses use as platforms for replication. These are thought to: i) protect their replication intermediates from immune detection; ii) provide a scaffold for viral replication machinery; and iii) concentrate nucleotides and other essential factors for genome replication [84]. Thus, RNA viruses have evolved diverse mechanisms to remodel host cellular membranes to form their replication complexes. The morphology of these replication complexes is distinct between viruses, and in the majority of cases the membrane source remains unclear [84]. For several viruses, the autophagy machinery has been suggested to play a role in the formation of these membrane structures.
Original studies in poliovirus suggested the role for autophagy in replicative DMV formation based off several distinct qualities of these structures. Under thin section EM analysis, these structures had the characteristic DMV morphology associated with autophagosomes, and autophagosome-like structures could be seen forming in several images [85]. Additionally, these structures – which are positive for the 2BC protein, among others – did not co-fractionate with the markers of other known organelles, suggesting that it was possibly a distinct structure from other organelles [85], [86]. The expression of poliovirus 2BC and 3A, which are sufficient to generate membrane structures like those during poliovirus replication, are also sufficient for the lipidation of LC3 as well as formation of autophagy-like DMVs [85], [87]. The accumulation of DMVs in other picornaviruses, such as CVB3, foot-and-mouth disease virus (FMDV), and enterovirus 71 (EV71) have a similar morphology and are positive for LC3 as well as viral components in vivo and in vitro [88], [89], [90]. These findings have lead to the suggestion that autophagosomes may serve as a conserved replication platform for picornaviruses.
However, recent work has demonstrated that autophagy is not a universal enhancer or byproduct of picornavirus replication [91], [92], [93]. In the human rhinovirus family (HRV), it has been shown that HRV-14 induces autophagy during infection, while HRV-1a does not, and its replication is not affected by autophagy modulators [91], [94]. Conflicting reports exist regarding whether HRV-2 induces autophagy and is sensitive to autophagy modulation [91], [92]. These inconsistencies may reflect differences in drug and virus concentrations used as well as the assays to measure autophagy induction.
The membrane composition of picornavirus replication complexes is heterogeneous and numerous other mechanisms have been proposed to influence their formation and composition. The contribution of COP-II coated vesicles, GBF-1, ARF1, BIG-1/2, and PI4KB to the formation and ability of these membranes to support viral replication suggests an intimate involvement of the secretory compartment in the formation and utility of these structures [94], [95], [96], [97], [98]. Furthermore, poliovirus, CVB3, FMDV, and EV71 can all replicate in autophagy-deficient cells, although for several of them there is a reduced production of infectious virus [88], [89], [93], [99]. Thus, autophagosomes are most likely not essential for picornavirus replication complex formation, although autophagy proteins appear to frequently contribute to the heterogeneity of the structures.
In the case of coronaviruses, it was initially proposed that autophagosomes were the sites of replication [100], however subsequent studies argued otherwise [101], [102], [103]. Initial studies showed that the viral replicase genes colocalized with LC3 and inhibition of autophagy via deletion of Atg5 or 3-MA treatment inhibited viral replication [100]. However, subsequent studies indicated that these requirements may be cell type specific [101]. Furthermore, cryo-EM tomography reconstructions revealed that the DMVs of the coronavirus replication complex were actually a convoluted network of contiguous ER invaginations [103]. Thus, the membrane structures were not distinct autophagosomes. However, the co-localization of LC3 with viral replicase machinery was still observed [102]. It was subsequently shown that LC3-I, but not LC3-II, colocalized with the replication machinery as well as the replication membranes, suggesting a role for the cytosolic form of LC3 and not the autophagosome associated LC3-II [102]. These structures were also positive for markers of the ER-associated degradation (ERAD) response, and remained tethered to the ER. Thus, coronavirus uses components of the autophagy pathway for its replication, but not autophagosomes.
For rotavirus infections, it was observed that LC3 localizes with the viral protein NSP4, a marker of rotavirus replication factories (“viroplasms”), and that these LC3-NSP4 structures do not colocalize with LAMP1 [104]. However, the requirement of autophagy for rotavirus replication remains to be tested [104].
Recent work in HCV has also suggested a role for autophagy in initiating viral replication [105], among other potential functions. Inhibition of autophagy by knockdown of Atg5, Atg7, and Beclin-1, and pharmacological agents blocked HCV replication. Subsequent experiments demonstrated that the autophagy genes were essential for the initiation of viral replication, but not its maintenance [105]. Whether autophagy is needed initially for bending ER membranes and providing a platform for incoming viral translation and replication, or some other role is necessary is yet to be determined. The HCV polymerase has been shown to interact with Atg5 at early points during infection, although the functional outcome of this interaction is ambiguous [106].
6.1.2. Virion assembly and egress
The late stages of the viral lifecycle can be broken into assembly of the viral particle, maturation of the capsid/envelope proteins, envelopment (if applicable) and exit (egress) from the cell. In nearly each of these late stages, the autophagy machinery has been shown to be important for distinct viruses [35], [78], [93], [99].
The proper maturation and envelopment of HIV and HBV, respectively, are linked to physical interactions between LC3 and their glycoproteins [35], [78]. In HIV-1 infection of macrophages, Gag was shown to co-precipitate and co-fractionate with LC3-II, and this association facilitated the processing of the Gag p24 subunit. Interestingly, confocal and EM studies of gag p17 (a marker of HIV-1 virions) and LC3 showed that the virions and LC3 colocalized in membranous compartments that were juxtaposed to the plasma membrane [78]. Furthermore, these membranes were suggested to be of plasma membrane origin, as clathrin-coated pits were seen forming from them. The authors note these structures are reminiscent of the proposed HIV-1 assembly and budding in macrophages [78]. It has been suggested that the assembly site of HIV-1 is distinct from the endocytic pathway, contiguous with the plasma membrane, and dependent upon the ESCRT machinery [107], [108]. It is possible that HIV-1 uses these autophagosomal structures to enhance the delivery of its virion components to the site of assembly. The ability of Nef to block autophagosome acidification is also essential for release of mature virions [78]. However, it is not clear how acidification affects the Gag-LC3 interaction and how this impacts the immune responses of macrophages to HIV-1.
For HBV, autophagy has been implicated in both replication and viral envelopment [35], [42]. While the extent of viral replication inhibition differed between the two studies, both showed that viral RNA is efficiently packaged into the nucleocapsid in the absence of autophagy. However, Li et al. also demonstrated that the efficient envelopment of HBV was associated with autophagy induction [35]. By immunoprecipitating the viral capsid (core) or major envelope protein (sHB) and quantifying the associated viral RNA, the authors showed that significantly less viral nucleic acid is associated with both intracellular and extracellular enveloped particles in autophagy-deficient cells [35]. Furthermore, sHB was shown to immunoprecipitate and co-localize with LC3-I and -II during HBV infection or ectopic expression of sHB, suggesting that this interaction may be important for the acquisition of viral envelope. Currently, the HBV envelope is thought to be acquired post-ER, but pre-Golgi, however the source of this membrane is unknown [109]. The autophagosome may be a potential membrane source or an intermediate compartment HBV uses to traffic through en route to envelopment. Autophagosomes induced by HBV do not mature, as evidenced by a lack of p62 turnover and LAMP co-localization [35], [42]. Whether this occurs through a similar mechanism to HIV-1 is unclear, however for both viruses the lack of acidification could be a result of redirecting autophagosomes along an alternative pathway.
The autophagosome has also been suggested to play a role in the non-lytic release of the non-enveloped picornaviruses [89], [93], [99]. The inhibition of autophagy via siRNA or drugs leads to a general decrease in infectious virus production, with a more pronounced effect on extracellular infectious virus. Furthermore, electron micrographs captured GFP-LC3+ or VP1+ vesicles budding from and fusing with the plasma membrane to release virions [93]. These images are reminiscent of the first study to suggest a non-lytic release of poliovirus from infected cells [110]. Further evidence from poliovirus suggested that disrupting the microtubule network, either via nocodazole or using a poliovirus mutated in the 3A protein (3A-2), lead to the increased mobility of PV-induced GFP-LC3 puncta and a coincident increase in extracellular infectious virions without increased cell lysis [99]. While it is currently unclear how virions may be escaping via this non-lytic release, several studies suggest a role for autophagy in the secretion of various substrates [111], [112], [113], [114].
An alternative hypothesis is that the release of picornavirus components from infected cells may be from fully acidified autolysosomes and reflect a dumping of degraded contents into the extracellular milieu. It is known that this is a way of stimulating resident phagocytic cells and alerting the immune system [115]. In support of this, Taylor et al, show that by the time these PV-carrying autophagosomes are being released from the cell they are Lamp2-positive, suggesting that they have fused with lysosomes [99]. However, whether these have acidified is unclear.Whether poliovirus is hijacking autophagosomes for efficient non-lytic spread, or the cell is packaging poliovirus into autolysosomes and dumping the contents into the extracellular space to alarm the immune system (or a combination of the two) requires further work.
6.2. Indirect roles of autophagy
Although most studies of proviral autophagy have focused on direct roles in the viral lifecycle, examples of indirect roles for autophagy in viral infection are emerging. In particular, the induction of a selective autophagy, which can modulate specific aspects of cellular metabolism, cell fate, or immune signaling, is an attractive mechanism to remodel the cell into an optimal environment for viral replication (Fig. 2C). Furthermore, autophagy may be linked with an inhibition of apoptosis such that cells survive longer, thus resulting in a longer window for virus production.
Autophagy can promote prolonged cell survival both by maintaining a metabolic homeostasis and through its regulatory links to the programmed cell death pathways. In the case of human parvovirus B19 infection, the inhibition of autophagy with 3-MA or siRNAs targeting autophagy gene expression lead to the accelerated death of B19 infected cells [116]. While the effect of autophagy inhibition on viral replication and titer was not explored, apoptosis is a well-known cellular defense response that can restrict viral replication and spread within a host. The mechanistic details remain unclear, however, EM micrographs suggest that B19 infection may induce a selective autophagy that degrades mitochondria (Figs. 2C and 5) [116]. The elimination of mitochondria may provide short-term cell survival and immune evasion, since many pro-apoptotic and immune signals are transmitted through the mitochondria. Although not linked to a potential selective autophagy, infection of immortalized human hepatocytes with HCV suggest that autophagy has anti-apoptotic properties in these cells which may be important for establishing chronic infection in vivo [117].
Robust dengue virus (DENV) replication requires autophagy, and this was initially proposed to play a direct role in viral replication complex formation [118], [119], [120]. However, similarly to coronaviruses, EM tomography studies found that DENV replication complexes are contiguous with the ER and not distinct autophagosomes [121]. This suggested that autophagy might play an indirect role in viral replication. Recent work in many viral systems has defined the importance of cellular metabolism for viral infection [122], [123], [124], [125], [126]. Because viral replication is an energy intensive process, viruses elaborate a variety of mechanisms to stimulate metabolism.
DENV remodels cellular lipid metabolism by inducing autophagy (Figs. 2C and 5) [127]. DENV-induced autophagy specifically targets lipid droplets for depletion in hepatocytes by a selective autophagy termed lipophagy. Autophagosomes deplete lipid droplet triacylglycerides, which are processed by lipases and released as free fatty acids. These are transported to the mitochondria for β-oxidation and ATP generation [127]. Supplementing infected cells with exogenous free fatty acids completely restored the replication defect associated with autophagy inhibition to wild-type levels. Thus, despite the many roles for autophagy in cellular homeostasis, the stimulation of lipid metabolism is the critical function for DENV replication [127].
Future studies into the mechanism by which DENV induces lipophagy will be important in understanding how viruses can trigger a selective autophagy to remodel the host cell. Additionally, as with B19, this could also be a deft immune evasion strategy by DENV. Redirecting autophagosomes to lipid droplets would gain the virus necessary substrates while avoiding autophagy-mediated immune detection and clearance of virions. Recent work has also suggested that DENV NS4A can induce autophagy that is cytoprotective, but it is unclear whether this NS4A-induced autophagy can recapitulate the other aspects of DENV mediated autophagy [34]. As the molecular mechanisms of DENV-induced lipophagy are unraveled, it will be interesting to test the hypothesis that directing autophagosomes to lipid droplets is an additional strategy to evade autophagy-mediated immune surveillance and activation [128].
Although autophagy is associated with stimulated endosomal TLR signaling, it has been noted that autophagy can down regulate antiviral immune signaling by the cytosolic RLRs [129], [130]. Studies in ATG5KO MEFs suggested a role for the ATG5-12 complex in the regulation of RLR signaling by competitively binding the CARD (caspase activation and recruitment domain) of RIG-I, MDA-5, and MAVS [130]. This binding prevents the interaction of the RLRs and MAVS, leading to blunted interferon-β production by either stimulated or infected cells [130]. While it is unclear whether this mechanism directly is being utilized by HCV, recent work has shown that knocking down autophagy, using siRNAs or pharmacological inhibitors, increases the level of interferon-β transcript produced by infected cells [40], [117]. Moreover transfecting HCV-infected cells with HCV and DENV PAMPs, known to stimulate RLR signaling, showed a decrease in interferon-β transcript as compared to uninfected controls in an autophagy-dependent manner [40]. Indeed, this did not appear to be a targeted role of autophagy during HCV infection, since the induction of autophagy via rapamycin or starvation led to a similar decrease in interferon-β transcript when cells were transfected with HCV and DENV PAMPs [40].
7. Conclusions
As our understanding of the cellular roles of autophagy expands, so does the number of ways that viruses interact with autophagy. The myriad antiviral roles of the autophagosome place a strong evolutionary pressure on viruses to develop ways to evade autophagy or subvert it for their own purposes. The autophagosome can degrade virions and shuttle those degraded components to endosomal PRRs and MHC-I and –II molecules. At the same time, viruses can directly exploit the autophagy machinery to potentially provide membranes for their replication or facilitate the maturation, envelopment, and egress of virions. Furthermore, the modulation of cell fate and metabolism, in addition to the negative regulation of RLR signaling, can enhance viral replication and immune evasion. The study of viral interactions with autophagy has been important in understanding how autophagy contributes to both innate and adaptive immunity, and can aid in the future dissection of the molecular pathways of general and selective autophagy.
Viral evasion or subversion of autophagy may also contribute to disease pathologies. For example, does the autophagy-dependent gp41-mediated killing of naïve CD4 T-cells contribute to the T lymphocyte depletion that leads to AIDS? In the case of HCV infection, an open question is whether the chronic impairment of autophagosome acidification seen in vitro is associated with some liver disorders, such as steatosis (an increase in lipid droplets in the liver). Since the lipid droplet is an important site of HCV assembly, one wonders whether HCV may block autophagosomal maturation to prevent the lipid droplet depletion observed in DENV infection. As a result, this could aid in the development of virally induced steatosis. Along similar lines, autophagy impairment may influence neurodegenerative pathologies of various viral pathogens known to both infect neurons and inhibit autophagy. Finally, it is unknown how the link between autophagy, cell survival, and programmed cell death impact different viral pathologies.
An emerging area is the role of selective autophagy in both anti- and pro-viral autophagy. It is clear that proteins of several viruses interact with LC3 either directly or through an autophagy adaptor, and for many other viruses, electron micrographs that show autophagosomes forming specifically around virions suggests that there is a mechanism by which these are being targeted. Furthermore, p62 and its Drosophila homolog, Ref(2)P, are important for the control of Sinbdis Virus and sigma virus infections, respectively, implicating a potential role for selective autophagy. Investigations into the recognition of virions by the autophagy machinery may open avenues to understanding novel mechanisms of targeting cargo for degradation. Additionally, the examples of DENV and human parvovirus B19 suggest that inducing a selective autophagy can benefit the pathogen by altering cell metabolism and survival, in addition to a possible immune evasion strategy. As we explore the roles of selective autophagy, it will be important to assess whether specific adaptors, in addition to p62, also recognize viruses and the associated mechanisms of their recognition.
Lastly, as researchers move past characterizing whether autophagy is important for a viral infection and what it may be doing, the careful characterization of how and what type of autophagy is induced will be important. As the tools to modulate and characterize virally induced autophagy expand, so should the rigor of these studies. Such mechanistic understanding of how autophagy is induced may help resolve controversies in fields such as HCV where numerous and sometimes conflicting proviral roles for autophagy have been reported. It is indeed likely that autophagy is performing a variety of functions, however, as the regulatory network of autophagy is unraveled it may be worth revisiting in a more fine tuned approach how disruption of particular signaling networks contribute to autophagosome formation and the many documented effects of autophagy. With the diverse physiological functions of autophagy and the growing appreciation of selective autophagies, it is likely that although the same core machinery may generate autophagosome, all autophagosomes are not created equally.
Several of the autophagy-dependent mechanisms used by these viruses divert the autophagosome from fusing with lysosomes and re-direct their trafficking. It is certainly possible, and probable, that viral proteins are playing an active role in this subversion of autophagy. However, they may also be enhancing natural cellular pathways by modulating existing components of the many protein complexes that regulate autophagy. Understanding the signals viruses manipulate and the pathways they initiate to generate autophagosomes and control their trafficking will undoubtedly expand our knowledge of autophagy in general and in the context of viral infection.
Acknowledgments
We thank Kristi Berger and Nicholas Heaton for critical reading of the manuscript. The authors wish to acknowledge membership within and support from the Region V ‘Great Lakes’ RCE (NIH award 1-U54-AI-057153). T.X.J. is funded by NIH training grant T32GM007183. G.R. is also supported by NIAID (1R01AI080703), the American Cancer Society (118676-RSG-10-059-01-MPC) and Susan and David Sherman.
References
- 1.Rabinowitz J.D., White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgado M., Singh S., De Haro S., Master S., Ponpuak M., Dinkins C., Ornatowski W., Vergne I., Deretic V. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Virgin H.W., Levine B. Autophagy genes in immunity. Nat. Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky D.J., Codogno P., Cuervo A.M., Deretic V., Elazar Z., Fueyo-Margareto J., Gewirtz D.A., Kroemer G., Levine B., Mizushima N., Rubinsztein D.C., Thumm M., Tooze S.A. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kroemer G., Marino G., Levine B. Autophagy and the integrated stress response. Mol. Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung C.H., Ro S.H., Cao J., Otto N.M., Kim D.H. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funderburk S.F., Wang Q.J., Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simonsen A., Tooze S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan W., Nassiri A., Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc. Natl. Acad. Sci. U S A. 2011;108:7769–7774. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itakura E., Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy. 2009;5:534–536. doi: 10.4161/auto.5.4.8062. [DOI] [PubMed] [Google Scholar]
- 14.Sun Q., Fan W., Chen K., Ding X., Chen S., Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U S A. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Y., Wang Q.J., Li X., Yan Y., Backer J.M., Chait B.T., Heintz N., Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsunaga K., Saitoh T., Tabata K., Omori H., Satoh T., Kurotori N., Maejima I., Shirahama-Noda K., Ichimura T., Isobe T., Akira S., Noda T., Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 17.Liang C., Lee J.S., Inn K.S., Gack M.U., Li Q., Roberts E.A., Vergne I., Deretic V., Feng P., Akazawa C., Jung J.U. Beclin-1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidberg H., Shvets E., Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 19.Weidberg H., Shpilka T., Shvets E., Abada A., Shimron F., Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev. Cell. 2011;20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Moreau K., Ravikumar B., Renna M., Puri C., Rubinsztein D.C. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–317. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakatogawa H., Ichimura Y., Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 22.Geng J., Klionsky D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu M., Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells. 2010;15:923–933. doi: 10.1111/j.1365-2443.2010.01433.x. [DOI] [PubMed] [Google Scholar]
- 24.Kirkin V., McEwan D.G., Novak I., Dikic I. A role for ubiquitin in selective autophagy. Mol. Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Waters S., Marchbank K., Solomon E., Whitehouse C., Gautel M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett. 2009;583:1846–1852. doi: 10.1016/j.febslet.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 26.Thurston T.L., Ryzhakov G., Bloor S., von Muhlinen N., Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 27.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Overvatn A., Bjorkoy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 28.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gal J., Strom A.L., Kwinter D.M., Kilty R., Zhang J., Shi P., Fu W., Wooten M.W., Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009;111:1062–1073. doi: 10.1111/j.1471-4159.2009.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shelly S., Lukinova N., Bambina S., Berman A., Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joubert P.E., Meiffren G., Gregoire I.P., Pontini G., Richetta C., Flacher M., Azocar O., Vidalain P.O., Vidal M., Lotteau V., Codogno P., Rabourdin-Combe C., Faure M. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 2009;6:354–366. doi: 10.1016/j.chom.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Espert L., Denizot M., Grimaldi M., Robert-Hebmann V., Gay B., Varbanov M., Codogno P., Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sir D., Chen W.L., Choi J., Wakita T., Yen T.S., Ou J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLean J.E., Wudzinska A., Datan E., Quaglino D., Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011;286:22147–22159. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J., Liu Y., Wang Z., Liu K., Wang Y., Liu J., Ding H., Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011;85:6319–6333. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denizot M., Varbanov M., Espert L., Robert-Hebmann V., Sagnier S., Garcia E., Curriu M., Mamoun R., Blanco J., Biard-Piechaczyk M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy. 2008;4:998–1008. doi: 10.4161/auto.6880. [DOI] [PubMed] [Google Scholar]
- 37.Espert L., Varbanov M., Robert-Hebmann V., Sagnier S., Robbins I., Sanchez F., Lafont V., Biard-Piechaczyk M. Differential role of autophagy in CD4 T-cells and macrophages during X4 and R5 HIV-1 infection. PLoS One. 2009;4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schroder M., Kaufman R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 39.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 40.Ke P.Y., Chen S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenter J.E., Jackson W., Benetti L., Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 2011;85:9414–9424. doi: 10.1128/JVI.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sir D., Tian Y., Chen W.L., Ann D.K., Yen T.S., Ou J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U S A. 2010;107:4383–4388. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang H., Da L., Mao Y., Li Y., Li D., Xu Z., Li F., Wang Y., Tiollais P., Li T., Zhao M. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of Beclin 1 expression. Hepatology. 2009;49:60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 44.Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Curr. Opin. Immunol. 2009;21:53–62. doi: 10.1016/j.coi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoneyama M., Fujita T. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 46.Lee H.K., Lund J.M., Ramanathan B., Mizushima N., Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 47.Gorbea C., Makar K.A., Pauschinger M., Pratt G., Bersola J.L., Varela J., David R.M., Banks L., Huang C.H., Li H., Schultheiss H.P., Towbin J.A., Vallejo J.G., Bowles N.E. A role for toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010;285:23208–23223. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanchet F.P., Moris A., Nikolic D.S., Lehmann M., Cardinaud S., Stalder R., Garcia E., Dinkins C., Leuba F., Wu L., Schwartz O., Deretic V., Piguet V. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y., Jagannath C., Liu X.D., Sharafkhaneh A., Kolodziejska K.E., Eissa N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talloczy Z., Virgin H.W.t., Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 51.Shi C.S., Kehrl J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008;283:33175–33182. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgado M.A., Elmaoued R.A., Davis A.S., Kyei G., Deretic V. Toll-like receptors control autophagy. Embo J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 54.He B., Gross M., Roizman B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 1998;273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- 55.He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U S A. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He B., Chou J., Brandimarti R., Mohr I., Gluzman Y., Roizman B. Suppression of the phenotype of gamma(1)34.5-herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J. Virol. 1997;71:6049–6054. doi: 10.1128/jvi.71.8.6049-6054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chou J., Kern E.R., Whitley R.J., Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 58.Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H., Ogawa S., Kaufman R.J., Kominami E., Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 59.Jensen P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007;8:1041–1048. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- 60.Schmid D., Pypaert M., Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmid D., Munz C. Localization and MHC class II presentation of antigens targeted for macroautophagy. Methods Mol. Biol. 2008;445:213–225. doi: 10.1007/978-1-59745-157-4_14. [DOI] [PubMed] [Google Scholar]
- 62.Paludan C., Schmid D., Landthaler M., Vockerodt M., Kube D., Tuschl T., Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 63.English L., Chemali M., Duron J., Rondeau C., Laplante A., Gingras D., Alexander D., Leib D., Norbury C., Lippe R., Desjardins M. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nimmerjahn F., Milosevic S., Behrends U., Jaffee E.M., Pardoll D.M., Bornkamm G.W., Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- 65.Lee H.K., Mattei L.M., Steinberg B.E., Alberts P., Lee Y.H., Chervonsky A., Mizushima N., Grinstein S., Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vyas J.M., Van der Veen A.G., Ploegh H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang X.H., Kleeman L.K., Jiang H.H., Gordon G., Goldman J.E., Berry G., Herman B., Levine B. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orvedahl A., MacPherson S., Sumpter R., Jr., Talloczy Z., Zou Z., Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Itakura E., Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ichimura Y., Kumanomidou T., Sou Y.S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 71.Nezis I.P., Simonsen A., Sagona A.P., Finley K., Gaumer S., Contamine D., Rusten T.E., Stenmark H., Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J. Cell Biol. 2008;180:1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carre-Mlouka A., Gaumer S., Gay P., Petitjean A.M., Coulondre C., Dru P., Bras F., Dezelee S., Contamine D. Control of sigma virus multiplication by the Ref(2)P gene of Drosophila melanogaster: an in vivo study of the PB1 domain of Ref(2)P. Genetics. 2007;176:409–419. doi: 10.1534/genetics.106.063826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wyers F., Petitjean A.M., Dru P., Gay P., Contamine D. Localization of domains within the Drosophila Ref(2)P protein involved in the intracellular control of sigma rhabdovirus multiplication. J. Virol. 1995;69:4463–4470. doi: 10.1128/jvi.69.7.4463-4470.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dezelee S., Bras F., Contamine D., Lopez-Ferber M., Segretain D., Teninges D. Molecular analysis of Ref(2)P, a Drosophila gene implicated in sigma rhabdovirus multiplication and necessary for male fertility. Embo J. 1989;8:3437–3446. doi: 10.1002/j.1460-2075.1989.tb08508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y., Schiff M., Czymmek K., Talloczy Z., Levine B., Dinesh-Kumar S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 76.Sinha S., Colbert C.L., Becker N., Wei Y., Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4:989–997. doi: 10.4161/auto.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ku B., Woo J.S., Liang C., Lee K.H., Hong H.S., E X., Kim K.S., Jung J.U., Oh B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kyei G.B., Dinkins C., Davis A.S., Roberts E., Singh S.B., Dong C., Wu L., Kominami E., Ueno T., Yamamoto A., Federico M., Panganiban A., Vergne I., Deretic V. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gannage M., Dormann D., Albrecht R., Dengjel J., Torossi T., Ramer P.C., Lee M., Strowig T., Arrey F., Conenello G., Pypaert M., Andersen J., Garcia-Sastre A., Munz C. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee J.S., Li Q., Lee J.Y., Lee S.H., Jeong J.H., Lee H.R., Chang H., Zhou F.C., Gao S.J., Liang C., Jung J.U. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chaumorcel M., Souquere S., Pierron G., Codogno P., Esclatine A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy. 2008;4:46–53. doi: 10.4161/auto.5184. [DOI] [PubMed] [Google Scholar]
- 82.Alirezaei M., Kiosses W.B., Flynn C.T., Brady N.R., Fox H.S. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One. 2008;3:e2906. doi: 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alirezaei M., Kiosses W.B., Fox H.S. Decreased neuronal autophagy in HIV dementia: a mechanism of indirect neurotoxicity. Autophagy. 2008;4:963–966. doi: 10.4161/auto.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.den Boon J.A., Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 85.Suhy D.A., Giddings T.H., Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schlegel A., Giddings T.H., Jr., Ladinsky M.S., Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taylor M.P., Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kemball C.C., Alirezaei M., Flynn C.T., Wood M.R., Harkins S., Kiosses W.B., Whitton J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010;84:12110–12124. doi: 10.1128/JVI.01417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang S.C., Chang C.L., Wang P.S., Tsai Y., Liu H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009;81:1241–1252. doi: 10.1002/jmv.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Donnell V., Pacheco J.M., LaRocco M., Burrage T., Jackson W., Rodriguez L.L., Borca M.V., Baxt B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology. 2011;410:142–150. doi: 10.1016/j.virol.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Klein K.A., Jackson W.T. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 2011;85:9651–9654. doi: 10.1128/JVI.00316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brabec-Zaruba M., Berka U., Blaas D., Fuchs R. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 2007;81:10815–10817. doi: 10.1128/JVI.00143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jackson W.T., Giddings T.H., Jr., Taylor M.P., Mulinyawe S., Rabinovitch M., Kopito R.R., Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quiner C.A., Jackson W.T. Fragmentation of the Golgi apparatus provides replication membranes for human rhinovirus 1A. Virology. 2010;407:185–195. doi: 10.1016/j.virol.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Belov G.A., Feng Q., Nikovics K., Jackson C.L., Ehrenfeld E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008;4:e1000216. doi: 10.1371/journal.ppat.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belov G.A., Altan-Bonnet N., Kovtunovych G., Jackson C.L., Lippincott-Schwartz J., Ehrenfeld E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2007;81:558–567. doi: 10.1128/JVI.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T., Cameron C.E., Ehrenfeld E., van Kuppeveld F.J., Altan-Bonnet N. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cherry S., Kunte A., Wang H., Coyne C., Rawson R.B., Perrimon N. COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog. 2006;2:e102. doi: 10.1371/journal.ppat.0020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Taylor M.P., Burgon T.B., Kirkegaard K., Jackson W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao Z., Thackray L.B., Miller B.C., Lynn T.M., Becker M.M., Ward E., Mizushima N.N., Denison M.R., Virgin H.W.t. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 102.Reggiori F., Monastyrska I., Verheije M.H., Cali T., Ulasli M., Bianchi S., Bernasconi R., de Haan C.A., Molinari M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berkova Z., Crawford S.E., Trugnan G., Yoshimori T., Morris A.P., Estes M.K. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 2006;80:6061–6071. doi: 10.1128/JVI.02167-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dreux M., Gastaminza P., Wieland S.F., Chisari F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U S A. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guevin C., Manna D., Belanger C., Konan K.V., Mak P., Labonte P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology. 2010;405:1–7. doi: 10.1016/j.virol.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carter C.A., Ehrlich L.S. Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 2008;62:425–443. doi: 10.1146/annurev.micro.62.081307.162758. [DOI] [PubMed] [Google Scholar]
- 108.Benaroch P., Billard E., Gaudin R., Schindler M., Jouve M. HIV-1 assembly in macrophages. Retrovirology. 2010;7:29. doi: 10.1186/1742-4690-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bruss V. Envelopment of the hepatitis B virus nucleocapsid. Virus Res. 2004;106:199–209. doi: 10.1016/j.virusres.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 110.Tucker S.P., Thornton C.L., Wimmer E., Compans R.W. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J. Virol. 1993;67:4274–4282. doi: 10.1128/jvi.67.7.4274-4282.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takenouchi T., Nakai M., Iwamaru Y., Sugama S., Tsukimoto M., Fujita M., Wei J., Sekigawa A., Sato M., Kojima S., Kitani H., Hashimoto M. The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 2009;182:2051–2062. doi: 10.4049/jimmunol.0802577. [DOI] [PubMed] [Google Scholar]
- 112.Manjithaya R., Anjard C., Loomis W.F., Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Duran J.M., Anjard C., Stefan C., Loomis W.F., Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cabral M., Anjard C., Malhotra V., Loomis W.F., Kuspa A. Unconventional secretion of AcbA in Dictyostelium discoideum through a vesicular intermediate. Eukaryot. Cell. 2010;9:1009–1017. doi: 10.1128/EC.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Blott E.J., Griffiths G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002;3:122–131. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 116.Nakashima A., Tanaka N., Tamai K., Kyuuma M., Ishikawa Y., Sato H., Yoshimori T., Saito S., Sugamura K. Survival of parvovirus B19-infected cells by cellular autophagy. Virology. 2006;349:254–263. doi: 10.1016/j.virol.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 117.Shrivastava S., Raychoudhuri A., Steele R., Ray R., Ray R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Panyasrivanit M., Khakpoor A., Wikan N., Smith D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009;90:448–456. doi: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 119.Lee Y.R., Lei H.Y., Liu M.T., Wang J.R., Chen S.H., Jiang-Shieh Y.F., Lin Y.S., Yeh T.M., Liu C.C., Liu H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374:240–248. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khakpoor A., Panyasrivanit M., Wikan N., Smith D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009;90:1093–1103. doi: 10.1099/vir.0.007914-0. [DOI] [PubMed] [Google Scholar]
- 121.Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu Y., Clippinger A.J., Alwine J.C. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011;19:360–367. doi: 10.1016/j.tim.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vastag L., Koyuncu E., Grady S.L., Shenk T.E., Rabinowitz J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011;7:e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heaton N.S., Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011;19:368–375. doi: 10.1016/j.tim.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Munger J., Bennett B.D., Parikh A., Feng X.J., McArdle J., Rabitz H.A., Shenk T., Rabinowitz J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Munger J., Bajad S.U., Coller H.A., Shenk T., Rabinowitz J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Heaton N.S., Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Heaton N.S., Randall G. Dengue virus and autophagy. Viruses. 2011;3:1332–1341. doi: 10.3390/v3081332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tal M.C., Sasai M., Lee H.K., Yordy B., Shadel G.S., Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jounai N., Takeshita F., Kobiyama K., Sawano A., Miyawaki A., Xin K.Q., Ishii K.J., Kawai T., Akira S., Suzuki K., Okuda K. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U S A. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]