

Abstract
Virchow’s triad is traditionally invoked to explain pathophysiologic mechanisms leading to thrombosis, alleging concerted roles for abnormalities in blood composition, vessel wall components, and blood flow in the development of arterial and venous thrombosis. Given the tissue-specific bleeding observed in hemophilia patients, it may be instructive to consider the principles of Virchow’s triad when investigating mechanisms operant in hemostatic disorders as well. Blood composition (the function of circulating blood cells and plasma proteins) is the most well-studied component of the triad. For example, increased levels of plasma procoagulant proteins such as prothrombin and fibrinogen are established risk factors for thrombosis, whereas deficiencies in plasma factors VIII and IX result in bleeding (hemophilia A and B, respectively). Vessel wall (cellular) components contribute adhesion molecules that recruit circulating leukocytes and platelets to sites of vascular damage, tissue factor, which provides a procoagulant signal of vascular breach, and a surface upon which coagulation complexes are assembled. Blood flow is often characterized by two key variables: shear rate and shear stress. Shear rate affects several aspects of coagulation, including transport rates of platelets and plasma proteins to and from the injury site, platelet activation, and the kinetics of fibrin monomer formation and polymerization. Shear stress modulates adhesion rates of platelets and expression of adhesion molecules and procoagulant activity on endothelial cells lining the blood vessels. That no one abnormality in any component of Virchow’s triad fully predicts coagulopathy a priori suggests coagulopathies are complex, multifactorial and interactive. In this review, we focus on contributions of blood composition, vascular cells, and blood flow to hemostasis and thrombosis, and suggests cross-talk among the three components of Virchow’s triad is necessary for hemostasis and determines propensity for thrombosis or bleeding. Investigative models that permit interplay among these components are necessary to understand the operant pathophysiology, and effectively treat and prevent thrombotic and bleeding disorders.
Introduction
Abnormalities in blood coagulation are the leading cause of death world-wide, with treatment costs estimated at more than $250 billion and projected to more than triple to $818.1billion by 2030.1 In addition to inherited bleeding and thrombotic disorders, therapeutic approaches to manage hemorrhagic and thrombotic episodes are expected to paradoxically increase the incidence of thrombotic and bleeding events, respectively. For example, while the hemostatic agent recombinant factor VIIa is highly effective at minimizing bleeding in hemophilic patients with inhibitors, its off-label use in nonhemophilic patients has been associated with thrombosis.2 Similarly, whereas the recently approved antithrombotic dabigatran exhibits improved safety and efficacy compared to warfarin, there is currently no rapid reversal agent, which may leave patients prone to bleeds.3 Understanding mechanism(s) of blood coagulation and its associated disorders will allow design of targeted, and therefore safer and more effective, therapeutics to treat both bleeding and thrombosis.
Tissue factor (TF), thrombin, and fibrin(ogen) in clot formation and stability
Procoagulant activities have been traditionally separated into extrinsic, intrinsic, and common pathways; however, the acknowledgement that thrombin generation must be localized to a site of injury, as well as complex presentation of certain factor deficiencies, has led to the conceptual integration of these pathways.4 Briefly, coagulation is initiated via extrinsic activity after exposure of cell-derived TF, formation of the factor VIIa/TF complex, and conversion of factor X to factor Xa. Thrombin generation is subsequently augmented by intrinsic (factors XI, IX, and VIII-dependent) activities. Though both the extrinsic (factor VIIa/TF) and intrinsic (factors IXa/VIIIa) tenase complexes produce factor Xa, the relative contributions of these activities are dictated, in part, by the local TF concentration and type of cell surface supporting enzyme/cofactor complex assembly (discussed below). Cellular and plasma-dependent mechanisms culminate in prothrombinase complex (factors Xa, Va, and prothrombin) assembly, and production of the enzyme thrombin. Most current antithrombotic agents target one or more of the active enzymes generated during the clotting cascade, including factors Xa, IXa, and thrombin.
Proteolytic conversion of circulating, soluble fibrinogen to an insoluble fibrin meshwork involves thrombin-mediated cleavage of N-terminal peptides from fibrinogen, end-to-end polymerization of fibrin monomers to protofibrils, and lateral aggregation of protofibrils to fibers. This sequence of events has been extensively studied and reviewed.5–7 Fibrin’s remarkable biophysical characteristics make it extraordinarily suited to provide structural support to the clot; individual fibers can be strained more than 330% without rupturing.8 As such, fibrin is an effective therapeutic target for both minimizing bleeding in hemostatic disorders9, and dissolving intravascular thrombi in myocardial infarction, ischemic stroke, and deep vein thrombosis.10–12
Determinants of fibrin network characteristics have been almost entirely studied in purified systems or in platelet-poor plasma. These studies have shown that the conditions under which fibrinogen is converted to fibrin determine the fiber thickness, branching, and network density of the resulting clot. These conditions include the local pH, ionic strength, and concentrations of calcium, polyphosphate, fibrin(ogen)-binding proteins (e.g. factor XIII), thrombin, and fibrinogen present during fibrin formation.13–19 Of these, the influences of thrombin, fibrinogen and factor XIII on fibrin structure and function have been the best characterized. At a constant fibrinogen concentration, low thrombin concentrations produce coarse, unbranched networks of thick fibrin fibers, whereas high thrombin concentrations produce dense, highly branched networks of thin fibers.15,16 Similarly, at a constant thrombin concentration, increasing the fibrinogen concentration produces denser, highly-branched fibrin networks.16–18 The structural composition of a fibrin clot is important because the structure determines its biochemical and mechanical properties (reviewed in 20). In general, coarse networks have a lower elastic modulus (ability to undergo elastic deformation, “stress/strain”) and increased susceptibility to fibrinolysis, whereas dense networks have a higher elastic modulus and are relatively resistant to fibrinolysis.18,21,22 Factor XIII activity provides additional mechanical and biochemical support for the fibrin network by cross-linking adjacent chains, as well as inhibitors (α2-antiplasmin) to the clot.19,23
Pathophysiologic mechanisms in thrombosis and bleeding
Although the origin is highly controversial24, the concept of concerted pathogenic roles for the triad of abnormalities in blood composition, vessel wall function, and blood flow/shear in venous thrombosis/thromboembolism is typically attributed to Rudolph Virchow in the 1700s.25 Regardless of its controversial origins, the conceptual integration of these functions has considerably advanced understanding of the pathogenesis of thrombosis as well as bleeding (Figure 1). Specific components of Virchow’s triad are illustrated in Figure 2. Abnormal levels of pro- and anticoagulant proteins26–44, thrombin generation39–45, clotting factor activity46 or resistance to inactivation47,48, markers of vascular cell damage or activation49,50, and fibrinolysis inhibitors51,52, have been correlated with venous and/or arterial coagulopathies in case reports and small and large epidemiologic studies. That no one abnormality predicts coagulopathy a priori highlights the complex, interconnected pathways in which these components regulate coagulation.
Figure 1.

Venn diagram illustrating the propensity of thrombosis and bleeding at the intersection of abnormalities in blood composition, vessel wall function, and blood flow/shear.
Figure 2. Schematic showing the elements of Virchow’s triad.

This conceptual model describes the three components (blood flow, blood composition, vascular function) that regulate coagulation. Abbreviations: IX, factor IX; VIII, factor VIII; II, prothrombin; Fgn, fibrinogen; TM, thrombomodulin
Arterial thrombosis is typically associated with atherosclerotic plaque rupture. This pathogenic process results in exposure or release of subendothelial cells and procoagulant material (e.g., TF, collagen) from within the plaque and activation and aggregation of platelets. The growing thrombus increases the degree of stenosis which can result in extremely high shear rates (up to 70,000 s−1) within the stenotic region.53 In some cases, depending on stenosis geometry and location in the vasculature, turbulent flow may develop downstream of the stenosis. Ultimately, platelet accumulation and fibrin deposition produces an occlusive platelet-rich intravascular thrombus (Figure 3).
Figure 3. Interplay between abnormalities in blood components, the vasculature, and blood flow contribute to the development of arterial thrombosis.

Arterial thrombosis involves the formation of platelet-rich "white clots" that form after rupture of atherosclerotic plaques and exposure of procoagulant material such as lipid-rich macrophages (foam cells), collagen, tissue factor and/or endothelial breach, in a high shear environment. Abbreviations: TM, thrombomodulin; II, prothrombin; IIa, thrombin; Fgn, fibrinogen; TF, tissue factor
In contrast, venous thrombosis/thromboembolism is typically associated with plasma hypercoagulability and thought to be triggered by expression of procoagulant activity on intact endothelium from inflammation and/or stasis/reduced blood flow resulting from prolonged immobility. Venous clots have regions or layers showing substantial erythrocyte incorporation (Figure 4).
Figure 4. Interplay between abnormalities in blood components, the vasculature, and blood flow contribute to the development of venous thrombosis.
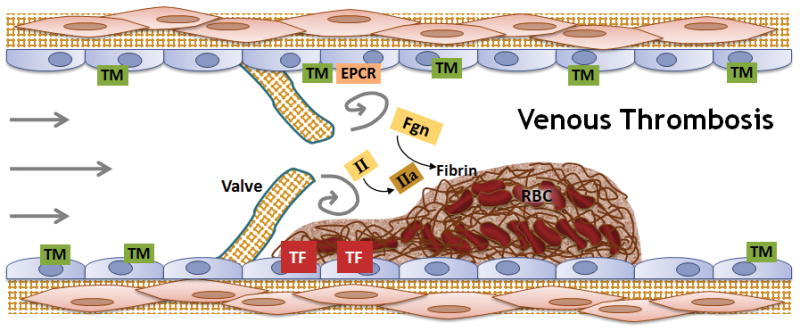
Venous thrombosis involves the formation of fibrin-rich “red clots” that result from exposure of procoagulant activity on intact endothelium plus plasma hypercoagulability, in reduced or static blood flow. Venous thrombi are thought to initiate behind valve pockets, where reduced or static flow decreases wall shear stress that normally regulates endothelial cell phenotype. Abbreviations: TM, thrombomodulin; EPCR, endothelial protein C receptor; II, prothrombin; IIa, thrombin; TF, tissue factor; Fgn, fibrinogen; RBC, red blood cells
Given the tissue-specific bleeding (predominantly joints and muscles) observed in hemophilia patients54–56, it may also be instructive to consider the principles of Virchow’s triad when interrogating mechanisms operant in hemostatic disorders. Hemostasis is the response to bleeding resulting from transection of the full thickness of the vessel wall and extravasation of blood into the extravascular space. Hemophiliacs rarely bleed into tissues with high subendothelial/extravascular TF expression, suggesting relative contributions from blood composition (levels of circulating coagulation factors) and tissue-specific coagulant activity (e.g., local TF or thrombomodulin expression) dictate hemostatic potential at an injury site.
Predicting thrombotic or hemorrhagic events and devising targeted approaches for minimizing these events requires thorough consideration of mechanisms that promote these pathologies. This review focuses on the procoagulant contributions of blood composition, vasculature, and flow/shear to hemostasis and thrombosis.
Contributions of plasma procoagulant activity in thrombosis and bleeding
Likely due to the ease of obtaining blood for ex vivo experiments, blood composition (circulating blood cells and plasma proteins) is the best-characterized facet of Virchow’s triad. Techniques to evaluate the isolated effects of abnormal plasma clotting factor levels in purified assays have given way to technologies that measure the global hemostatic potential of blood and plasma. These newer techniques permit translation of changes in individual factor levels to net changes in thrombin generation and clot formation and stability.
Deficiencies in either factor VIII or IX (hemophilia A or B, respectively) are well-characterized hypocoagulabilities in which patients produce a primary platelet plug in response to vascular injury, but typically re-bleed hours to days after injury. In vitro studies show hemophilic conditions result in reduced thrombin generation, with a prolonged onset and lower thrombin generation rate and peak level.39,41–43 This abnormal thrombin generation pattern causes deficiencies in fibrin production, fibrin network density and permeability, and susceptibility of the fibrin network to fibrinolysis.38–42,57–59 Both replacement and bypassing strategies used to treat and prevent bleeding in hemophilia improve thrombin generation variables and accelerate and stabilize the resulting fibrin network.40,42,58,60
The role(s) of elevated plasma factor levels (hypercoagulabilities) on clotting are less well understood. Elevated prothrombin levels, typically associated with a glycine to arginine mutation at residue 20210 (G20210A), increase risk of venous thrombosis.26 The mechanism for this correlation is currently unclear. Patients with the G20210A mutation do not have elevated levels of the prothrombin cleavage product fragment 1.2, suggesting coagulation is not activated at baseline.61 However, in vitro assays with reconstituted systems and patient plasma suggest once coagulation is triggered, hyperprothrombinemia increases the rate and peak of thrombin generation.61–65 This apparent paradox may be reconciled by the hypothesis that low levels of thrombin activation are functionally anticoagulant because thrombin in complex with thrombomodulin activates negative feedback pathways to limit coagulation, whereas the in vitro assays are triggered with higher TF concentrations that overwhelm anticoagulant mechanisms and promote higher procoagulant activity. In the in vitro assays, increased thrombin generation results in the formation of fibrin clots composed of a fine network of thin fibrin fibers63 and increased activation of the thrombin-activable fibrinolysis inhibitor (TAFI)66, mechanisms that have both been correlated with increased thrombosis risk. Thus, although still to be demonstrated in vivo, these data suggest a mechanism correlating elevated prothrombin, thrombin generation, and clot stability.
Abnormalities in fibrin(ogen) synthesis, formation and function are implicated in both bleeding and thrombotic disorders. More than 500 fibrinogen gene mutations have been identified that result in expression of low (afibrinogenemia) or abnormally functioning (dysfibrinogenemia) fibrinogen chains. In addition, posttranslational modification (e.g., oxidation, nitration, homocysteinylation, and glycation) of circulating fibrinogen has been implicated as an “acquired dysfibrinogenemia” in prothrombotic disorders, including acute coronary syndrome, diabetes, hyperhomocysteinemia, and thrombosis associated with acute cigarette smoke exposure, presumably because it alters fibrinogen cleavage, incorporation of fibrin into the fibrin network, and/or fibrin network stability67. Although an elevated fibrinogen level (hyperfibrinogenemia) is a well-established risk factor for both arterial and venous thrombosis29–35,49,68, its etiologic role has been controversial. Transgenic mice that over-express murine fibrinogen show elevated fibrin degradation products (D-dimer) and spontaneous fibrin deposition in the spleen, but only marginal shortening of the time to 75% occlusion after 20% ferric chloride (FeCl3) application to the carotid artery.69–71 Using a different hyperfibrinogenemia model in which wild type mice were infused with human fibrinogen, we showed that hyperfibrinogenemia shortens the time to occlusion after application of 10% and 5% FeCl3 to the carotid artery and saphenous vein, respectively.18 Furthermore, hyperfibrinogenemia increases thrombus fibrin content, promotes faster fibrin formation, and increases fibrin network density, strength, and stability. Importantly, hyperfibrinogenemia increases resistance to pharmacologically induced thrombolysis in vivo. These data demonstrate a direct, etiologic link between hyperfibrinogenemia, thrombosis, and thrombolysis in acute settings.18
Contributions of cellular procoagulant activity in thrombosis and bleeding
Vascular cells contribute at least two essential functions during coagulation. First, cellular adhesion molecules including P-selectin glycoprotein ligand-1 (PSGL-1) exposed during vascular injury recruit leukocytes and platelets to sites of vascular damage72. Second, accumulation of these cells, as well as exposure of active cellular TF (normally sequestered from blood in healthy humans) provides a procoagulant signal of vascular breach and a surface upon which to assemble procoagulant complexes. Evidence suggests extravascular TF exists in complex with factor VIIa73, such that vascular injury exposes the fully formed factor VIIa/TF complex, enabling immediate activation of factor X to factor Xa. Subsequent assembly of the prothrombinase complex (factors Xa, Va, and prothrombin) takes place on the negatively charged lipid (phosphatidylserine) surface provided by TF-bearing cells and activated platelets. Cells also provide a site for factor XI(a) binding and activation of factor IX, as well as assembly of the intrinsic tenase complex (factors IXa, VIIIa, and X) to augment thrombin generation.
We and others have directly compared the procoagulant activity of intravascular and extravascular cells and their ability to support fibrin formation. Whereas quiescent intravascular cells have little to no TF activity, extravascular cells (e.g., fibroblasts and smooth muscle cells) express considerable TF and are primed to trigger coagulation upon contact with blood. Because thrombin dictates fibrin network formation and network properties, the highly procoagulant extravascular cells support the rapid production of a dense fibrin network that is relatively resistant to fibrinolysis.74–76 Such networks stabilize the primary platelet plug at a site of vascular injury. The relative ability of extravascular cells to trigger clotting in blood escaping from a ruptured vessel can be implicated in tissue-specific hemostasis. Vascular breach into a site of high TF activity would not require an intact intrinsic pathway; thrombin generation could take place entirely via the TF-driven extrinsic pathway. In contrast, vessel disruption into tissues with lower extravascular TF expression would necessitate intrinsic pathway activity to augment thrombin generation and promote stable fibrin formation. For example, hemophilia patients frequently bleed into muscles and joints. Although this presentation has been related to physical stress placed on these tissues54–56, hemophilia patients do not typically bleed into other tissues that endure high physical activity such as the heart. These observations suggest that attributing bleeding solely to physical stress oversimplifies the pathology. Cardiomyocytes express high levels of TF, which can promote substantial thrombin generation independently of intrinsic factors VIII and IX, suggesting tissue-specific extrinsic activity can compensate for reduced intrinsic activity. This scenario illustrates the required integration of two components of Virchow’s triad, blood and tissues, in maintaining hemostasis.
Extravascular TF expression/activity is implicated in arterial thrombosis resulting from atherosclerotic plaque rupture. The amount of TF found in atherosclerotic plaques in humans is positively correlated with the thrombogenicity of the lesions after plaquerupture.77 The importance of TF in the progression of arterial thrombosis is underscored by observations that treatment of human plaques with TF pathway inhibitor significantly reduces adhesion of both fibrinogen and platelets to the plaque, indicating the thrombotic potential of atherosclerotic lesions may be decreased by inhibiting TF-dependent procoagulant activity.78
Inappropriate expression of intravascular procoagulant activity is hypothesized to trigger venous thromboembolism (VTE). Proinflammatory mediators including bacterial lipopolysaccharide, and the cytokines interleukin-1β and tumor necrosis factor-alpha (TNF-α), induce procoagulant activity (thrombin generation) in cultured endothelial cells via de novo TF synthesis and decreased thrombomodulin expression in a time- and dose-dependent manner.18,76,79,80 TNF-α-stimulated, cultured endothelial cells support robust fibrin formation.76 Laser-induced injury of cultured endothelial cells has also been shown to induce procoagulant activity and fibrin formation, although the time course of those experiments suggests TF activity is released from intracellular stores and is not synthesized de novo.81 In vivo, endothelial cells residing in different vascular beds (e.g., aorta versus pulmonary microcirculation) have different phenotypes. Even within a given vessel, endothelial cells vary in their expression of coagulation proteins. Brooks et al. elegantly showed increased expression of thrombomodulin and endothelial cell protein C receptor, and decreased expression of von Willebrand factor in the valve sinus relative to the immediately adjacent vein lumen in saphenous veins harvested during cardiac bypass surgery.82 Although these studies convincingly show vascular bed-specific expression of anticoagulant proteins on endothelial cells, evidence for endothelial cell TF expression in vivo remains controversial.
Vascular bed-specific function is best illustrated clinically by the specific presentations of thrombosis in either arterial or venous circulation, but rarely both. Additionally, risk factors strongly associated with arterial thrombosis (e.g., hypertension, diabetes mellitus, smoking, hypercholesterolemia) only modestly increase risk of venous thrombosis/thromboembolism83, and meta-analysis suggests history of unprovoked VTE only slightly increases (~1.5-fold) risk of arterial cardiovascular events over long-term followup.84 Because any abnormality in the blood is present systemically, these events reflect local abnormalities in vascular bed-specific pro- or anticoagulant activity and/or blood flow (discussed below). Vascular bed-specific activities are further illustrated by animal models of coagulation dysfunction that show bleeding and/or fibrin deposition in specific tissues. For example, mice with partial TFPI deficiency and decreased thrombomodulin function demonstrate fibrin deposition in the brain and liver, but not other tissues.85 These observations suggest unique regulatory mechanisms are present in specific tissues.
Neither “cell” nor “plasma protein,” cell-derived microparticles express cell-specific markers and retain procoagulant properties derived from their parent cell86,87, and can circulate and evade normal spatial restrictions on cellular procoagulant activity75. Although circulating microparticles derived from leukocytes, platelets, erythrocytes, endothelial cells, megakaryocytes, and tumors have been identified in numerous studies (Figure 5), their precise role(s) in hemostasis and thrombosis is poorly understood. Scott syndrome patients have a defect that impairs the translocation of phosphatidylserine from the inner to the outer leaflet of the membrane of erythrocytes and platelets. Although the bleeding tendency of these patients has been primarily attributed to decreased platelet procoagulant activity, platelets from Scott syndrome patients also demonstrate defective generation of phosphatidylserine-positive microparticles in vitro, suggesting these microparticles are necessary for hemostasis.88,89 However, it is difficult to separate the role(s) of reduced expression of phosphatidylserine on platelets from reduced production of phosphatidylserine-bearing microparticles. Healthy humans have few circulating leukocyte-derived microparticles; however, the numbers of leukocyte-derived microparticles increase in certain diseases, including cancer90,91, diabetes92, and sickle cell disease93. Microparticles derived directly from tumors have also been specifically associated with VTE in cancer patients, and are thought to contribute to the thrombotic presentation.94,95 Expression of procoagulant activity on microparticles (TF and/or phosphatidylserine) has been implicated in microparticle function in vitro and in vivo.96–99 One prospective study demonstrated a sharp increase in microparticle TF activity immediately before venous thrombosis in two patients91, suggesting a causative role of microparticles in the thrombotic event. Murine studies support this observation; human monocyte-derived microparticles promote thrombus formation100,101 and increase thrombus weight98 in intravascular thrombosis models. Additional theories regarding the mechanistic role(s) of microparticles in thrombosis invoke their expression of cellular adhesion molecules and ability to activate endothelial cells102 and leukocytes.
Figure 5.

Circulating microparticles are derived from a variety of cell types including leukocytes, platelets, megakaryocytes, red blood cells, endothelial cells, and tumors. Microparticles (MP) carry cell-specific markers and functional properties of their parent cell.
Contributions of blood flow in thrombosis and bleeding
The effects of blood flow on clotting are perhaps the least interrogated aspect of Virchow’s triad. Blood flow in the vascular system is typically characterized by two key variables: shear stress and shear rate. In laminar flow (fluid that flows in concentric, parallel layers), shear stress (τ) is the force per unit area applied between adjacent layers of fluid, and shear rate (γ, units of inverse time) is the relative velocity gradient between adjacent layers of fluid. For a Newtonian fluid, such as water or plasma, shear stress is the product of fluid viscosity and shear rate. The term “shear stress” when referring to blood flow typically means wall shear stress, which is the tangential force that the fluid imposes directly on the endothelium. For laminar flow in a straight vessel, wall shear stress is directly proportional to the volumetric flow rate (volume per time) and inversely proportional to the third power of the inner vessel diameter. In much of the arterial system, blood vessels adapt their caliber via endothelium-mediated regulation to maintain a mean wall shear stress of approximately 5–20 dynes/cm2.103,104 This maintenance is thought to be crucial to normal vascular function, including the promotion of antiinflammatory, antithrombotic, anticoagulant, profibrinolytic and antihypertrophic states.104,105
Under venous conditions, static or severely reduced flow (“stasis”) leads to decreased wall shear stress. Stasis induced by immobility (hospitalization or long-haul air travel) or trauma is associated with venous thrombosis risk, perhaps by modulating the endothelial cell phenotype. The shear stress-responsive Krüppel-like transcription factors (KLF) have been implicated in endothelial cell function.106–108 Shear stress upregulates KLF expression, inducing expression of antiinflammatory and antithrombotic proteins, including thrombomodulin; whereas, stasis- or proinflammatory cytokine-induced loss of KLF expression leads to enhanced expression of vascular cell adhesion molecule-1 and TF.109,110 As such, KLF is an intriguing target for novel antithrombotics aimed at combating stasis-induced thrombosis.
Vascular endothelial cells are also subjected to pressure, a force per area that acts perpendicularly to the vessel wall and modulates endothelial function. Hypertension increases pressure and mechanical damage to arteries, increasing circulating levels of both endothelial- and platelet-derived microparticles.111 Hypertension may also increase TF exposure in vivo; application of pressure to cultured endothelial cells increases TF activity112 and release of endothelin-1113.
Flow conditions near a vessel wall are best characterized by wall shear rate. Wall shear rate describes how fast solutes and platelets are delivered within a short, radial distance from the vessel wall, but also the rate at which they are carried downstream as they diffuse away from the wall. Model systems have shown that local flow conditions strongly regulate the delivery of both platelets to a subendothelial surface, and factor X to a wall containing the factor VIIa/TF complex.114 Typical arterial and venous shear rates are 500–1500 s−1 and 10–100 s−1, respectively. However, the shear rate and shear stress within a stenotic region significantly increase as the stenosis grows; shear rates may reach 70,000 s−1 to 250,000 s−1 in severely stenotic vessels.53,115 Depending on stenosis geometry and location in the vasculature, a recirculation zone (illustrated in Figure 3) may form immediately downstream from the stenotic region. Recirculation zones are often characterized by low shear conditions and long residence times for materials contained within them.116 Notably, areas of disturbed flow such as branch points and curvatures, especially where wall shear stress is low or oscillatory, correlate with regions of atherosclerotic lesion initiation and stenosis.104,117
Shear-dependent expression of platelet P-selectin and formation of monocyte-platelet aggregates suggest that both platelets and leukocytes are activated when they traverse stenotic regions (estimated peak wall shear stress of 2.64 to 281.5 Pa, correlated with stenosis severity), priming these cells for deposition on dysfunctioning or ruptured vasculature and/or contribution to thrombotic events if they become entrapped in the recirculation zone.116,118 Moreover, soluble agonist-independent accumulation of platelets at a stenotic region is attributed to flow (rheology)-dependent aggregation.119 As such, flow-dependent changes induced by the stenosis, itself, further increase the risk of thrombosis upon plaque rupture.
The shear rate also dramatically modulates the kinetics of both fibrin monomer formation and polymerization.120,121 At a constant, given shear rate, initiation of clotting is a function of the size of the exposed region of TF.120 Once initiated, the shear rate dictates both influx of procoagulant reactants including prothrombin and fibrinogen, and efflux of reaction products including thrombin and fibrin. In in vitro assays, increasing shear from 10 to 100 s−1 depletes local fibrin monomer concentrations, limiting lateral aggregation and protofibril extension. However, decreasing the shear rate or increasing thrombin generation at a given shear rate increases local monomer concentrations, permitting protofibril and subsequent fibrin fiber formation.121 Although fibrin networks formed under static (no flow) conditions show an isotropic distribution of fibers with relatively uniform diameters, fibrin produced in flowing blood is oriented along flow vectors121–123, with a prominent network of thick fibers as well as fiber “aggregates” in which multiple individual fibers are coalesced into bundles.123 Because the fibrin network’s ability to withstand both biochemical dissolution and mechanical disruption is a function of its fiber thickness and branching20, these studies indicate networks produced under flow would demonstrate significantly different stability than isotropic networks formed under stasis.123 In fact, using mechanical stress to recapitulate the effects of blood flow on fiber orientation, a recent study found that stretched fibrin networks are resistant to fibrinolysis124, underscoring the importance of the effects of blood flow/shear rate on thrombus formation, structure and function.
Multiple, inter-related “hits” in thrombosis and bleeding
The proposition that multiple “hits” culminate in abnormal coagulation suggests simultaneous consideration of soluble, cellular, and mechanical biomarkers is required to determine clinical risk of thrombosis and bleeding. Indeed, concerted dysfunction in the blood, vessel wall, and blood flow may increase clinical risk via additive or even synergistic mechanisms. Two recent studies have highlighted the need to understand the extent of vessel wall injury when interpreting thrombotic risk related to platelet and plasma protein functions. Hechler et al.125 addressed the controversy regarding the role of GPVI-collagen interactions in thrombosis by showing that thrombi resulting from superficial laser injury depended on platelet GPVI but not procoagulant activity, whereas thrombosis after deeper laser injury that both denuded the endothelium and exposed the adventitia was less dependent on GPVI, but instead could be blocked by hirudin. We have shown that the procoagulant/prothrombotic effects of elevated plasma factor VIII levels can be observed after mild vascular injury (short exposure to FeCl3), but not severe injury to the carotid artery.126 These studies demonstrate the interdependent relationship between plasma procoagulant activity and vascular function.
Epidemiologic studies in humans support these observations. For example, the prognostic importance of hyperfibrinogenemia appears to be independent of, but additive to, myocardial damage (assessed by troponin-T levels) in patients with unstable coronary artery disease.35 However, few diagnostic algorithms simultaneously consider markers of vascular damage and plasma hypercoagulability when determining thrombosis risk. Acevedo et al.30 showed that elevated levels of both homocysteine (a trigger of endothelial damage) and fibrinogen contributed to an increased hazard ratio (HR) of 3.29 for 3-year mortality, more than the HR for either homocysteine (HR=2.14) or fibrinogen (HR=2.28), alone. This observation supports the development and use of diagnostic algorithms that incorporate multiple biomarkers that indicate not only blood composition, but also vascular function, when determining clinical interventions and duration of anticoagulant therapy.
Clearly then, delineating the operant pathophysiologic mechanisms necessitates understanding the substantial cross-talk and feedback pathways that connect the aspects of Virchow’s triad. However, a significant impediment to these investigations is the difficulty in simulating and collecting data on multiple biochemical, cellular and mechanical processes simultaneously. Mathematical and computational modeling is being used to circumvent this limitation. Indeed, models of coagulation kinetics have been instrumental in delineating TF regulatory mechanisms127,128, sensitivity of this pathway to initial TF concentration129, and threshold behavior of thrombin production and the importance of binding site density on the reactive surface of platelets.130,131 Fewer models have incorporated the effects of flow in coagulation, and only a subset have generated hypotheses that were subsequently tested and validated experimentally132–134. Of note, theoretical work by Kuharsky and Fogelson130 led to the hypothesis that platelets physically inhibit subendothelial activity. This hypothesis was later given experimental support135 and incorporated into the biological literature136,137.
Two models aimed to capture interplay between the biochemistry, blood cells and fluid environment and predict clot behavior in multiple parameter regimes. Xu et al.138 developed a computational model that invoked the cellular potts model to represent platelets in flow, which allowed for tracking of individual discrete platelet behaviors. Coagulation biochemistry was based on the system described by Jones and Mann128 and did not include surface-dependent reactions. The model predicted that red blood cell entrapment within the growing platelet thrombus led to thrombus heterogeneity and structural instability. Leiderman and Fogelson139 developed a model that included detailed biochemistry, including surface-dependent reactions, chemical activation and deposition of platelets. The model tracked platelet concentrations rather than discrete objects and treated the growing platelet mass as a porous material. This feature allowed analysis of fluid and solute transport within the growing mass, and showed that diffusive transport within the mass promotes its upstream growth. Furthermore, this model showed thrombus growth was strongly influenced by wall shear rate and the near-wall excess of platelets. Although both models substantially advance simulation of the complex and coupled blood clotting system under flow, the predictions made by these models remain to be tested in biological assays. Moreover, all of these models still lack features known to affect coagulation including effects of platelet-platelet cohesion and clot dissolution. However, improved computing power, continued refinement of computational models and incorporation of new experimental data into the models are expected to identify novel therapeutic targets for the treatment of thrombosis and bleeding.
Conclusions
Understanding the interplay between the components of Virchow’s triad is necessary to effectively diagnose and treat bleeding and thrombotic disorders. Increasing awareness of the complexities of these presentations, along with increasingly sophisticated technologies to analyze soluble, cellular, and physical dysfunctions in concert will shed new light on these pathologies and identify novel therapeutic targets. With such targets, we may achieve the long-sought, but thus far elusive, goal for coagulation treatment: safe and effective antithrombotic and hemostatic approaches that are not complicated by bleeding or thrombotic risks.
Acknowledgments
Funding: The authors are supported by funding from the National Institutes of Health (R01HL094740 to ASW, T32ES007017 to MMA), the National Science Foundation (DMS-0943760 to KL), and the American Heart Association (10PRE3720011 to KRM).
Footnotes
The authors declare no conflicts of interest.
Reprints will not be available from the authors.
DISCLOSURES:
Name: Alisa S. Wolberg, PhD, FAHA
Contribution: Alisa S. Wolberg helped write and reviewed the manuscript.
Name: Maria M. Aleman, BS
Contribution: Maria M. Aleman helped write and reviewed the manuscript.
Name: Karin Leiderman, PhD
Contribution: Karin Leiderman helped write and reviewed the manuscript.
Name: Kellie R. Machlus, PhD
Contribution: Kellie R. Machlus helped write and reviewed the manuscript.
This manuscript was handled by: Jerrold H. Levy, MD, FAHA
Contributor Information
Alisa S. Wolberg, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC.
Maria M. Aleman, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC
Karin Leiderman, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC.
Kellie R. Machlus, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC.
References
- 1.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–44. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 2.Yank V, Tuohy CV, Logan AC, Bravata DM, Staudenmayer K, Eisenhut R, Sundaram V, McMahon D, Olkin I, McDonald KM, Owens DK, Stafford RS. Systematic review: benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med. 2011;154:529–40. doi: 10.7326/0003-4819-154-8-201104190-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz NE, Albers GW. Dabigatran challenges warfarin's superiority for stroke prevention in atrial fibrillation. Stroke. 2010;41:1307–9. doi: 10.1161/STROKEAHA.110.584557. [DOI] [PubMed] [Google Scholar]
- 4.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 5.Lord ST. Fibrinogen and fibrin: scaffold proteins in hemostasis. Curr Opin Hematol. 2007;14:236–41. doi: 10.1097/MOH.0b013e3280dce58c. [DOI] [PubMed] [Google Scholar]
- 6.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5 (Suppl 1):116–24. doi: 10.1111/j.1538-7836.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 7.Chernysh IN, Nagaswami C, Weisel JW. Visualization and identification of the structures formed during early stages of fibrin polymerization. Blood. 2011;117:4609–14. doi: 10.1182/blood-2010-07-297671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Jawerth LM, Sparks EA, Falvo MR, Hantgan RR, Superfine R, Lord ST, Guthold M. Fibrin fibers have extraordinary extensibillity and elasticity. Science. 2006;313:634. doi: 10.1126/science.1127317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh K, Shetty S, Jijina F, Mohanty D. Role of epsilon amino caproic acid in the management of haemophilic patients with inhibitors. Haemophilia. 2004;10:58 – 62. doi: 10.1046/j.1351-8216.2003.00839.x. [DOI] [PubMed] [Google Scholar]
- 10.White HD, Rivers JT, Maslowski AH, Ormiston JA, Takayama M, Hart HH, Sharpe DN, Whitlock RM, Norris RM. Effect of intravenous streptokinase as compared with that of tissue plasminogen activator on left ventricular function after first myocardial infarction. N Engl J Med. 1989;320:817–21. doi: 10.1056/NEJM198903303201301. [DOI] [PubMed] [Google Scholar]
- 11.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 12.Popuri RK, Vedantham S. The role of thrombolysis in the clinical management of DVT. Arterio Thromb Vasc Biol. 2011;31:479–84. doi: 10.1161/ATVBAHA.110.213413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair CH, Shah GA, Dhall DP. Effect of temperature, pH and ionic strength and composition on fibrin network structure and its development. Thromb Res. 1986;42:809–16. doi: 10.1016/0049-3848(86)90117-9. [DOI] [PubMed] [Google Scholar]
- 14.Smith SA, Morrissey JH. Polyphosphate enhances fibrin clot structure. Blood. 2008;112:2810–6. doi: 10.1182/blood-2008-03-145755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blomback B, Carlsson K, Fatah K, Hessel B, Procyk R. Fibrin in human plasma: gel architectures governed by rate and nature of fibrinogen activation. Thromb Res. 1994;75:521–38. doi: 10.1016/0049-3848(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 16.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophys J. 1999;77:2813–26. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glover CJ, McIntire LV, Brown CH, 3rd, Natelson EA. Rheological properties of fibrin clots. Effects of fibrinogen concentration, factor XIII deficiency, and factor XIII inhibition. J Lab Clin Med. 1975;86:644–56. [PubMed] [Google Scholar]
- 18.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117:4953–63. doi: 10.1182/blood-2010-11-316885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francis CW, Marder VJ. Increased Resistance to Plasmic Degradation of Fibrin with Highly Crosslinked Alpha-Polymer Chains Formed at High Factor-Xiii Concentrations. Blood. 1988;71:1361–5. [PubMed] [Google Scholar]
- 20.Weisel JW, Litvinov RI. The biochemical and physical process of fibrinolysis and effects of clot structure and stability on the lysis rate. Cardiovasc Hematol Agents Med Chem. 2008;6:161–80. doi: 10.2174/187152508784871963. [DOI] [PubMed] [Google Scholar]
- 21.Collet JP, Park D, Lesty C, Soria J, Soria C, Montalescot G, Weisel JW. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterio Thromb Vasc Biol. 2000;20:1354–61. doi: 10.1161/01.atv.20.5.1354. [DOI] [PubMed] [Google Scholar]
- 22.Carr ME, Jr, Alving BM. Effect of fibrin structure on plasmin-mediated dissolution of plasma clots. Blood Coag Fibrinol. 1995;6:567–73. doi: 10.1097/00001721-199509000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through alpha-antiplasmin cross-linking. Blood. 2011;117:6371–4. doi: 10.1182/blood-2011-02-333203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bagot CN, Arya R. Virchow and his triad: a question of attribution. Br J Haematol. 2008;143:180–90. doi: 10.1111/j.1365-2141.2008.07323.x. [DOI] [PubMed] [Google Scholar]
- 25.Virchow R. Gesammalte abhandlungen zur wissenschaftlichen medtzin. Frankfurt: Medinger Sohn & Co; 1856. pp. 219–732. [Google Scholar]
- 26.Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–703. [PubMed] [Google Scholar]
- 27.Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B, Weltermann A, Speiser W, Lechner K, Eichinger S. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med. 2000;343:457–62. doi: 10.1056/NEJM200008173430702. [DOI] [PubMed] [Google Scholar]
- 28.Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:731–8. doi: 10.1161/01.atv.21.5.731. [DOI] [PubMed] [Google Scholar]
- 29.Wilhelmsen L, Svardsudd K, Korsan-Bengsten K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501–5. doi: 10.1056/NEJM198408233110804. [DOI] [PubMed] [Google Scholar]
- 30.Acevedo M, Pearce GL, Kottke-Marchant K, Sprecher DL. Elevated fibrinogen and homocysteine levels enhance the risk of mortality in patients from a high-risk preventive cardiology clinic. Arterioscler Thromb Vasc Biol. 2002;22:1042–5. doi: 10.1161/01.atv.0000020007.25154.62. [DOI] [PubMed] [Google Scholar]
- 31.Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, Wilson AC, Folsom AR, Wu K, Benderly M, Goldbourt U, Willeit J, Kiechl S, Yarnell JW, Sweetnam PM, Elwood PC, Cushman M, Psaty BM, Tracy RP, Tybjaerg-Hansen A, Haverkate F, de Maat MP, Fowkes FG, Lee AJ, Smith FB, Salomaa V, Harald K, Rasi R, Vahtera E, Jousilahti P, Pekkanen J, D'Agostino R, Kannel WB, Wilson PW, Tofler G, Arocha-Pinango CL, Rodriguez-Larralde A, Nagy E, Mijares M, Espinosa R, Rodriquez-Roa E, Ryder E, Diez-Ewald MP, Campos G, Fernandez V, Torres E, Marchioli R, Valagussa F, Rosengren A, Wilhelmsen L, Lappas G, Eriksson H, Cremer P, Nagel D, Curb JD, Rodriguez B, Yano K, Salonen JT, Nyyssonen K, Tuomainen TP, Hedblad B, Lind P, Loewel H, Koenig W, Meade TW, Cooper JA, De Stavola B, Knottenbelt C, Miller GJ, Bauer KA, Rosenberg RD, Sato S, Kitamura A, Naito Y, Palosuo T, Ducimetiere P, Amouyel P, Arveiler D, Evans AE, Ferrieres J, Juhan-Vague I, Bingham A, Schulte H, Assmann G, Cantin B, Lamarche B, Despres JP, Dagenais GR, Tunstall-Pedoe H, Woodward M, Ben-Shlomo Y, Davey Smith G, Palmieri V, Yeh JL, Rudnicka A, Ridker P, Rodeghiero F, Tosetto A, Shepherd J, Ford I, Robertson M, Brunner E, Shipley M, Feskens EJ, Kromhout D, Dickinson A, Ireland B, Juzwishin K, Kaptoge S, Memon A, Sarwar N, Walker M, Wheeler J, White I, Wood A. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 32.van Hylckama Vlieg A, Rosendaal FR. High levels of fibrinogen are associated with the risk of deep venous thrombosis mainly in the elderly. J Thromb Haemost. 2003;1:2677–8. doi: 10.1111/j.1538-7836.2003.0543b.x. [DOI] [PubMed] [Google Scholar]
- 33.Yarnell JW, Baker IA, Sweetnam PM, Bainton D, O'Brien JR, Whitehead PJ, Elwood PC. Fibrinogen, viscosity, and white blood cell count are major risk factors for ischemic heart disease. The Caerphilly and Speedwell collaborative heart disease studies. Circulation. 1991;83:836–44. doi: 10.1161/01.cir.83.3.836. [DOI] [PubMed] [Google Scholar]
- 34.Kannel WB, Wolf PA, Castelli WP, D'Agostino RB. Fibrinogen and risk of cardiovascular disease. The Framingham Study. JAMA. 1987;258:1183–6. [PubMed] [Google Scholar]
- 35.Toss H, Lindahl B, Siegbahn A, Wallentin L. Prognostic influence of increased fibrinogen and C-reactive protein levels in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. Circulation. 1997;96:4204–10. doi: 10.1161/01.cir.96.12.4204. [DOI] [PubMed] [Google Scholar]
- 36.Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–3. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pabinger I, Schneider B. Thrombotic risk in hereditary antithrombin III, protein C, or protein S deficiency. A cooperative, retrospective study. Gesellschaft fur Thrombose- und Hamostaseforschung (GTH) Study Group on Natural Inhibitors. Arterioscler Thromb Vasc Biol. 1996;16:742–8. doi: 10.1161/01.atv.16.6.742. [DOI] [PubMed] [Google Scholar]
- 38.Biggs R, Douglas AS, Macfarlane RG, Dacie JV, Pitney WR, Merskey Christmas disease: a condition previously mistaken for haemophilia. Br Med J. 1952;2:1378–82. doi: 10.1136/bmj.2.4799.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macfarlane RG, Biggs R. A thrombin generation test; the application in haemophilia and thrombocytopenia. J Clin Pathol. 1953;6:3–8. doi: 10.1136/jcp.6.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen GA, Persson E, Campbell RA, Ezban M, Hedner U, Wolberg AS. A variant of recombinant factor VIIa with enhanced procoagulant and antifibrinolytic activities in an in vitro model of hemophilia. Arterioscler Thromb Vasc Biol. 2007;27:683–9. doi: 10.1161/01.ATV.0000257204.82396.2b. [DOI] [PubMed] [Google Scholar]
- 41.Butenas S, van 't Veer C, Cawthern K, Brummel KE, Mann KG. Models of blood coagulation. Blood Coag Fibrinol. 2000;11 (Suppl 1):S9–13. doi: 10.1097/00001721-200004001-00003. [DOI] [PubMed] [Google Scholar]
- 42.Wolberg AS, Allen GA, Monroe DM, Hedner U, Roberts HR, Hoffman M. High dose factor VIIa enhances clot stability in a model of hemophilia B. Brit J Haematol. 2005;131:645–55. doi: 10.1111/j.1365-2141.2005.05820.x. [DOI] [PubMed] [Google Scholar]
- 43.Dargaud Y, Beguin S, Lienhart A, Al Dieri R, Trzeciak C, Bordet JC, Hemker HC, Negrier C. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost. 2005;93:475–80. doi: 10.1160/TH04-10-0706. [DOI] [PubMed] [Google Scholar]
- 44.Al Dieri R, Peyvandi F, Santagostino E, Giansily M, Mannucci PM, Schved JF, Beguin S, Hemker HC. The thrombogram in rare inherited coagulation disorders: its relation to clinical bleeding. Thromb Haemost. 2002;88:576–82. [PubMed] [Google Scholar]
- 45.Hron G, Kollars M, Binder BR, Eichinger S, Kyrle PA. Identification of patients at low risk for recurrent venous thromboembolism by measuring thrombin generation. JAMA. 2006;296:397–402. doi: 10.1001/jama.296.4.397. [DOI] [PubMed] [Google Scholar]
- 46.Simioni P, Tormene D, Tognin G, Gavasso S, Bulato C, Iacobelli NP, Finn JD, Spiezia L, Radu C, Arruda VR. X-linked thrombophilia with a mutant factor IX (factor IX Padua) N Engl J Med. 2009;361:1671–5. doi: 10.1056/NEJMoa0904377. [DOI] [PubMed] [Google Scholar]
- 47.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–7. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 48.Zoller B, Svensson PJ, He X, Dahlback B. Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest. 1994;94:2521–4. doi: 10.1172/JCI117623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N Engl J Med. 2000;343:1139–47. doi: 10.1056/NEJM200010193431602. [DOI] [PubMed] [Google Scholar]
- 50.Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, Horstman LL, Soriano AO, Zambrano JP, Ahn YS. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. 2005;45:1467–71. doi: 10.1016/j.jacc.2004.12.075. [DOI] [PubMed] [Google Scholar]
- 51.Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, Rosendaal FR. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. 2010;116:113–21. doi: 10.1182/blood-2010-02-267740. [DOI] [PubMed] [Google Scholar]
- 52.Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90:204–8. [PubMed] [Google Scholar]
- 53.Bark DL, Jr, Ku DN. Wall shear over high degree stenoses pertinent to atherothrombosis. J Biomech. 2010;43:2970–7. doi: 10.1016/j.jbiomech.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 54.Roosendaal G, Lafeber FP. Blood-induced joint damage in hemophilia. Semin Thromb Hemost. 2003;29:37–42. doi: 10.1055/s-2003-37938. [DOI] [PubMed] [Google Scholar]
- 55.Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801–9. doi: 10.1016/S0140-6736(03)13405-8. [DOI] [PubMed] [Google Scholar]
- 56.Soucie JM, Cianfrini C, Janco RL, Kulkarni R, Hambleton J, Evatt B, Forsyth A, Geraghty S, Hoots K, Abshire T, Curtis R, Forsberg A, Huszti H, Wagner M, White GC., 2nd Joint range-of-motion limitations among young males with hemophilia: prevalence and risk factors. Blood. 2004;103:2467–73. doi: 10.1182/blood-2003-05-1457. [DOI] [PubMed] [Google Scholar]
- 57.Bettigole RE, Hampton JW, Bird RM. Abnormal plasma clots in hemophilia. Thromb Diath Haemorrh. 1964;12:331–7. [PubMed] [Google Scholar]
- 58.He S, Blomback M, Jacobsson Ekman G, Hedner U. The role of recombinant factor VIIa (FVIIa) in fibrin structure in the absence of FVIII/FIX. J Thromb Haemost. 2003;1:1215–9. doi: 10.1046/j.1538-7836.2003.00242.x. [DOI] [PubMed] [Google Scholar]
- 59.Brummel-Ziedins KE, Branda RF, Butenas S, Mann KG. Discordant fibrin formation in hemophilia. J Thromb Haemost. 2009;7:825–32. doi: 10.1111/j.1538-7836.2009.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sorensen B, Persson E, Ingerslev J. Factor VIIa analogue (V158D/E296V/M298Q-FVIIa) normalises clot formation in whole blood from patients with severe haemophilia A. Br J Haematol. 2007;137:158–65. doi: 10.1111/j.1365-2141.2007.06534.x. [DOI] [PubMed] [Google Scholar]
- 61.Kyrle PA, Mannhalter C, Beguin S, Stumpflen A, Hirschl M, Weltermann A, Stain M, Brenner B, Speiser W, Pabinger I, Lechner K, Eichinger S. Clinical studies and thrombin generation in patients homozygous or heterozygous for the G20210A mutation in the prothrombin gene. Arterio Thromb Vasc Biol. 1998;18:1287–91. doi: 10.1161/01.atv.18.8.1287. [DOI] [PubMed] [Google Scholar]
- 62.Butenas S, van't Veer C, Mann KG. "Normal" thrombin generation. Blood. 1999;94:2169–78. [PubMed] [Google Scholar]
- 63.Wolberg AS, Monroe DM, Roberts HR, Hoffman M. Elevated prothrombin results in clots with an altered fiber structure: a possible mechanism of the increased thrombotic risk. Blood. 2003;101:3008–13. doi: 10.1182/blood-2002-08-2527. [DOI] [PubMed] [Google Scholar]
- 64.Allen GA, Wolberg AS, Oliver JA, Hoffman M, Roberts HR, Monroe DM. Impact of procoagulant concentration on rate, peak and total thrombin generation in a model system. J Thromb Haemost. 2004;2:402–13. doi: 10.1111/j.1538-7933.2003.00617.x. [DOI] [PubMed] [Google Scholar]
- 65.Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effect of tissue factor, thrombomodulin, and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thromb Haemost. 2009;102:936–44. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colucci M, Binetti BM, Tripodi A, Chantarangkul V, Semeraro N. Hyperprothrombinemia associated with prothrombin G20210A mutation inhibits plasma fibrinolysis through a TAFI-mediated mechanism. Blood. 2004;103:2157–61. doi: 10.1182/blood-2003-06-2169. [DOI] [PubMed] [Google Scholar]
- 67.Hoffman M. Alterations of fibrinogen structure in human disease. Cardiovasc Hematol Agents Med Chem. 2008;6:206–11. doi: 10.2174/187152508784871981. [DOI] [PubMed] [Google Scholar]
- 68.Kamphuisen PW, Eikenboom JC, Vos HL, Pablo R, Sturk A, Bertina RM, Rosendaal FR. Increased levels of factor VIII and fibrinogen in patients with venous thrombosis are not caused by acute phase reactions. Thromb Haemost. 1999;81:680–3. [PubMed] [Google Scholar]
- 69.Gulledge AA, Rezaee F, Verheijen JH, Lord ST. A novel transgenic mouse model of hyperfibrinogenemia. Thromb Haemost. 2001;86:511–6. [PubMed] [Google Scholar]
- 70.Gulledge AA, McShea C, Schwartz T, Koch G, Lord ST. Effects of hyperfibrinogenemia on vasculature of C57BL/6 mice with and without atherogenic diet. Arterioscler Thromb Vasc Biol. 2003;23:130–5. doi: 10.1161/01.atv.0000041037.06509.c2. [DOI] [PubMed] [Google Scholar]
- 71.Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S, Weiler H. Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood. 2004;103:1728–34. doi: 10.1182/blood-2003-08-2886. [DOI] [PubMed] [Google Scholar]
- 72.Vandendries ER, Furie BC, Furie B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost. 2004;92:459–66. doi: 10.1160/TH04-05-0306. [DOI] [PubMed] [Google Scholar]
- 73.Hoffman M, Colina CM, McDonald AG, Arepally GM, Pedersen L, Monroe DM. Tissue factor around dermal vessels has bound factor VII in the absence of injury. J Thromb Haemost. 2007;5:1403–8. doi: 10.1111/j.1538-7836.2007.02576.x. [DOI] [PubMed] [Google Scholar]
- 74.Ovanesov MV, Ananyeva NM, Panteleev MA, Ataullakhanov FI, Saenko EL. Initiation and propagation of coagulation from tissue factor-bearing cell monolayers to plasma: initiator cells do not regulate spatial growth rate. J Thromb Haemost. 2005;3:321–31. doi: 10.1111/j.1538-7836.2005.01128.x. [DOI] [PubMed] [Google Scholar]
- 75.Campbell RA, Overmyer KA, Bagnell CR, Wolberg AS. Cellular procoagulant activities dictate clot structure and stability as a function of distance from the cell surface. Arterio Thromb Vasc Biol. 2008;28:2247–54. doi: 10.1161/ATVBAHA.108.176008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114:4886–96. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toschi V, Gallo R, Lettino M, Fallon JT, Gertz SD, Fernandez-Ortiz A, Chesebro JH, Badimon L, Nemerson Y, Fuster V, Badimon JJ. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation. 1997;95:594–9. doi: 10.1161/01.cir.95.3.594. [DOI] [PubMed] [Google Scholar]
- 78.Badimon JJ, Lettino M, Toschi V, Fuster V, Berrozpe M, Chesebro JH, Badimon L. Local inhibition of tissue factor reduces the thrombogenicity of disrupted human atherosclerotic plaques: effects of tissue factor pathway inhibitor on plaque thrombogenicity under flow conditions. Circulation. 1999;99:1780–7. doi: 10.1161/01.cir.99.14.1780. [DOI] [PubMed] [Google Scholar]
- 79.Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA., Jr Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci U S A. 1986;83:4533–7. doi: 10.1073/pnas.83.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herbert JM, Savi P, Laplace MC, Lale A. IL-4 inhibits LPS-, IL-1 beta- and TNF alpha-induced expression of tissue factor in endothelial cells and monocytes. FEBS Lett. 1992;310:31–3. doi: 10.1016/0014-5793(92)81139-d. [DOI] [PubMed] [Google Scholar]
- 81.Atkinson BT, Jasuja R, Chen VM, Nandivada P, Furie B, Furie BC. Laser-induced endothelial cell activation supports fibrin formation. Blood. 2010;116:4675–83. doi: 10.1182/blood-2010-05-283986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brooks EG, Trotman W, Wadsworth MP, Taatjes DJ, Evans MF, Ittleman FP, Callas PW, Esmon CT, Bovill EG. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114:1276–9. doi: 10.1182/blood-2009-03-209981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ageno W, Becattini C, Brighton T, Selby R, Kamphuisen PW. Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circulation. 2008;117:93–102. doi: 10.1161/CIRCULATIONAHA.107.709204. [DOI] [PubMed] [Google Scholar]
- 84.Becattini C, Vedovati MC, Ageno W, Dentali F, Agnelli G. Incidence of arterial cardiovascular events after venous thromboembolism: a systematic review and a meta-analysis. J Thromb Haemost. 2010;8:891–7. doi: 10.1111/j.1538-7836.2010.03777.x. [DOI] [PubMed] [Google Scholar]
- 85.Maroney SA, Cooley BC, Sood R, Weiler H, Mast AE. Combined tissue factor pathway inhibitor and thrombomodulin deficiency produces an augmented hypercoagulable state with tissue specific fibrin deposition. J Thromb Haemost. 2008;6:111–7. doi: 10.1111/j.1538-7836.2007.02817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31:15–26. doi: 10.1161/ATVBAHA.109.200956. [DOI] [PubMed] [Google Scholar]
- 87.Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–97. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem. 1989;264:17049–57. [PubMed] [Google Scholar]
- 89.Castaman G, Yu-Feng L, Rodeghiero F. A bleeding disorder characterised by isolated deficiency of platelet microvesicle generation. Lancet. 1996;347:700–1. doi: 10.1016/s0140-6736(96)91259-3. [DOI] [PubMed] [Google Scholar]
- 90.Kanazawa S, Nomura S, Kuwana M, Muramatsu M, Yamaguchi K, Fukuhara S. Monocyte-derived microparticles may be a sign of vascular complication in patients with lung cancer. Lung Cancer. 2003;39:145–9. doi: 10.1016/s0169-5002(02)00441-5. [DOI] [PubMed] [Google Scholar]
- 91.Khorana AA, Francis CW, Menzies KE, Wang JG, Hyrien O, Hathcock J, Mackman N, Taubman MB. Plasma tissue factor may be predictive of venous thromboembolism in pancreatic cancer. J Thromb Haemost. 2008;6:1983–5. doi: 10.1111/j.1538-7836.2008.03156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tripodi A, Branchi A, Chantarangkul V, Clerici M, Merati G, Artoni A, Mannucci PM. Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis. 2011;31:165–72. doi: 10.1007/s11239-010-0506-0. [DOI] [PubMed] [Google Scholar]
- 93.Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–83. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- 94.Tesselaar ME, Romijn FP, van der Linden IK, Bertina RM, Osanto S. Microparticle-associated tissue factor activity in cancer patients with and without thrombosis. J Thromb Haemost. 2009;7:1421–3. doi: 10.1111/j.1538-7836.2009.03504.x. [DOI] [PubMed] [Google Scholar]
- 95.Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, Furie B. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. 2009;15:6830–40. doi: 10.1158/1078-0432.CCR-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, Celi A, Croce K, Furie BC, Furie B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197:1585–98. doi: 10.1084/jem.20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gross PL, Furie BC, Merrill-Skoloff G, Chou J, Furie B. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J Leukoc Biol. 2005;78:1318–26. doi: 10.1189/jlb.0405193. [DOI] [PubMed] [Google Scholar]
- 98.Ramacciotti E, Hawley AE, Farris DM, Ballard NE, Wrobleski SK, Myers DD, Jr, Henke PK, Wakefield TW. Leukocyte- and platelet-derived microparticles correlate with thrombus weight and tissue factor activity in an experimental mouse model of venous thrombosis. Thromb Haemost. 2009;101:748–54. [PMC free article] [PubMed] [Google Scholar]
- 99.Aleman MM, Gardiner C, Harrison P, Wolberg AS. Unique contributions of monocyte- and platelet-derived microparticles to thrombin generation and fibrin formation and stability. J Thromb Haem. 2011 doi: 10.1111/j.1538-7836.2011.04488.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Biro E, Sturk-Maquelin KN, Vogel GM, Meuleman DG, Smit MJ, Hack CE, Sturk A, Nieuwland R. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J Thromb Haemost. 2003;1:2561–8. doi: 10.1046/j.1538-7836.2003.00456.x. [DOI] [PubMed] [Google Scholar]
- 101.Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP, Mackman N. Monocytic microparticles activate endothelial cells in an IL-1(beta)-dependent manner. Blood. 2011 doi: 10.1182/blood-2011-01-330878. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wootton DM, Ku DN. Fluid mechanics of vascular systems, diseases, and thrombosis. Ann Rev Biomed Eng. 1999;01:299–329. doi: 10.1146/annurev.bioeng.1.1.299. [DOI] [PubMed] [Google Scholar]
- 104.Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 2005;85:9–23. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 105.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, Garcia-Cardena G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 107.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–63. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 108.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–79. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 109.Bhattacharya R, Senbanerjee S, Lin Z, Mir S, Hamik A, Wang P, Mukherjee P, Mukhopadhyay D, Jain MK. Inhibition of vascular permeability factor/vascular endothelial growth factor-mediated angiogenesis by the Kruppel-like factor KLF2. J Biol Chem. 2005;280:28848–51. doi: 10.1074/jbc.C500200200. [DOI] [PubMed] [Google Scholar]
- 110.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Preston RA, Jy W, Jimenez JJ, Mauro LM, Horstman LL, Valle M, Aime G, Ahn YS. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension. 2003;41:211–7. doi: 10.1161/01.hyp.0000049760.15764.2d. [DOI] [PubMed] [Google Scholar]
- 112.Silverman MD, Samet MM, Lelkew PI. Pressure modulates tissue factor expression in human aortic and vena endothelial cells. J Vasc Res. 1996;33:93. [Google Scholar]
- 113.Hishikawa K, Nakaki T, Marumo T, Suzuki H, Kato R, Saruta T. Pressure enhances endothelin-1 release from cultured human endothelial cells. Hypertension. 1995;25:449–52. doi: 10.1161/01.hyp.25.3.449. [DOI] [PubMed] [Google Scholar]
- 114.Hathcock JJ. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2006;26:1729–37. doi: 10.1161/01.ATV.0000229658.76797.30. [DOI] [PubMed] [Google Scholar]
- 115.Siegel JM, Markou CP, Ku DN, Hanson SR. A scaling law for wall shear rate through an arterial stenosis. J Biomech Eng. 1994;116:446–51. doi: 10.1115/1.2895795. [DOI] [PubMed] [Google Scholar]
- 116.Turitto VT, Hall CL. Mechanical factors affecting hemostasis and thrombosis. Thromb Res. 1998;92:S25–31. doi: 10.1016/s0049-3848(98)00157-1. [DOI] [PubMed] [Google Scholar]
- 117.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yong AS, Pennings GJ, Chang M, Hamzah A, Chung T, Qi M, Brieger D, Behnia M, Krilis SA, Ng MK, Lowe HC, Kritharides L. Intracoronary shear-related up-regulation of platelet P-selectin and platelet-monocyte aggregation despite the use of aspirin and clopidogrel. Blood. 2011;117:11–20. doi: 10.1182/blood-2010-04-278812. [DOI] [PubMed] [Google Scholar]
- 119.Nesbitt WS, Westein E, Tovar-Lopez FJ, Tolouei E, Mitchell A, Fu J, Carberry J, Fouras A, Jackson SP. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–73. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 120.Shen F, Kastrup CJ, Liu Y, Ismagilov RF. Threshold response of initiation of blood coagulation by tissue factor in patterned microfluidic capillaries Is controlled by shear rate. Arterioscler Thromb Vasc Biol. 2008;28:2035–41. doi: 10.1161/ATVBAHA.108.173930. [DOI] [PubMed] [Google Scholar]
- 121.Neeves KB, Illing DA, Diamond SL. Thrombin flux and wall shear rate regulate fibrin fiber deposition state during polymerization under flow. Biophys J. 2010;98:1344–52. doi: 10.1016/j.bpj.2009.12.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wielders SJ, Broers J, ten Cate H, Collins PW, Bevers EM, Lindhout T. Absence of platelet-dependent fibrin formation in a patient with Scott syndrome. Thromb Haemost. 2009;102:76–82. doi: 10.1160/TH08-11-0719. [DOI] [PubMed] [Google Scholar]
- 123.Campbell RA, Aleman MM, Gray LD, Falvo MR, Wolberg AS. Flow profoundly influences fibrin network structure: Implications for fibrin formation and clot stability in hemostasis. Thromb Haemost. 2010;104:1281–4. doi: 10.1160/TH10-07-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Varju I, Sotonyi P, Machovich R, Szabo L, Tenekedjiev K, Silva MM, Longstaff C, Kolev K. Hindered dissolution of fibrin formed under mechanical stress. J Thromb Haemost. 2011;9:979–86. doi: 10.1111/j.1538-7836.2011.04203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hechler B, Nonne C, Eckly A, Magnenat S, Rinckel JY, Denis CV, Freund M, Cazenave JP, Lanza F, Gachet C. Arterial thrombosis: relevance of a model with two levels of severity assessed by histologic, ultrastructural and functional characterization. J Thromb Haemost. 2010;8:173–84. doi: 10.1111/j.1538-7836.2009.03666.x. [DOI] [PubMed] [Google Scholar]
- 126.Machlus KR, Lin F-C, Wolberg AS. Procoagulant activity induced by vascular injury determines contribution of elevated factor VIII to thrombosis and thrombus stability in mice. Blood. 2011 doi: 10.1182/blood-2011-06-362814. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269:23357–66. [PubMed] [Google Scholar]
- 128.Jones KC, Mann KG. A model for the tissue factor pathway to thrombin. II. A mathematical simulation. J Biol Chem. 1994;269:23367–73. [PubMed] [Google Scholar]
- 129.Hockin MF, Jones KC, Everse SJ, Mann KG. A model for the stoichiometric regulation of blood coagulation. Journal of Biological Chemistry. 2002;277:18322–33. doi: 10.1074/jbc.M201173200. [DOI] [PubMed] [Google Scholar]
- 130.Kuharsky AL, Fogelson AL. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys J. 2001;80:1050–74. doi: 10.1016/S0006-3495(01)76085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fogelson AL, Tania N. Coagulation under flow: the influence of flow-mediated transport on the initiation and inhibition of coagulation. Pathophysiol Haemost Thromb. 2005;34:91–108. doi: 10.1159/000089930. [DOI] [PubMed] [Google Scholar]
- 132.Diamond SL. Systems biology to predict blood function. J Thromb Haemost. 2009;7 (Suppl 1):177–80. doi: 10.1111/j.1538-7836.2009.03463.x. [DOI] [PubMed] [Google Scholar]
- 133.Panteleev MA, Ananyeva NM, Ataullakhanov FI, Saenko EL. Mathematical models of blood coagulation and platelet adhesion: clinical applications. Curr Pharm Des. 2007;13:1457–67. doi: 10.2174/138161207780765936. [DOI] [PubMed] [Google Scholar]
- 134.Xu Z, Kamocka M, Alber M, Rosen ED. Computational approaches to studying thrombus development. Arterioscler Thromb Vasc Biol. 2011;31:500–5. doi: 10.1161/ATVBAHA.110.213397. [DOI] [PubMed] [Google Scholar]
- 135.Hathcock JJ, Nemerson Y. Platelet deposition inhibits tissue factor activity: in vitro clots are impermeable to factor Xa. Blood. 2004;104:123–7. doi: 10.1182/blood-2003-12-4352. [DOI] [PubMed] [Google Scholar]
- 136.Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol. 2006;26:41–8. doi: 10.1161/01.ATV.0000193624.28251.83. [DOI] [PubMed] [Google Scholar]
- 137.Gailani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007;5:1106–12. doi: 10.1111/j.1538-7836.2007.02446.x. [DOI] [PubMed] [Google Scholar]
- 138.Xu Z, Chen N, Kamocka MM, Rosen ED, Alber M. A multiscale model of thrombus development. J R Soc Interface. 2008;5:705–22. doi: 10.1098/rsif.2007.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Leiderman K, Fogelson AL. Grow with the flow: a spatial-temporal model of platelet deposition and blood coagulation under flow. Math Med Biol. 2011;28:47–84. doi: 10.1093/imammb/dqq005. [DOI] [PMC free article] [PubMed] [Google Scholar]