

Abstract
Background
Mild traumatic brain injury (mTBI; cerebral concussion) results in cognitive and emotional dysfunction. These injuries are a significant risk factor for the development of anxiety disorders including post-traumatic stress disorder (PTSD). However, because physically traumatic events typically occur in a highly emotional context, it is unknown whether TBI itself is a cause of augmented fear and anxiety.
Methods
Rats were trained with one of five fear conditioning procedures (N = 105) two days after concussive brain trauma. Fear learning was assessed over subsequent days and chronic changes in fear learning and memory circuitry were assessed by measuring NMDA receptor subunits and GAD-67 protein levels in the hippocampus and basolateral amygdala complex (BLA).
Results
Injured rats exhibited an overall increase in fear conditioning regardless of whether fear was retrieved via discrete or contextual-spatial stimuli. Moreover, injured rats appeared to over generalize learned fear to both conditioned and novel stimuli. Although no gross histopathology was evident, injury resulted in a significant up-regulation of excitatory NMDA receptors in the BLA. There was a trend toward decreased GABA related inhibition (GAD-67) in the BLA and hippocampus.
Conclusions
These results suggest that mTBI predisposes the brain toward heightened fear learning during stressful post-injury events and provides a potential molecular mechanism by which this occurs. Furthermore, these data represent a novel rodent model that can help advance the neurobiological and therapeutic understanding of the co-morbidity of PTSD and TBI.
Keywords: traumatic brain injury, concussion, PTSD, fear conditioning, NMDA, GAD-67
Introduction
Traumatic brain injury (TBI) is a major cause of disability and death, with the majority of reported cases being classified as mild TBI (mTBI or concussion) (1). Many symptoms of mTBI are characterized as ‘psychological’ rather than ‘physical’ and diagnosis is predominantly based upon subjective symptoms, therefore mTBI is often not properly diagnosed. For these reasons mTBI has been labeled a “silent epidemic”, as well as the “signature injury” of the current theaters of combat (1, 2). Anxiety disorders are often diagnosed in people with a history of mTBI (3). Also, given that the traumatic events that produce post-traumatic stress disorder (PTSD) can incorporate TBI, it is not surprising that there is a high correlation between diagnoses of mTBI and PTSD (2). Therefore, it remains unknown whether cerebral concussion alone is a predisposing factor for the development of the fear and anxiety related symptoms of PTSD.
To investigate the potential neurobiological link between concussion and the acquisition of PTSD, we combined the use of lateral fluid percussion injury (LFPI) with Pavlovian fear conditioning. Lateral fluid percussion models concussion in rodents and is one of the most extensively validated models of experimental TBI (4). The primary biomechanical injury sets in motion a neurochemical and neurometabolic cascade (5) and neurons that survive the initial mechanical trauma may function sub-optimally, resulting in dysfunctional neural networks. Pavlovian fear conditioning is a well accepted tool for modeling symptoms of exaggerated, dysfunctional fear and to investigate the neurobiological basis of PTSD (6, 7). The hippocampus and amygdala play key roles in Pavlovian fear conditioning (8) and there is substantial evidence of prolonged hippocampal dysfunction following LFPI (4). The effect of LFPI on the amygdala is not well understood, however there is evidence to suggest that this region exhibits dysfunction after injury (9, 10). Within these regions excitatory NMDA (N-methyl-D-aspartate) receptors and inhibitory GABA (γ-Aminobutyric acid) are crucial for normal fear learning and memory (11–16). Antagonism of NMDA receptors blocked the acquisition of conditional fear (11–13) and each receptor subunit confers a receptor with different functional properties. The NR1 subunit is necessary for normal NMDA receptor function and therefore its quantification reflects the total number of receptors within a region (17). The NR2B subunit is thought to be important for the formation of fear memories (i.e. synaptic plasticity) while the NR2A subunit is not only involved in learning but may be particularly important for the expression of fear (i.e. synaptic transmission) (14, 16–19). The BLA contains a system of GABAergic interneurons and receptors that are necessary for normal fear learning and plasticity (15, 20). Likewise, GABA receptor knockouts that targeted the hippocampus produced enhanced fear conditioning under certain conditions (21–24). Changes in these neurotransmitter and receptor systems have been reported following LFPI (25–28) and an optimal balance between excitatory and inhibitory neurotransmission is necessary for normal learning and memory processes (29); this balance may be disrupted following LFPI (30).
Here we describe the first rodent model of mTBI induced PTSD-like symptoms. Two days following brain trauma (LFPI) rats experienced a behavioral trauma. The traumatic event consisted of multiple footshocks as part of Pavlovian fear conditioning and five different conditioning procedures were employed. How fearful the animals had become of the traumatic environment (context) and a cue paired with trauma (an auditory cue) were investigated on subsequent testing days. Furthermore, we hypothesized that injury induced changes in excitatory and inhibitory processes involved in normal neurotransmission could produce a susceptibility to psychological stressors encountered after brain trauma. To address this possibility, chronic, stable alterations in the amount of GABA related inhibition (i.e., GAD-67 levels) and the number and type of NMDA receptors (NR1, NR2A and NR2B subunits) were investigated within the hippocampus and basolateral amygdala complex (BLA).
Methods and Materials
Subjects
Subjects were adult male Sprague-Dawley rats (N = 105) from Charles River Laboratories (Hollister, California). Animals were maintained on a 12 hour light/dark cycle in standard vivarium housing (2 rats/cage), with food and water ad libitum. All procedures were conducted with approval from the University of California at Los Angeles Chancellor’s Animal Research Committee and were in conformity with the NIH Guide for the Care and Use of Laboratory Animals.
Surgery and Histology
Lateral fluid percussion injury was induced using a previously published protocol (31). Rats were anesthetized with a 1–2% isoflurane-O2 mixture. Following a midline incision, a 3 mm diameter craniotomy was made over the left parietal cortex, centered at 3 mm posterior to bregma and 6 mm lateral to midline. A plastic injury cap was adhered to the edges of the craniotomy using silicone gel and then dental cement. The injury cap was filled with saline and attached to the fluid percussion device (Virginia Commonwealth University, Richmond, Virginia) (Figure 1A). A brief fluid pulse (~20 ms) of saline was applied to the intact dura mater of the brain through the injury cap by allowing a mallet suspended at the opposite end to strike the saline filled, cylindrical body of the device. Although inflicted laterally, the injury traumatizes both hemispheres, with greater involvement of the ipsilateral hemisphere (32).
Figure 1.

(A) Timeline representing the experimental design followed by schematic timeline of fear conditioning behavior in relation to day of surgery (PID = post-injury day). The lateral fluid percussion injury device is pictured on PID0. Note (*) the weak delay procedure had a second day of context exposure prior to cued fear testing. (B) Five similar yet distinct training procedures were used to investigate the effects of TBI on fear conditioning in detail. Testing procedures were homogenous (see timeline and note above).
Anesthesia was removed immediately before attachment to the device and injury was induced when the animal exhibited a reflex to toe-pinch. Immediately after injury, resuscitation was performed with gentle positive pressure with O2 if injury resulted in apnea > 30 s. Once the LOC time was measured, rats were placed back under anesthesia to remove the injury cap and suture the incision site. Injury severity was determined by the loss of consciousness time, which was defined as the absence of a post-injury toe-pinch withdrawal reflex; only animals that exhibited a lack of reflex for ≥2 min were included in behavioral work. Sham animals were given a craniotomy. Upon the completion of surgery, animals were placed in a heated recovery chamber until the resumption of normal behavior (e.g. fully awake, exploring, grooming). Following approximately an hour of recovery, animals were placed in their home cages and returned to the vivarium. Although the topical analgesic bupivacaine was applied to the incision site at the time of suturing, no post-operative, systemic analgesic or antibiotic was administrated so that recovery from head injury progressed naturally. Besides daily health checks, animals were left undisturbed until the beginning of the behavioral phase.
Two days after surgery the majority of animals went on for behavioral training and testing while a sub-set was perfused for histology. The two days post-injury time point was selected for these initial experiments because TBI effects on learning and behavior were most likely to be found during the acute recovery period.
Perfused brains (one sham, two injured with loss of consciousness times of 90 s and 300 s) were sectioned coronally at 40 microns thick using a cryostat. The tissue was Nissl stained with thionine and visually examined by light microscopy for damage at the injury site and to the hippocampus and amygdala.
Fear Conditioning
Following five days of handling (1 min/day), rats underwent surgery and were trained two days later on one of five fear conditioning tasks. Testing of learned fear began 24 hours after training (Figure 1A). In the ‘strong delay’ conditioning procedures rats were given either three or ten presentations of an auditory cue paired with a strong footshock (0.9 mA). In a ‘weak delay’ condition, a relatively mild footshock (0.5 mA) was paired with an auditory cue. The ‘trace’ condition was identical to the delay condition except that the noise cue was shortened to allow for a 28 s long gap or ‘trace interval’ between termination of the auditory cue and footshock presentation. A ‘context’ conditioning group was included, which matched the delay and trace groups with the exception that auditory cues were omitted. (Figure 1B)
Training and testing occurred in two distinct conditioning chambers (Box A and Box B) that differed in smell, lighting, chamber shape and flooring (Med Associates Inc., Georgia, Vermont). An animal was placed in the chamber (Box A) for a 192 s baseline, acclimation period, next a 70 dB white noise cue was repeatedly presented in all relevant procedures (i.e. not in the context conditioned group) that co-terminated with a 2 s long scrambled footshock administered through the grid floor. The noise was 46 s long with exception of the trace procedure, in which the noise was shortened to 16 s to allow for a 28 s trace interval. The interval between the shock termination and the presentation of the next cue was 210 s. Note that the baseline of the context conditioned group was actually 236 s (see Figure 1B), however the first 192 s were used in statistical analyses to make equivalent comparisons to other procedures.
Freezing to the discrete, auditory stimulus and static training environment were taken as measures of auditory and contextual fear learning, respectively. One day after training, each animal was placed back into the training chamber and context fear related freezing to Box A was quantified over 8 min. Twenty-four hours later, rats that had been conditioned to the noise were individually placed in novel Box B for 192 s, prior to seven presentations of the noise cue from training. Animals from the weak delay condition received an additional contextual fear test one day prior to cued fear testing, so that the cued test was administered on post-injury day five instead of four. The percentages of time spent freezing during the baseline acclimation periods, to noise cues and to the training context were electronically quantified using software provided by Med Associates Inc. (Georgia, Vermont). Electronic scoring was calibrated to human scoring prior to the experiments.
The sample sizes of the different training conditions were as follows: strong delay conditioning with 10 (sham: n = 10, injured: n = 10) or 3 (sham: n = 10, injured: n = 11) pairings, weak delay conditioning with mild footshock (sham: n = 9, injured: n = 11), context conditioned group (sham: n = 8, injured: n = 8) and trace group (acquisition: sham n = 12, injured n = 12; context test: sham n = 14, injured n = 14; cued test: sham n = 6, injured n = 7). Some sample sizes vary between trials due to equipment malfunction during data collection.
Western Blotting
Seventeen days following surgery the strong ten shock delay group was sacrificed for immunoblotting of NMDA receptor subunit and GAD-67 (glutamic acid decarboxylase, 67 kDa isoform)protein levels (sham: n = 10; TBI: n = 9)(Figure 1A). NMDA receptors bind the excitatory neurotransmitter glutamate (17) while the inhibitory neurotransmitter GABA is synthesized by GAD (33). The 17 days post-injury time point was selected for this initial study in an attempt to identify chronic, stable and substantial molecular changes that could have impacted fear learning or vice versa. Acute pathophysiology has been shown to largely resolve by ten days post-LFPI (5) and an additional week was provided so that sampling occurred well outside of the acute recovery period. It was also not known how the stress of multiple trial fear conditioning with strong footshock would interact with the standard recovery period and 17 days allowed for an additional week of recovery from any stress effects related to fear learning and recall.
Fresh hippocampus and BLA were dissected bilaterally and homogenized for 20 s in buffer (0.137 M NaCl, 2.7 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 10 mM HEPES, 10 mM glucose, pH 7.4). Immunoblots were performed according to a previously published protocol (34) on crude homogenates using an antibody concentration of 1:1000 for NR1, NR2A, NR2B NMDA subunits and GAD-67 (Millipore, Temecula, California). Blots were exposed and the protein signal was quantified in a Biorad Fluor-S Max Imager (Hercules, California) for 1–5 min. Total protein per lane was determined after transfer using Biorad Ruby Red Sypro Stain exposed in the imager. Raw values for the proteins of interest were controlled to total protein from the same lane on the same blot. Total protein controlled subunit and GAD-67 values from each individual sample were then normalized to the average of the shams, such that the mean sham value equaled 100%. Receptor subunit and GAD-67 levels are represented as the percent change from sham levels.
Results
Fear Conditioning
Acquisition trial data were analyzed separately for each of the five conditioning procedures. For the procedures incorporating noise cue conditioning, repeated measures ANOVAs were run looking at the percentage of time spent freezing (%Freezing) during the baseline period and to each of the cues (within-subjects factor), with treatment condition (sham vs. injured) as a between-subjects factor. Repeated measures ANOVAs were also run for the trace procedure with trace interval as a within-subjects factor and similarly for the context conditioned group using the post-shock intervals. Injury did not significantly affect baseline freezing rates prior to conditioning, nor did injury affect acquisition curves (Figure 2A-E). The amount of behavioral reactivity to each of the shock presentations was analyzed in the strong ten shock delay group to examine pain sensitivity and changes in conditioned analgesia (Repeated measures ANOVA, shock presentation as the within-subjects factor, treatment as the between-subjects factor); no injury effects were found (Figure 2F).
Figure 2.

Performance during acquisition of Pavlovian fear conditioning for each of the five training procedures.Graphs represent freezing to the novel context prior to shock presentation (Baseline), freezing during the post-shock inter-trial intervals (ITIs) (context conditioned group only, A), to the white noise cues (B-E) and freezing during the trace intervals (trace group only, B). Behavioral reactivity to 0.9 mA footshock was also examined in the strong ten shock delay procedure (F). Data are represented as the mean ± s.e.m.
Context test data were analyzed with ANOVA by examining the average %Freezing across the 8 min trial; treatment and training procedure were fixed factors. There was a significant main effect of training procedure, F(4, 95) = 8.40, p < 0.01, whereby different procedures produced differing amounts of conditional context fear (context: 88.3%; trace: 59.1%; strong 10 shock delay: 48.2%; strong 3 shock delay: 46.9%; weak delay: 36.9%). There was also a significant main effect of treatment, F(1, 95) = 7.09, p < 0.01, whereby injured rats exhibited greater context fear than shams (61.7% vs. 47.8%), regardless of training procedure. Although the injury effect was clearest following strong ten shock delay, the interaction term was non-significant, indicating that the effect of injury was nonetheless consistent across training conditions (Figure 3A). The injury effect did not vary according to individual injury severity, as LOC did not significantly correlate with level of context fear (t-test conducted on Pearson correlation coefficient).
Figure 3.

Effects of concussive head injury on learned fear. Graphs depict conditional fear to contextual (A) and discrete (B) stimuli at testing. Both context and discrete cue fear were significantly enhanced following brain trauma, regardless of procedure. Data are represented as the mean ± s.e.m.
The cued fear test data were analyzed by ANOVA by examining the %Freezing at baseline or averaged across the seven non-reinforced white noise presentations; treatment and training procedure were fixed factors. There was a significant main effect of training procedure, F(3, 66) = 10.03, p < 0.01, whereby different procedures produced differing amounts of conditional cue fear (strong 10 shock delay: 83.1%; strong 3 shock delay: 73.6%; weak delay: 53.8%; trace: 43.1%). There was also a significant main effect of treatment, F(1, 66) = 10.69, p < 0.01, whereby injured rats exhibited greater cued fear than shams (73.2% vs. 56.8%), regardless of training procedure (Figure 3B). The interaction term was non-significant, signifying that the injury effect was not dependent upon the type of training, although strong ten shock delay and trace procedures yielded the largest injury effects (Figure 3B). There was no significant correlation between an individual animal’s injury severity (i.e. LOC) and noise cue fear.
Injured animals also exhibited significantly greater baseline freezing compared to shams in the novel testing context (15.6% vs. 5.8%), prior to presentation of the conditional noise cue, F(1, 66) = 6.42, p < 0.05. The main effect of training procedure and interaction term were not significant. However, there was evidence against confounding of the cued test results by the simple summation of baseline and cued fear levels. Although the level of context test fear correlated with baseline cued fear in both injured, r(38) = +.38, p < 0.05, and sham groups, r(34) = +.53, p < 0.01, context test fear and baseline fear correlated with cued fear at testing only in injured animals [injured: r(38) = +.49, p < 0.01, r(38) = +.43, p < 0.05; sham: r = +.21, r = +.24]. In addition to enhanced cued fear expression, these analyses were suggestive of a post-injury increase in fear generalization.
Histology
The examination of Nissl stained brain sections at the injury site (over the dorsal hippocampus) and from the BLA revealed no gross histopathology (i.e. no cavernous lesions or pervasive cell loss) two days post-surgery, at the time of fear learning. The only apparent signs of traumatic brain injury were at the injury site after the more severe injury; some cortical thinning was noted at the craniotomy site, as well as some shearing and hemorrhaging at the gray-white matter junction along the dorsal hippocampus (Figure 4). This pattern and range of histopathology was typical of LFPI generated in our laboratory.
Figure 4.

Representative histological sections from the injury site and from the ipsilateral basolateral amygdala complex (BLA). Loss of consciousness time (LOC) denotes injury severity. Two days following lateral fluid percussion brain injury (corresponding to the day of fear training) structures proximal to the injury site, such as the dorsal hippocampus, were intact, as was the BLA. Some cortical thinning at the injury site (
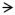 ) and white matter tearing and hemorrhage at the gray-white matter junction (
) and white matter tearing and hemorrhage at the gray-white matter junction (
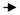 ) under the injury site were noted after more severe injury (i.e. LOC = 300 s).
) under the injury site were noted after more severe injury (i.e. LOC = 300 s).
Western Blots
Independent samples t-tests were used to compare relative injured protein levels to sham levels 17 days after the final fear test. Following behavior, injury was found to cause a chronic, 17% increase in the NR1 subunit of the NMDA receptor in the BLA ipsilateral to the injury site, t(17) = 2.82, p < 0.05 (Figure 5A), but not in the ipsilateral hippocampus (Figure 5B). Injury did not significantly affect the other subunits, although post-injury NR2A and NR2B tended to increase in parallel with NR1 in the ipsilateral BLA (+10% and +12%, respectively). The increase in NR1 denoted an overall increase in NMDA receptor levels (17). The level of GAD-67, an enzyme used to synthesize the inhibitory neurotransmitter GABA (33), was not significantly affected by injury, however there were trends for post-injury reductions in the ipsilateral BLA and hippocampus of 20% and 24%, respectively (Figure 6C, D). Representative blots of actual protein levels for ipsilateral NR1 and GAD-67 are provided in Figure 6E. No injury effects were detected contralateral to the injury site.
Figure 5.

Chronic changes in NMDA subunit and GAD-67 protein levels within the basolateral amygdala complex (BLA) and hippocampus following concussive brain injury. Graphs depict the percent change in injured from sham protein levels from structures ipsilateral to the injury site, 17 days following the last test of learned fear. The NR1 subunit was significantly increased in the BLA (A) but not in the hippocampus (B). There was no significant effect of injury on GABA related GAD-67, however levels tended to be reduced in both the BLA (C) and hippocampus (D). Representative bands depicting GAD-67 and NR1 protein levels for sham (S) vs. injured (TBI) rats (E). Data are represented as the mean ± s.e.m and *p < 0.05 (sham: n = 10; injured: n = 9).
Discussion
There is a high correlation between diagnoses of mild traumatic brain injury (mTBI) and post-traumatic stress disorder (PTSD), and the current increase in soldiers exposed to active combat has generated further interest in this dual diagnosis as a public health problem (2, 35, 36). Yet there has been controversy surrounding this comorbidity. Brain trauma may result in memory impairment and it has been argued that the lack of a traumatic memory could protect against the development of PTSD (37–39). This has led some to question whether TBI and PTSD can co-occur at all. Furthermore, symptoms of mTBI may be subtle and largely transient, which have led some to argue that patient’s lasting symptoms stem from PTSD alone (36). The simple rodent model developed here has helped address these clinical questions by showing that PTSD symptoms can be modeled with Pavlovian fear conditioning acutely after mTBI. The development of a successful rodent model further allows for the investigation of neural mechanisms, which we have begun here.
Beginning two days after lateral fluid percussion injury (LFPI), which models aspects of concussion (4), rats were trained with one of five, multiple-trial, fear conditioning procedures. Animals were then tested on subsequent days for learned fear of the traumatic environment (i.e., training context) and a discrete noise cue. Regardless of conditioning procedure, injured animals showed an overall significant increasein context and cued fear e xpression, although the effect was greatest following delay conditioning with ten strong shocks. This general enhancement in fear conditioning was not attributable to obvious sources of interference, such as fear sensitization resulting from the pain and stress experienced as part of the initial trauma of surgery. Learning curves and performance during training were normal following mTBI. Injured animals were not more fearful of novelty, as there were no differences in initial baseline freezing rates to the chamber prior to conditioning. Performance differences were also not likely due to differing pain sensitivities, as behavioral reactivity to shock (40) was equivalent. Similar to PTSD, a disorder that produces exaggerated fear conditioning (41, 42), these data suggest that cerebral concussion can produce a state whereby fear conditioning is enhanced in stressful situations.
Injured animals also exhibited significantly greater baseline freezing in the novel context, prior to presentation of the noise cue at testing. Therefore although cued fear expression was enhanced by brain trauma, understanding of the specific effect of TBI on cued fear memory, in isolation, remains to be studied. However recent work that directly tested the effect of baseline freezing on auditory cue fear found that it was possible to have a considerable amount of baseline fear that did not interact with cued fear expression, although the most accurate quantification occurred when baseline fear was negligible (43). In line with this published work, the present data showed that context fear from training predicted the amount of generalized context fear, however context fear did not predict the amount of cued fear in control animals. In contrast, context fear predicted cued fear post-mTBI. So in addition to the specific effect of concussion on context fear memory and a possible effect on auditory fear memory, the data suggested that injured animals exhibited increased generalization of conditional fear across multiple aspects of the task. People with PTSD may overreact or panic in environments not specifically related to their past trauma (44), thus the inappropriate generalization of fear or stress intolerance detected here was akin to this generalized response.
Unlike all prior studies demonstrating either intact or impaired cognitive abilities following LFPI/experimental brain trauma (4, 45–48) we reported learning and memory enhancements. This finding is particularly noteworthy considering that the small literature on fear conditioning and LFPI has found impaired and intact context fear (30, 49–52) and intact auditory cue fear (52). An explanation for this resides in the small number of trials used during training. Previous publications employed single (30, 49–51) or double trial (52) procedures, which may not have been “traumatizing” enough to produce the consistent phenotype we observed. And indeed the amount of freezing behavior increases with both shock number and intensity (53), as does an animal’s physiological stress response (54).
Secondly, although the previous TBI literature presented behavioral and physiological evidence of hippocampal dysfunction, the range of injury effects we observed across a variety of multiple-trial procedures was suggestive, rather, of primary dysfunction within the amygdala. The molecular evidence supported this hypothesis. The amygdala is broadly responsible for context and cued fear conditioning (55, 56), while the hippocampus is recruited into the fear circuitry to process contextual stimuli (8, 55). No gross histopathology was noted in the amygdala or hippocampus at the time of learning two days post-injury, nonetheless approximately two weeks after injury NR1 was significantly increased in the ipsilateral BLA. The NR1 subunit is necessary for functional NMDA receptors and is an indicator of the number of receptors within a region (17). In support of an overall increase in receptor number, the other subunits NR2A and NR2B tended to increase in parallel with NR1 in the ipsilateral BLA.
Together these data suggest that the mechanically traumatized brain is vulnerable to psychological trauma. Animal models can not completely capture the complexity of human psychological disorders, nonetheless following concussion rats showed enhanced fear learning and generalization, which are suggestive of a PTSD-like phenotype. Although other brain regions may play a role, such as the hippocampus, the general enhancement in fear across conditioning procedures implicated dysfunction in the amygdala. Along with these behavioral data, injured animals exhibited receptor changes within the amygdala that would be consistent with an increase in NMDA-mediated excitation and plasticity. Strong evidence indicates that NMDA receptor mediated long-term potentiation in the BLA may underlie fear learning and memory (57). There were no significant injury effects on an enzyme used to produce the inhibitory neurotransmitter GABA, GAD-67 (33), however there was a trend toward a reduction in the ipsilateral BLA and hippocampus. GAD-67 is just one marker of inhibitory processes and these data warrant further study of the role of GABA receptors and inhibitory neurotransmission in post-mTBI fear learning. Also, this initial molecular work was suggestive of long lasting changes in fear circuitry and these results should be substantiated through subsequent profiling of the acute recovery period during fear learning and recall. In conclusion, these data suggest that mTBI can predispose the brain toward enhanced fear learning. Furthermore a concussion induced increase in the net excitability of emotional learning and memory circuitry could underlie the co-morbidity of mTBI and PTSD.
Acknowledgments
We thank Yan Cai and Naomi Santa Maria for their technical assistance. This research was supported by the UCLA Brain Injury Research Center, NIH grants 1PO1NS058489 (D.A.H.), 1RO1NS27544 (D.A.H), RC1MH088184 (M.S.F.), KO2NS057420 (C.C.G.) and the Department of Defense grant DR080372 (C.C.G.). Preliminary data from the current study were previously presented at the Society for Neuroscience Meeting, November 2010, San Diego, California, at the National Neurotrauma Society Meeting, June 2010, Los Gatos, California, at the Combined Meeting of the National and International Neurotrauma Societies, September 2009, Santa Barbara, California and at the Society for Neuroscience Meeting, November 2008, Washington, D.C.
Footnotes
Financial Disclosures
All authors reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Centers for Disease Control and Prevention, National Center for Injury Prevention and Control Website. Traumatic Brain Injury page. 2011 October 13; http://www.cdc.gov/ncipc/tbi/TBI.htm.
- 2.Center for Military Health Policy Research, RAND Corporation. Tanielian T, Jaycox LH, editors. RAND Report: Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. 2008 Available Online as of October 13, 2011: http://www.rand.org/pubs/monographs/MG720/
- 3.Moore EL, Terryberry-Spohr L, Hope DA. Mild traumatic brain injury and anxiety sequelae: A review of literature. Brain Inj. 2006;2:117–132. doi: 10.1080/02699050500443558. [DOI] [PubMed] [Google Scholar]
- 4.Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, et al. Lateral fluid percussion brain injury: A 15-year review and evaluation. J Neurotrauma. 2005;27:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- 5.Giza CC, Hovda DA. The neurometabolic cascade of concussion. J Athl Train. 2001;36:228–235. [PMC free article] [PubMed] [Google Scholar]
- 6.Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: An animal of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29(8):1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Stam R. PTSD and stress sensitization: A tale of brain and body Part 2: Animal models. Neurosci Biobehav Rev. 2007;31:558–584. doi: 10.1016/j.neubiorev.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- 9.Hovda DA, Yoshino A, Kawamata T, Katayama Y, Becker DP. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 1991;567:1–10. doi: 10.1016/0006-8993(91)91429-5. [DOI] [PubMed] [Google Scholar]
- 10.Prins ML, Hovda DA. Mapping cerebral glucose metabolism during spatial learning: Interaction of development and traumatic brain injury. J Neurotrauma. 2001;18:31–46. doi: 10.1089/089771501750055758. [DOI] [PubMed] [Google Scholar]
- 11.Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 12.Fanselow MS, Kim JJ. Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist D, L-2-amino-5-phosphonovaleric acid to basolateral amygdala. Behav Neurosci. 1994;108(1):210–212. doi: 10.1037//0735-7044.108.1.210. [DOI] [PubMed] [Google Scholar]
- 13.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 14.Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 16.Makkar SR, Shirley QZ, Cranney J. Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology. 2010;35:1625–1652. doi: 10.1038/npp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 18.Shin RM, Tsvetkov E, Bolshakov VY. Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron. 2006;52:883–896. doi: 10.1016/j.neuron.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker DL, Davis M. Amygdala infusions of an NR2B-selective of an NR2A-preferring NMDA receptor antagonist differentially influence fear conditioning and expression in the fear-potentiated startle test. Learning Mem. 2008;15:67–74. doi: 10.1101/lm.798908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiltgen BJ, Godsil BP, Peng Z, Saab F, June HL, Linn ML, et al. The alpha1 subunit of the GABA(A) receptor modulates fear learning and plasticity in the lateral amygdala. Front Behav Neurosci. 2009;3:37. doi: 10.3389/neuro.08.037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, et al. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- 22.Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, et al. Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc Natl Acad Sci. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crestani F, Keist R, Fritsch JM, Benke D, Vogt K, Prut L, et al. Trace fear conditioning involves hippocampal α5 GABAA. Proc Natl Acad Sci. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiltgen BJ, Sanders MJ, Ferguson C, Homanics GE, Fanselow MS. Trace fear conditioning in enhanced in mice lacking δ subunit of the GABAA receptor. Learn Mem. 2005;12:327–333. doi: 10.1101/lm.89705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller LP, Lyeth BG, Jenkins LW, Oleniak L, Panchision D, Hamm RJ, et al. Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Res. 1990;526:103–107. doi: 10.1016/0006-8993(90)90254-9. [DOI] [PubMed] [Google Scholar]
- 26.Santhakumar V, Razliff AD, Jeng J, Toth Z, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. 2001;50:708–717. doi: 10.1002/ana.1230. [DOI] [PubMed] [Google Scholar]
- 27.Kumar A, Zou L, Yuan X, Long Y, Yang K. N-methyl-D-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J Neurosci Res. 2002;67:781–786. doi: 10.1002/jnr.10181. [DOI] [PubMed] [Google Scholar]
- 28.Osteen CL, Giza CC, Hovda DA. Injury-induced alterations in N-Methyl-D-aspartate receptor subunit composition contribute to prolonged 45calcium accumulation following lateral fluid percussion. Neuroscience. 2004;125:305–322. doi: 10.1016/j.neuroscience.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 29.Liu G. Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat Neurosci. 2004;7:373–379. doi: 10.1038/nn1206. [DOI] [PubMed] [Google Scholar]
- 30.Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang S-L, Grady MS, Cohen AS. Regional hippcampal alteration associated with cognitive deficit following experimental brain injury: A systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 31.Fineman I, Giza CC, Nahed BV, Lee SM, Hovda DA. Inhibition of neocortical plasticity during development by a moderate concussive brain injury. J Neuortrauma. 2000;17:739–749. doi: 10.1089/neu.2000.17.739. [DOI] [PubMed] [Google Scholar]
- 32.Raghupahti R, McIntosh TK. Regionally and temporally distinct patterns of induction of c-fos, c-jun, and junB mRNSs following experimental brain injury in the rat. Brain Res Mol Brain Res. 1996;37:134–144. doi: 10.1016/0169-328x(95)00289-5. [DOI] [PubMed] [Google Scholar]
- 33.Soghomonian J-J, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- 34.Giza CC, Maria NS, Hovda DA. N-methyl-D-aspartate receptor subunit changes after traumatic injury to the developing brain. J Neurotrauma. 2006;23:950–961. doi: 10.1089/neu.2006.23.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kennedy JE, Jaffee MS, Leskin GA, Stokes JW, Leal FO, Fitzpatrick PJ. Posttraumatic stress disorder and posttraumatic stress disorder-like symptoms and mild traumatic brain injury. J Rehabil Res Dev. 2007;44:895–920. doi: 10.1682/jrrd.2006.12.0166. [DOI] [PubMed] [Google Scholar]
- 36.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med. 2008;358(5):453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 37.Sbordone R, Liter J. Mild traumatic brain injury does not produce post traumatic stress disorder. Brain Inj. 1995;9:405–412. doi: 10.3109/02699059509005780. [DOI] [PubMed] [Google Scholar]
- 38.Sbordone R, Seyranina GD, Ruff RM. Are the subjective complaints of traumatically brain injured patients reliable? Brain Inj. 1998;12:505–515. doi: 10.1080/026990598122467. [DOI] [PubMed] [Google Scholar]
- 39.Koenigs M, Huey ED, Raymont V, Cheon B, Solomon J, Wasserman EM, Grafman J. Focal brain damage protects against post-traumatic stress disorder in combat veterans. Nat Neurosci. 2008;11(2):232–237. doi: 10.1038/nn2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godsil BP, Spooner JR, Anagnostaras SG, Fanselow MS. Quantification of the unconditional response to footshock in the rat: measurement of the activity burst with the use of computer-based image analysis. Society for Neuroscience. 1997;23:1612. [Abstract] [Google Scholar]
- 41.Foa E, Huppert J, Cahill S. Emotional processing theory: An Update. In: Rothbaum B, editor. Pathological anxiety: emotional processing in etiology and treatment. New York: Guilford Press; 2006. pp. 3–24. [Google Scholar]
- 42.Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol. 2000;109:290–298. [PubMed] [Google Scholar]
- 43.Jacobs NS, Cushman JD, Fanselow MS. The accurate measurement of fear memory in Pavlovian conditioning: Resolving the baseline issue. J Neurosci Methods. 2010;190:235–239. doi: 10.1016/j.jneumeth.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothbaum BO, Kozak MJ, Foa EB, Whitaker DJ. Posttraumatic stress disorder in rape victims: autonomic habituation to auditory stimuli. J Trauma Stress. 2001;14:283–293. doi: 10.1023/A:1011160800958. [DOI] [PubMed] [Google Scholar]
- 45.Hogg S, Moser PC, Sanger DJ. Mild traumatic lesion of the right parietal cortex of the rat: Selective behavioural deficits in the absence of neurological impairment. Behav Brain Res. 1998;93:143–155. doi: 10.1016/s0166-4328(97)00146-0. [DOI] [PubMed] [Google Scholar]
- 46.Fujimoto ST, Longhi L, Saatman KE, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–378. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Säljö A, Svensson B, Mayorga M, Hamberger A, Bolouri H. Low-level blasts raise intracranial pressure and impair cognitive function in rats. J Neurotrauma. 2009;26(8):1345–1352. doi: 10.1089/neu.2008-0856. [DOI] [PubMed] [Google Scholar]
- 48.Schwarzbold ML, Rial D, De Bem T, Machado DG, Cunha MP, dos Santos AA, et al. Effects of traumatic brain injury of different severities on emotional, cognitive, and oxidative stress-related parameters in mice. J Neurotrauma. 2010;27(10):1883–1892. doi: 10.1089/neu.2010.1318. [DOI] [PubMed] [Google Scholar]
- 49.Hogg S, Moser PC, Sanger DJ. Mild traumatic lesion of the right parietal cortex of the rat: Characterisation of a conditioned freezing deficit and its reversal by dizocilpine. Behav Brain Res. 1998;93:157–165. doi: 10.1016/s0166-4328(97)00145-9. [DOI] [PubMed] [Google Scholar]
- 50.Hogg S, Perron C, Barneoud P, Sanger DJ, Moser PC. Neuroprotective effect of eliprodil: Attenuation of a conditioned freezing deficit induced by traumatic injury of the right parietal cortex in the rat. J Neurotrauma. 1998;15(7):545–553. doi: 10.1089/neu.1998.15.545. [DOI] [PubMed] [Google Scholar]
- 51.Abrous DN, Rodriguez J, le Moal M, Moser PA, Barneoud P. Effects of mild traumatic brain injury on immunoreactivity for the inducible transcription factors of c-Fos, c- Jun, JunB, and Krox-24 in cerebral regions associated with conditioned fear responding. Brain Res. 1999;826:181–192. doi: 10.1016/s0006-8993(99)01259-7. [DOI] [PubMed] [Google Scholar]
- 52.Lifshitz J, Witgen BM, Grady MS. Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: evaluation by conditioned fear response. Behav Brain Res. 2007;177(2):347–357. doi: 10.1016/j.bbr.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fanselow MS, Bolles RC. Triggering of the endorphin analgesic reaction by a cue previously associated with shock: reversal by naloxone. Bull Psychon Soc. 1979;14:88–90. [Google Scholar]
- 54.Cordero MI, Merino JJ, Sandi C. Correlational relationship between shock intensity and corticosterone secretion on the establishment and subsequent expression of contextual fear conditioning. Behav Neurosci. 1998;112(4):885–891. doi: 10.1037//0735-7044.112.4.885. [DOI] [PubMed] [Google Scholar]
- 55.Maren S. Overtraining does not mitigate contextual fear conditioning deficits produced by neurotoxic lesions of the basolateral amygdala. J Neurosci. 1998;18(8):3088–3097. doi: 10.1523/JNEUROSCI.18-08-03088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of far memories across the adult lifetime of rats. J Neurosci. 2004;24(15):3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JK, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: A critical review. Neurosci Biobehav Rev. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]