

Abstract
Cardiac arrhythmias can cause sudden cardiac death (SCD) and add to the current heart failure (HF) health crisis. Nevertheless, the pathological processes underlying arrhythmias are unclear. Arrhythmic conditions are associated with systemic and cardiac oxidative stress caused by reactive oxygen species (ROS). In excitable cardiac cells, ROS regulate both cellular metabolism and ion homeostasis. Increasing evidence suggests that elevated cellular ROS can cause alterations of the cardiac sodium channel (Nav1.5), abnormal Ca2+ handling, changes of mitochondrial function, and gap junction remodeling, leading to arrhythmogenesis. This review summarizes our knowledge of the mechanisms by which ROS may cause arrhythmias and discusses potential therapeutic strategies to prevent arrhythmias by targeting ROS and its consequences.
Keywords: reactive oxygen species, sodium channel, Ca2+ handling, mitochondria, connexin, arrhythmia
1. Introduction
The majority of sudden cardiac death (SCD) results from the occurrence of potentially lethal ventricular tachycardia (VT) or ventricular fibrillation (VF), only two of many types of arrhythmia. Arrhythmia is an irregular heart rhythm and is classified by rate as either tachycardia or bradycardia (resting heart rate >100 beats/min or < 60 beats/min, respectively). Arrhythmias are also mechanistically classified as automatic, reentrant, and triggered. Reentry is favored by slow, inhomogeneous conduction. Types of arrhythmia include (1) premature beats; (2) supraventricular arrhythmias (e.g., atrial fibrillation (AF), atrial flutter, and paroxysmal supraventricular tachycardia); (3) ventricular arrhythmias (e.g., VT and VF); and (4) bradyarrhythmias.
SCD occurs in approximately 180,000 – 250,000 cases annually in the United States, and an estimated 4-5 million cases worldwide [1]. SCD occurs in hypertrophic cardiomyopathy, dilated cardiomyopathies, arrhythmogenic right ventricular dysplasia, myocardial infiltrative diseases, and other related disease states [2]. The prevalence of cardiovascular diseases potentially associated with lethal ventricular arrhythmia is estimated at approximately 13 million US individuals, which is about 5% of the middle-aged population [3].
Paroxysmal or persistent AF afflicts approximately 2.2 million Americans in addition to 4.5 million people in the European Union. AF is a supraventricular tachyarrhythmia characterized by uncoordinated atrial activation with consequential deterioration of atrial mechanical function. It is the most common arrhythmia clinically encountered, accounting for over 30% of hospital admissions for cardiac rhythm disturbances [4] and is associated with increased risks for stroke, heart failure (HF), and death [5, 6]. The incidence of AF noticeably increases over the age of 60, afflicts 3-5 % of the population 65 to 75 years old and occurs in up to 8% of those older than 80 years [7-9]. The prevalence of this arrhythmia has significantly increased recently, and the number of Americans with AF is expected to surpass 5 million by 2050 [10].
Despite the high prevalence and significance of arrhythmias, the mechanisms of arrhythmogenesis are not fully understood. Some molecular mechanisms known to contribute to arrhythmias include genetic alterations of ion channels leading to electrophysiological dysregulations and structural remodeling of the left ventricle (LV) in hypertrophy and HF [11-14]. Increasing evidence suggests that altered cardiac ion homeostasis and structural remodeling are highly associated with elevated reactive oxygen species (ROS) and metabolic stress [15, 16]. In this review, we summarize possible mechanisms whereby the imbalanced cellular redox state may cause arrhythmogenesis by ROS-induced alterations of ion homeostasis and ion channel behavior.
2. Cardiac conditions associated with metabolic stress, ROS, and arrhythmias
Cardiac metabolism is reflected by adenosine-5′-triphosphate (ATP), which is the source of energy for maintenance of ion homeostasis as well as repetitive mechanical contraction and relaxation. Approximately 60-70% of ATP is used for cardiac muscle contraction, and the remaining 30-40% is used for Ca2+ uptake into the sarcoplasmic reticulum (SR) to initiate diastolic relaxation and to sustain ion current homeostasis including the maintenance of Na+ and K+ gradients across the plasma membrane [17, 18].
Mitochondria occupy approximately 30% of the volume of ventricular cardiomyocytes and form a network around the myofilaments resulting in the location of ATP production sites being adjacent to ATP consumption sites [19]. Although there are several sources of ROS in cardiac muscle including NADPH oxidase, xanthine oxidase, and uncoupled NOS, mitochondria are the major ROS source. Electron leakage from complex I and III is associated with the generation of ROS in the failing heart [20], ischemia/reperfusion [21] and arrhythmia [16]. Complex I (i.e. NADH and ubiquinone oxidoreductase) produces superoxide (O2·−) in the mitochondrial matrix whereas complex III (i.e. Q-cycle, cytochrome bc1 complex and coenzyme Q) produces superoxide in the matrix and intermembrane space [22]. Oxidative phosphorylation is tightly linked to mitochondrial regulation so that the cellular ATP content remains constant even with large increases in cardiac ATP consumption [23]. The electron transport chain (ETC) of the mitochondria matrix transfers electrons from carbon substrates (e.g., fatty acids and pyruvate) to nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) in order to synthesize ATP. During the occurrence of abnormal pathophysiological conditions that cause arrhythmia (when the balances of blood flow, oxygen delivery, mitochondrial metabolism, NADH formation, and ATP synthesis are disrupted), NADH, protons, and lactate accumulate, potentially contribute to arrhythmic risk. Moreover, when disrupted, mitochondria produce ROS via electrons leaking from the ETC that react with molecular oxygen to form superoxide. The accumulation of ROS in the mitochondria can lead to oscillations in the mitochondrial membrane potential (ΔΨm) and changes in matrix concentrations of Ca2+, NADH, ADP, and tricarboxylic acid (TCA) cycle intermediates [24]. Hypoxia also decreases the rate of metabolism, resulting in the decrease of intracellular NADH/NAD+ ratio and an increase in ROS production [25-27]. Accumulating ROS further increases the rate of ROS production, a phenomenon known as ROS induced ROS release [28].
ROS, also called reactive oxygen intermediates, are comprised of superoxide, hydrogen peroxide (H2O2), hydroxyl radicals (·OH) and peroxynitrite (ONOO−) [29]. ROS are highly reactive and unstable molecules leading to irreversible, deleterious reactions with lipids and proteins. Cardiac conditions such as hypertension [30], diabetes mellitus [31], coronary artery disease [32, 33], cardiomyopathies, and HF [34, 35] are associated with altered metabolism and arrhythmias. In diabetes mellitus, arrhythmias and oxidative stress are well correlated [31]. In ischemia/reperfusion injury, the mitochondrial ETC serves as the major source of ROS [21]. In chronic HF, ROS levels increase [20, 36] and myocardial antioxidant reserve decreases [37, 38].
Trace amount of ROS serve as signaling molecules in physiological conditions, but the excessive production of ROS elicits pathologic cellular processes [39]. To protect cellular functions from ROS, cells have two defense mechanisms, enzymatic and nonenzymatic pathways. The enzymatic pathway includes three superoxide dismutases, catalase, and glutathione peroxidase [40]. The dismutation of superoxide by superoxide dismutase results in the generation of H2O2, which catalase further metabolizes to water and oxygen. In the nonenzymatic pathways, there are a variety of redox-defense system including antioxidant scavengers, such as vitamins E and C [41], the ratio of reduced glutathione (GSH) to oxidized glutathione disulfide (GSSH), the ratio of oxidized and reduced forms nicotinamide adenine dinucleotide, NADH/NAD+, and the thioredoxin [14, 42]
One of the main consequences of oxidative stress is the depletion of key intracellular antioxidants such as glutathione (GSH), the largest antioxidant pool in the heart [43]. In ischemia [44-46], HF [47], and type 2 diabetes [48], conditions associated with arrhythmia, the oxidized GSH ratio (GSH/GSSH) of ~200-300 to 1 is decreased by 50-70%. During AF, there is biochemical evidence of oxidation by peroxynitrite and hydroxyl radicals [49]. Systemically, serum oxidative markers are elevated in individuals with AF [50]. Other oxidative serum markers including malondialdehyde and nitrotyrosine are also increased in arrhythmias [20, 51].
Another important cellular redox defense system is a cytosolic NADH/NAD+ level [43]. HF is associated with reduced NAD+ and increased NADH [52-54]. The balance of oxidized and reduced NAD forms varies some between species and tissues. In the monkey, the estimated ratio between free NADH to NAD+ in the cytoplasm is ~0.002. In rodents and swine, the ratio of total NADH/NAD+ is ranges from 0.05 to 4, under normal aerobic conditions [27, 43, 55, 56]. These numbers are in the range of the cytosolic NAD redox potential of −250 mV, which implies a NAD+/NADH ratio of ~500:1 [57]. In the mitochondrion, the concentration of NAD+ is similar to that in the cytosol [58]. More than 80% of mitochondrial NADH is bound to proteins, so the free concentration is much lower [59] and the ratio of oxidized to reduced forms may be different in the mitochondria. In our previous work, an increase in cellular NADH by 1.7 fold increased mitochondrial ROS production by 2 fold [60]. This is change in the NAD+/NADH ratio is on the order of that seen in heart failure [61, 62]. O’Rourke et al. have demonstrated repetitive and self-sustaining oscillations in NADH during mitochondrial injury [63]. Moreover, these changes in pyridine nucleotide levels are associated with ROS, and ROS may explain some arrhythmic effects. ROS bursts with NAD(P)H oxidation in single mitochondria [64], and isolated cardiac myocytes from guinea pigs [65]. Therefore, the NADH/NAD+ ratio, a measure of both redox balance and metabolic activity, can directly affect ROS production and arrhythmic risk as discussed below. Moreover, O’Rourke and colleagues have posited that oxidative stress can occur at either extreme of redox potential, either highly reduced or highly oxidized [66]. They point out that ROS overflow can increase because of ROS production, as the redox environment becomes more reduced, or at oxidized redox potentials as a result of scavenger pool depletion.
Although there are many reports suggesting that oxidative stress is related to the pathogenesis of arrhythmias, the major sources of ROS and their regulating mechanisms are not fully understood. ROS can be generated through electron leakage from mitochondria during oxidative phosphorylation and through the activation of several cellular enzymes, including the NADPH oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (NOS) (Fig. 1) [29, 39, 67, 68]. The NADPH oxidase complex is a significant cardiac ROS source [69]. In a swine rapid atrial pacing model of AF, increased activity of the NADPH oxidase and xanthine oxidase are associated with the production of superoxide and reduced nitric oxide (NO) bioavailability in both of the left atria (LA) and left atrial appendage (LAA) [70, 71]. Apocynin, a NADPH oxidase inhibitor, reduced LAA superoxide production by 91%, supporting the activation of NADPH oxidase in AF. Active Rac1, a small G-protein associated with NADPH oxidase activation, is increased by 6.9 fold in LAA of the AF model [70, 71]. Rapid atrial pacing has been shown to elevate myocardial peroxynitrite formation and leads to a shortening of the atrial effective refractory period, which can be reversed by treatment with ascorbate and statins. These drugs are known to decrease NADPH oxidase activation [72, 73]. Increased NADPH oxidase activity has been observed in pressure-overload LV hypertrophy, myocardial infarction, HF, and AF [74-78]. Moreover, NADPH oxidase activation and NOS uncoupling could be important factors in the initiation of mitochondrial ROS generation. Rotenone, a mitochondrial complex I inhibitor, lowers the basal levels of superoxide detected in atria in patients of AF [79].
Figure 1.
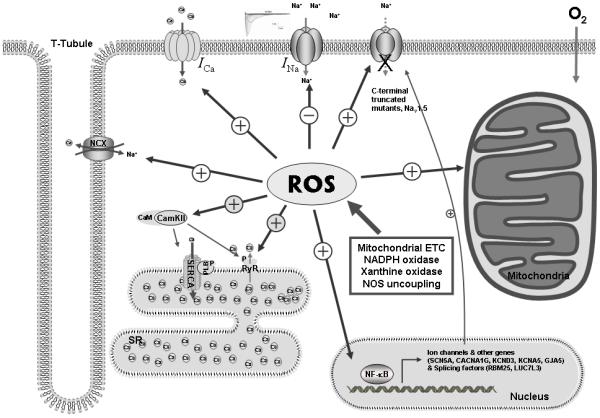
Schematic illustration of ROS-mediated ion channel alterations. ROS are generated from mitochondrial ETC electron leakage, NADPH oxidases, xanthine oxidase and NOS uncoupling in the heart. ROS induces functional and structural alteration of Nav1.5 by both transcriptional and posttranslational mechanisms. Elevated ROS mediates SERCA inhibition, enhanced SR Ca2+ release from RyR, enhanced inward L-type Ca2+ current, and increased NCX activity to increase intracellular Ca2+ level. ROS-mediated CaMKII activation stimulates hyperphosphorylation of RyR, resulting SR Ca2+ leak. In mitochondria, stimulated ROS are release by the IMAC and the membrane permeability transition pore, causing KATP activation, the mitochondrial calcium uniporter activity, and NADH accumulation. This ROS-mediated network is likely to contribute to the pathogenesis of arrhythmias.
3. Mechanisms whereby ROS can affect arrhythmias
Arrhythmogenesis and oxidative stress are correlated, but how do ROS cause arrhythmia? Possibilities include Na+, Ca2+, and K+ ion homeostasis changes and altered voltage-gated ion channel activity.
A. Regulation of Nav1.5 by ROS
Nav1.5 cardiac Na+ channel encoded by the SCN5A gene is one of the key ion channels for excitability and rapid impulse propagation. An appropriate number of functional Nav1.5 channels is critical for normal cardiac function [80]. Either excess or reduced channel current results in increased arrhythmic risk, as demonstrated in the inherited sudden death syndromes Long QT type 3 [81] and Brugada syndromes [82].
Potentially pro-arrhythmic transcription factors such as, peroxisome proliferator-activated receptor, c-fos and NF-κB, are known to be regulated by redox signalling pathways [83-85]. Among these transcriptional factors, NF-κB is one of key transcriptional regulator that mediates alterations of genes during biological states of stress such as injury, inflammation [78], hypertrophy [86], renin-angiotensin system (RAS) activation [87, 88], and oxidative stress [89]. Recently, it was shown that the promoter region of the cardiac sodium channel gene (SCN5A) contains a NF-κB binding sequence, suggesting that SCN5A is a gene regulated by NF-κB [90]. Angiotensin II (AngII) or H2O2 treatment of cardiomyocytes results in increased NF-κB binding to SCN5A promoter with subsequent reduction in transcriptional activity [87]. Simultaneous overexpression of the p50 and p65 NF-κB subunits recapitulates the effects of AngII or oxidative stress. Furthermore, a mutation in the NF-κB consensus sequence prevented this transcriptional downregulation. The reduction in INa seen with AngII is similar to its effect on other cardiac ion channels, including the transient outward current K+ channel α-subunit (Kv4.3) [91], the gap junction protein connexin (Cx) 43 [92], Cx40 [92, 93], and the Ca2+ current [93], which may be mediated by comparable mechanisms and may also contribute to enhanced arrhythmic risk in states of increased oxidative stress. Inspecting the promoter regions of other channel and subunit genes reveals that the T-type Ca2+ channel, the transient outward current, the ultra-rapid delayed rectifier, and Cx40 also have NF-κB consensus binding sequences [14]. All appear to undergo transcriptional regulation during arrhythmias, but experimental evidence currently exists only for SCN5A to be regulated by NF-κB [87].
Aside from the direct NF-κB regulation on ion channel promoters, SCN5A gene expression is also regulated by alternative mRNA splicing. In human HF, a condition linked with increased risk of arrhythmia, three C-terminal truncated splice variants are observed [80, 94]. These variants do not form functional channels and result in a reduction in INa. Stimuli for this alternative splicing included hypoxia and AngII, factors associated with ROS and arrhythmic risk. This mechanism is mediated by RBM 25 and LUC7L3, SCN5A splicing factors which are regulated by HIFα [95, 96].
As discussed above arrhythmic conditions with mitochondrial dysfunction and an accumulation of NADH [25, 26]. NADH increases mitochondrial ROS [25, 26]. Glycerol-3-phosphate dehydrogenase 1-like protein (GPD1-L) is involved in NAD-dependent energy metabolism, and mutations in the gene are associated with Brugada and sudden infant death syndromes [60, 97, 98]. Mutation of GPD1-L results in elevated NADH and Nav1.5 downregulation, suggesting a mechanism that links metabolism, ROS, and arrhythmic risk [16, 60, 97, 98]. Intracellular NADH level leads to activation of protein kinase C and subsequent increased mitochondrial ROS production [60]. The immediacy of the NADH effect on reducing INa and the lack of change in mRNA abundance suggests that this effect of NADH is post-transcriptional. Biological significance was implied when programmed electrical stimulation (PES)-induced VT was demonstrated after pyruvate/lactate buffer perfusion, a condition under which intracellular [NADH]i was increased 1.7-fold. On the other hand, the oxidize intracellular pyridine nucleotide, NAD+, ameliorated the mitochondrial ROS production, reduced INa, and inducibility in a mouse model in which one allele of the cardiac Nav1.5 channel was ablated (SCN5A+/−) (Fig. 2) [99]. Nevertheless, the NAD+ effect did not seem to occur by the same signaling mechanism as did the NADH effect, and the NAD+ effect could be recapitulated by a protein kinase A (PKA) activator or prevented by a PKA inhibitor (Fig. 3). This is consistent with the known effect of external NAD+ to activate PKA in human granulocytes [100] and osteoblastic cells [101] and of PKA activation to increase INa [102, 103]. Although these findings suggest that the balance of oxidized and reduced NAD(H) may be a critical mechanism in the regulation of INa in the metabolic state of myocytes, there are still not fully investigated that redox potential differences between intracellular and mitochondria NADH affect to mitochondrial ROS elevation directly or indirectly via signaling pathways.
Figure 2.
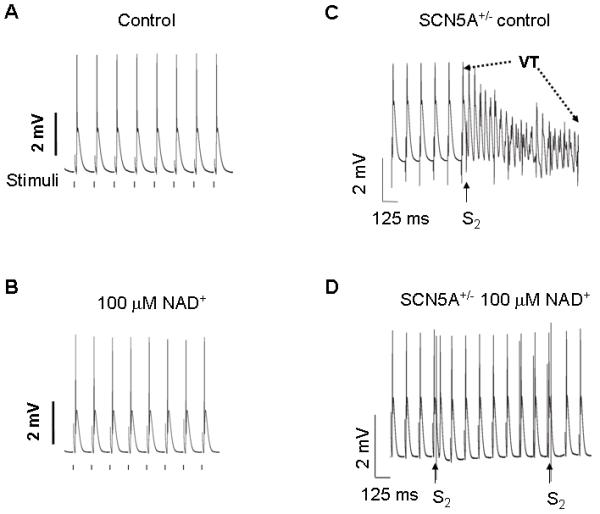
The effects of altering NAD(H) on arrhythmic risk in a mouse model of reduced Na+ current. Representative traces of MAPs from LV epicardium of (A) Langendorff-perfused SCN5A+/− heart during standard pacing at BCL of 125 ms in the control condition. Vertical lines below the monophasic action potentials (MAPs) represent the times when electrical stimulations were delivered. (B) MAPs after 20 min of perfusion with 100 μmol/L [NAD+]o. (C) Representative MAPs recorded during programmed electrical stimulation (PES) showing PES-induced ventricular tachycardia in SCN5A+/− hearts under control condition. The final six paced beats at 125 BCL (S1) were followed by an extra stimulus (S2) delivered at a S1-S2 interval of 42 ms. PES induced a polymorphic ventricular tachycardia of frequency, 20-40 Hz, sustained for ≈19 seconds. (D) Representative trace of PES-induced MAP recording in same SCN5A+/− heart after 20 min perfusion with 100 μM [NAD+]o. S2 stimuli delivered at a 35 ms S1-S2 interval produced a single MAP but failed to induce any arrhythmia. (Modified from reference [60])
Figure 3.
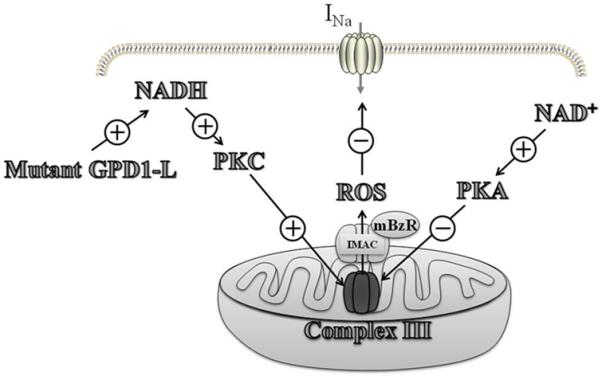
The illustration of the effects of mitochondrial ROS induced by NADH on the INa. A proposed signaling pathway by which mutant GPD1-L and NADH downregulate Nav1.5 by causing PKC activation and ROS overproduction from the mitochondrial ETC. ROS are released from the mitochondria by the IMAC, which is modulated by the mitochondrial benzodiazepine receptor. NAD+ inhibits mitochondrial ROS overproduction through PKA activation followed by the upregulation of Nav1.5. (Modified from reference [16])
Changes in the INa induced by NADH are consistent with alterations observed in other channels. Tipparaju et al. [104, 105] have reported that NAD(P)H to NAD(P)+ ratio regulates K+ currents, although the regulation mostly affects gating rather than peak current. Some transient receptor potential currents are increased by NAD+ in a manner similar to that seen in our experiments [106]. A non-selective cation channel conductance is increased by NAD+ [107].
B. Mitochondrial ROS and the metabolic sink theory
O’Rourke and colleagues suggests that the mitochondria have an important role in the development of arrhythmia through metabolic stress that causes ADP levels to rise and ATP to be depleted. This leads to opening of sarcoplasmic potassium-sensitive KATP (sarcKATP) channels and a decreased cardiac action potential (AP) duration, a reduced Ca2+ transient, and an increased K+ conductance [19, 26, 63, 108]. If enough channels open, cells can be rendered inexcitable by clamping the membrane potential close to potassium’s Nernst equilibrium potential, thus creating a current sink for propagating depolarization waves [109]. This phenomenon produces cellular regions of inexcitability that have been termed “metabolic sinks” [110, 111], and they occur when a threshold of sarcKATP channels open [109]. Because current sinks create regions of inhomogeneous excitability, the development of reentrant arrhythmias is favored [112-114].
Even without complete conduction block, the opening of these channels can significantly shorten the cardiac AP, creating heterogeneity suitable for reentrant arrhythmia [110, 111, 115]. The behavior of the mitochondria and oxidative stress has been linked to fluctuations in AP duration [26, 63]. During oxidative stress, increases in the sarcKATP current have been shown to fluctuate in phase with the ΔΨm [26, 113], demonstrating that arrhythmia may be interconnected with mitochondrial function. In a study conducted by Aon et al., increases of superoxide anions were induced by a laser flash that also caused oscillations in the ΔΨm [26], which could be prevented by blocking the activity of mitochondrial inner membrane anion channels (IMAC). These observed collapses in the ΔΨm may be caused by ROS-induced ROS release [116]. Further confirmation for the role of IMAC involved ROS release from mitochondria and subsequent arrhythmia was observed in a series of studies where the inhibition of IMAC prevented arrhythmia in intact mammalian hearts [110, 112, 117]. Furthermore, we have demonstrated that IMAC is involved in NADH/ROS regulation of INa [16].
Other mitochondrial ion channels may also influence arrhythmias. The mitochondrial Ca2+ uniporter modulates mitochondrial Ca2+ homeostasis [118]. Mitochondrial Ca2+ signaling is important in the control of fundamental energy functions including energy metabolism and apoptotic cell death [119]. The Ca2+ influx, mediated by the mitochondrial Ca2+ uniporter, is associated with opening the mitochondrial membrane permeability transition pore [120], and impairment in intracellular ion homeostasis and mitochondrial function have been implicated in the development of cardiomyopathy and arrhythmia [121].
Improper regulation of mitochondrial ion channels can lead to metabolic stress that produces local regions of elevated ROS, triggering slow oscillations of ROS, the ΔΨm, NADH and ATP. ROS increase the probability of opening the inner mitochondrial membrane ion channels, depolarize the ΔΨm, and result in a reduction of the ATP/ADP ratio. This activates repetitive and self-sustaining oscillations of the sarcKATP channels. Additionally, increases in NADH would be expected to cause decreases in Na+ current, resulting in the formation of spatially and temporally varying islands of inexcitability that can lead to arrhythmias.
C. Intracellular Ca2+ handling and oxidative stress in arrhythmia
In the process of EC coupling, cardiac contraction is triggered by membrane depolarization followed by the AP and subsequent Ca2+ entry through L-type Ca2+ channels. This initial Ca2+ influx drives the SR Ca2+ release via ryanodine receptor (RyR) opening, so called Ca2+-induced Ca2+ release (CICR). In contrast, cardiac relaxation is mediated by Ca2+ removal through the reuptake of the cytosolic Ca2+ into sarcoplasmic reticulum Ca2+-ATPase (SERCA) and extrusion by Na+-Ca2+ exchanger (NCX) [122]. Abnormal Ca2+ handling contributes to a variety of pathophysiological disease conditions, such as HF and hypertrophy. Many reports demonstrated that alterations in intracellular Ca2+ concentration ([Ca2+]i) can also affect arrhythmias through altered ion channel and gap junction conductances [123, 124].
Metabolic stress, intracellular acidification, reduction in ATP, and a rise in ADP concentrations have been shown to increase cytoplasmic free [Ca2+]i and inhibit both SR Ca2+ uptake and release [125]. Among 5 subunits that make up the voltage-gated, dihydropyridine-sensitive L-type Ca2+ channel (ICa,L), the α1C pore-forming subunit, is redox sensitive. ICa,L decreases when exposed to thiol oxidizing agents (e.g. thimerosal), and this decrease is reversed by dithiothreitol (DTT) [126]. Furthermore, in ferret ventricular myocytes, redox-sensitive thiols in L-type Ca2+ channel diminish ICa,L under oxidizing conditions, and this mechanism is regulated by NO and S-nitrosothiols [127]. Metabolic inhibition with increased [Ca2+]i causes a reduction of the amplitude of ICa,L in the ventricular myocytes of guinea pigs, rats, and rabbits, which contributes to shortening of the APD and an increased potential of reentrant arrhythmias [128-132].
In cardiac muscle, SERCA2a removes cytosolic Ca2+ into SR. SERCA also contains redox-sensitive cysteine residues in the active site, and oxidative stress impairs SERCA activity by oxidation of these sulfhydryl groups [133]. Reducing agents (e.g. DTT and GSH) preserve SERCA activity in the face of oxidative stress [134]. SERCA dysregulation is accompanied by concomitant AP prolongation [135], a potential source of triggered activity.
RyR is involved in cardiac E-C coupling by allowing SR Ca2+ release during CICR. RyR2 is the cardiac muscle specific isoform. RyRs consist of four large subunits, comprised of numerous regulatory subunits including calmodulin, the FK506-binding protein FKBP12.6 (also known as calstabin2), PKA, Ca2+/calmodulin kinase II (CaMKII), protein phosphatases 1 and 2A, phosphodiesterase, junctin, triadin, and calsequestrin [136]. A number of studies have shown that various ROS mediate RyR sulfhydryl oxidation (reviewed in ref [137]). An altered redox balance leads to reduced Ca2+ transients and enhanced SR Ca2+ leak in the failing heart, and these changes can be reversed by reducing agents [138]. This leak may be mediated by oxidative alteration of RyR [139]. Among the 89 cysteine residues of RyR, there are a few sulfhydryl groups that seem to mediate redox-sensitivity of the channel [140]. Free SH oxidizing reagents, 2.2′-dithiodipyridine (DTDP) or 5.5′-dithio-bis(2-nitrobenzoic acid) (DTNB), activate the RyR channel by formation of disulfide bonds within the RyR complex [141]. Oxidized GSSG stimulates RyR2 activity by decreasing calmodulin binding affinity [141, 142]. Exposure to superoxide stimulates RyR2 causing pro-arrhythmogenic SR Ca2+ releases [42]. Zima et al. also show that cytosolic NADH inhibited cardiac SR Ca2+ release channels, while NAD+ had activating effects on this channel [143]. Clinical significance of these RyR changes is reinforced by the observation that genetic alterations in RyR2 are linked to an autosomal-dominant form of inherited cardiac arrhythmias, known as catecholaminergic polymorphic VT [144]. Catecholaminergic polymorphic VT-associated variants sensitize RyR2 to activation by cytosolic Ca2+ [145].
Another important Ca2+ handling protein, NCX, is also sensitive to redox-sensitive regulation. NCX is the main Ca2+ efflux pathway, removing intracellular Ca2+ using the Na+ transmembrane gradient. Typically delayed afterdepolarization-type triggered activity is induced when high intracellular Ca2+ promotes NCX activity and associated depolarizing currents (reviewed in [136]). Consistent with this, upregulated of NCX enhances delayed afterdepolarizations [146]. Moreover, the reverse mode of NCX can lead to Ca2+ overload and potentially to early afterdepolarizations [147]. Hinata and colleagues demonstrated that H2O2 exposure stimulated NCX activity by activating mitogen-activated protein kinase and Src tyrosine kinase signaling pathways [148].
Altered Ca2+ homeostasis can contribute to arrhythmias by ROS-dependent CaMKII activation that enhances late INa, leading to cellular Na+ and Ca2+ overload and afterdepolarizations [149]. Morita and colleagues demonstrated that oxidative stress promotes early afterdepolarizations and triggered activity leading to VT and VF in aged fibrotic rat and rabbit ventricular myocytes. These arrhythmias were suppressed by the antioxidant, NAC, and CaMKII inhibitor, consistent with the oxidative activation of CaMKII [150, 151].
Stretch activated channels may play a role in redox-dependent Ca2+ alterations and arrhythmogenesis. The combination of mechanical stretch and low level oxidative stress triggers Ca2+ overload and prolong APD through premature beat resulting early afterdepolarizations [152]. This may explain the reduction of ventricular arrhythmias with intra-aortic balloon counterpulsation [153, 154].
D. Potassium channels in arrhythmogenesis
The metabolic stress and oxidative stress have been shown to affect repolarizing K+ currents, potentially causing arrhythmias. APD is regulated by a balance between inward depolarizing and K+ outward currents. The importance of ADP for arrhythmogenesis is highlighted by the large number of hERG (human ether-a-go-go-related gene) K+ channel mutations associated with long-QT syndrome and the case of congenital short QT syndrome with a gain-of-function in the delayed rectifier K+ channel, IKr [155]. Oxidative stress levels result in decreased hERG protein levels and arrhythmias in diabetic rabbits, and the abnormal QT prolongation is restored by the insulin treatment [156]. Direct treatment with H2O2 triggers an initial hERG channel activation and subsequent acceleration of channel deactivation, increasing the risks of ectopy [157]. This effect is mediated by thiol oxidation [158, 159].
Another important outward K+ current is the transient outward current (Ito), which is responsible for the rapid repolarization phase (phase 1) in ventricular AP. This K+ channel current is composed of several α subunit isoforms, Kv1.4, Kv4.2, and Kv4.3 and the β subunits, KChIP2 and Kvβ1.2. Downregulation of Ito has been shown in the diabetic heart, and this change could be reversed by the insulin treatment [160]. Recently, Marionneau and colleagues suggested peroxisome proliferator-activated receptor alpha (PPARα), a key regulator of fatty acid and glucose metabolism, mediated most of this change in current [161]. Nevertheless, a increase ROS induced downregulation of Ito in diabetic heart, and this was reversed by reduced antioxidant, GSH [160]. The channel subunit, Kvβ1.2, has been implicated in the redox control of K+ channels [162, 163].
Redox-dependent regulation has been reported in the inward rectifying K+ channel, IK1. S-nitrosylation of Cys76 residue in α subunit of Kir2.1 encoding this current increases the channel opening probability. Alternatively, NO increases the amplitude of this current [164]. Since this channel sets resting membrane potential and influences APD, redox-dependent changes would be expect to influence arrhythmic risk.
E. Gap Junction Remodeling
ROS has been implicated in gap junction remodeling. Impulse propagation is dependent on several factors, including intrinsic membrane excitability and cell-to-cell coupling. Cardiac cells are connected by gap junction channels that are low resistance pathways for electrical coupling of myocytes and are located within the intercalated disks [165]. Gap junction channels are made of Cxs, a family of proteins designated by molecular weight [166, 167]. In the adult heart, ventricular gap junctions are predominantly formed by Cx43 protein, and atrial gap junctions are formed mainly by Cx40 and Cx43 [167].
Abnormalities in gap junctional communication have been shown in cardiac ischemia [168, 169]. Intracellular acidosis associated with cardiac ischemia causes the direct interaction of the carboxyl terminal domain of Cx43 with the second half of the cytoplasmic loop of Cx43. This in turn results in closure of the channel [170], a reduction in conduction velocity in the ischemic area. In addition, cardiac ischemia affects the balance of protein kinase and phosphatases in a way that favors the dephosphorylation of Cx43, which results in the impairment of gap junction [171-173]. Lateralization involves the diffusion of Cx43 away from the intercalated disks and toward the lateral myocyte walls. A significant portion of Cx43 is lateralized and is thus nonfunctional after cardiac ischemia [169, 174] which can result in dysfunction of gap junctions. These changes are associated with increased arrhythmic risk.
In another mechanism that may alter gap junctions and arrhythmic risk, Kieken and colleagues showed that the phosphorylated form of c-Src tyrosine kinase competes with Cx43 to bind to the scaffolding protein zonula occludens-1 at the intercalated disks and that elevated levels of phosphorylated c-Src at the border zone of ischemia reduce the Cx43 [169]. The c-Src tyrosine kinase has been linked primarily to tumor growth, and the inhibition of c-Src has been shown to be effective in controlling cancers [175-179]. Elevated levels of ROS and AngII can result in the upregulation of c-Src [180-182]. In cardiac myocytes, Aikawa and colleagues have shown that treatment with H2O2 activates Src family tyrosine kinases in a concentration dependent manner [181]. Also, it has been shown that, in a rat model of Adult Respiratory Distress Syndrome, oxidative stress results in increased level of c-Src [182]. Some studies support the existence of a positive feedback loop between ROS and c-Src in which increase in ROS upregulates c-Src and the c-Src upregulation increases oxidative stress further [183].
The homozygous cardiac angiotensin converting enzyme (ACE) gene-targeted mice (ACE8/8) show normal LV function and structure [92]. Nevertheless, these animals show a high rate of SCD due to VT, VF and bradyarrhythmia. AngII results in elevation of ROS [69, 184] and oxidative stress in the cardiac tissue of the ACE8/8 [185]. Western blotting of the ventricular tissue of the ACE8/8 mice shows higher level of c-Src and a reduction in the total amount of Cx43 to 30% of controls. Administration of PP1 ((1-(1, 1-Dimethylethyl)-1-(4-methylphenyl)-1H-pyrazolo [3, 4-d] pyrimidin-4-amine), a specific c-Src inhibitor, reduced the total and phosphorylated form of c-Src, improved the total amount of Cx43 and corrected the gap junctional conduction impairment. These effects of PP1 were associated with correction of phenotype and prevention of sudden arrhythmic death. Since oxidative stress activates c-Src, c-Src downregulates Cx43, and Cx43 levels are inversely related to arrhythmic risk, this represents another ROS-dependent mechanism that may increase arrhythmic risk.
4. Therapeutic implications
Conventional antiarrhythmic drugs target ion channels and reduce ion currents. Nevertheless, in many cases, the targeted ion channels are already downregulated. The role of ROS in arrhythmogenesis, as outlined above, opens new and potentially safer therapeutic options to treat arrhythmias using antioxidants. Administration of either GSH or N-acethylcysteine significantly reduces reperfusion arrhythmias [186]. Additionally, altering ratios of GSH/GSSH can affect the ΔΨm and the occurrence of arrhythmias [113]. ROS-mediated collapses of the ΔΨm is prevented by the addition of exogenous GSH [187]. Ascorbate (vitamin C) reduces the incidence of atrial pacing-induced peroxynitrite formation as well as postoperative AF [72]. Vitamin E analogues scavenge free radicals and reduce the incidence of ischemia/reperfusion-induced VF [188]. Another synthetic ROS scavenger, 6,6-methylene bis 2,2-dimethyl-4-methane sulphonic acid: Na-1,2-dihydroquinoline (MTDQ-A), significantly reduces the incidence of VF after myocardial infarction following coronary ligation in a dog [189].
Nevertheless, general antioxidant strategies have not always been successful in antiarrhythmic therapies. Explanations for the strong association of oxidative stress and arrhythmia but a more qualified success of antioxidant strategies to prevent arrhythmias may be explained by the idea that redox stress is compartmentalized. As we discussed above, mitochondrial oxidative stress contributes to a wide range of damage in cardiac metabolism and ion homeostasis. Therefore, reducing mitochondria oxidative stress could be a more effective anti-oxidant approach. Indeed, several mitochondria-targeted ROS scavengers have been developed and are ripe for study of their potential anti-arrhythmic properties [112, 114, 190-192].
Strategies targeting mitochondrial ROS release pathways may be antiarrhythmic [114, 193]. IMAC inhibitors, DIDS and PK11195, can prevent the cell-wide accumulation of ROS and reversible collapses in the ΔΨm [114, 193]. Furthermore, 4′chlorodiazepam, a mitochondrial benzodiazepine receptor modulator, can prevent reperfusion arrhythmias in isolated rabbit heart [112]. This inhibitor, as well as DIDS and PK11195, can prevent the INa decrease induced by elevated NADH and ROS [16]. Additionally, other mitochondrial targeted antioxidants such as peptides, SS-02 (Dmt-D-Arg-Phe-Lys-NH2), SS-31 (D-Arg-Dmt-Lys-Phe-NH2) and SS-20 (Phe-D-Arg-Phe-Lys-NH2) are also effective in ischemia-reperfusion [194, 195]. Arrhythmias were significantly reduced in ischemia-reperfusion injured rat hearts when treated SS-02 and SS-31 directly before reperfusion [196, 197].
The mitochondrial anti-oxidant, MitoQ, is targeted to the mitochondria by covalent conjugation with a lipophilic triphenylphosphonium cation. As previously discussed, CoQ (ubiquinone) accepts electrons from complex I or II, and donates to complex II by the formation of reduced product, ubiquinol [191]. The protective effects of MitoQ have been demonstrated by the administration to rats in their drinking water [191]. Under ischemia-reperfusion injury, MitoQ shows significant protection against heart dysfunction, tissue damage, and mitochondrial dysfunction [191]. A similar study also has shown that MitoQ has the protective effects on cardiac ischemia-reperfusion injury [190]. MitoQ shows a protective effect against an increased blood pressure in a hypertensive rat model, which has mitochondrial oxidative damage in endothelial cells [192]. Therefore, it is likely to have antiarrhythmic properties.
Taken together, these findings suggest that reduced cardiac oxidation may be a promising therapeutic strategy for treatment of arrhythmias.
5. Summary
Oxidative stress is highly associated with cardiac arrhythmogenesis. Among other sources, altered mitochondrial metabolism can lead to ROS. Targeting oxidative changes, especially mediated by mitochondria, may represent a new strategy to prevent arrhythmias that could be safer than the conventional ion channel blockers used now.
Highlights.
Cardiomyopathies are associated with metabolic stress, oxidative stress, and arrhythmic risk.
Oxidative stress alters ion channels, Ca2+ handling, and gap junctions, possibly explaining the arrhythmic risk
Anti-oxidants may be useful antiarrhythmic drugs.
Highlights.
ROS mediate arrhythmogenesis through the alteration of ion homeostasis and structural remodeling.
Acknowledgement
This study was funded by National Institutes of Health grants, R01 HL085558, R01 HL073753, P01 HL058000, and a Veterans Affairs MERIT grant (SCD). Dr. Sovari received an American Heart Association Midwest Affiliate Postdoctoral Fellowship #AHA10POST4450037.
Abbreviations
- ΔΨm
mitochondrial membrane potential
- ACE
angiotensin converting enzyme
- ADP
adenosine diphosphate
- AF
atrial fibrillation
- AngII
angiotensin II
- AP
action potential
- ATP
adenosine-5′-triphosphate
- [Ca2+]i
intracellular Ca2+ concentration
- CICR
Ca2+-induced Ca2+ release CICR
- Cx
connexins
- DTT
dithiothreitol
- E-C coupling
excitation-contraction coupling
- ETC
electron transport chain
- GSH
glutathione
- GPD1-L
glycerol-3-phosphate dehydrogenase 1-like protein
- hERG
human ether-a-go-go-related gene
- HF
heart failure
- HIFα
Hypoxia inducing factor-1α
- IMAC
inner membrane anion channel
- INa
sodium current
- IKr
delayed rapid rectifier K+ current
- IKs
delayed slow rectifier K+ current
- MAP
monophasic action potential
- mitoKATP
mitochondrial ATP-sensitive potassium channel
- NAD
nicotinamide adenine dinucleotide
- Nav1.5
cardiac sodium channel
- NCX
Na+-Ca2+ exchanger
- NO
nitric oxide
- NOS
nitric oxide synthase
- PES
programmed electrical stimulation
- PK
protein kinase
- RAS
renin-angiotensin system
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- sarcKATP
sarcolemmal ATP-sensitive potassium channel
- SCD
sudden cardiac death
- SCN5A
cardiac sodium channel gene
- SERCA
sarcoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- TCA
tricarboxylic acid
- TdP
torsades de pointes
- VT
ventricular tachycardia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
SCD has patents pending on 1) Mitochondrial anti-oxidants for prevention of sudden death, 2) a method for modulating or controlling sodium channel current by ROS originating from mitochondria, 3) Activation of the renin-angiotensin system and SCD, 4) Oxidative stress markers predict AF, and 5) Modulation of sodium channels by NAD. Dudley SC is sole owner of ROS Technologies, Inc., a medical diagnostics company developing a blood test to predict sudden death risk in patients with heart failure.
References
- [1].Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al SM, et al. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–28. doi: 10.1016/j.pcad.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stecker EC, Vickers C, Waltz J, Socoteanu C, John BT, Mariani R, et al. Population-based analysis of sudden cardiac death with and without left ventricular systolic dysfunction: two-year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol. 2006;47:1161–6. doi: 10.1016/j.jacc.2005.11.045. [DOI] [PubMed] [Google Scholar]
- [3].Arking DE, Chugh SS, Chakravarti A, Spooner PM. Genomics in sudden cardiac death. Circ Res. 2004;94:712–23. doi: 10.1161/01.RES.0000123861.16082.95. [DOI] [PubMed] [Google Scholar]
- [4].Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, et al. ACC/AHA/ESC 2006 Guidelines for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association task force on practice guidelines and the European Society of Cardiology Committee for practice guidelines (Writing committee to revise the 2001 Guidelines for the management of patients with atrial fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e257–e354. doi: 10.1161/CIRCULATIONAHA.106.177292. [DOI] [PubMed] [Google Scholar]
- [5].Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med. 1982;306:1018–22. doi: 10.1056/NEJM198204293061703. [DOI] [PubMed] [Google Scholar]
- [6].Feinberg WM, Cornell ES, Nightingale SD, Pearce LA, Tracy RP, Hart RG, et al. Stroke Prevention in Atrial Fibrillation Investigators Relationship between prothrombin activation fragment F1.2 and international normalized ratio in patients with atrial fibrillation. Stroke. 1997;28:1101–6. doi: 10.1161/01.str.28.6.1101. [DOI] [PubMed] [Google Scholar]
- [7].Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–5. doi: 10.1001/jama.285.18.2370. [DOI] [PubMed] [Google Scholar]
- [8].Furberg CD, Psaty BM, Manolio TA, Gardin JM, Smith VE, Rautaharju PM. Prevalence of atrial fibrillation in elderly subjects (the Cardiovascular Health Study) Am J Cardiol. 1994;74:236–41. doi: 10.1016/0002-9149(94)90363-8. [DOI] [PubMed] [Google Scholar]
- [9].Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–8. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- [10].Lakshminarayan K, Anderson DC, Herzog CA, Qureshi AI. Clinical epidemiology of atrial fibrillation and related cerebrovascular events in the United States. Neurologist. 2008;14:143–50. doi: 10.1097/NRL.0b013e31815cffae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95:717–25. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- [12].Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, et al. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H1223–H1230. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- [13].Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–29. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- [14].Gao G, Dudley SC., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid Redox Signal. 2009;11:2265–77. doi: 10.1089/ars.2009.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barth AS, Tomaselli GF. Cardiac Metabolism and Arrhythmias. Circulation: Arrhythmia and Electrophysiology. 2009;2:327–35. doi: 10.1161/CIRCEP.108.817320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu M, Liu H, Dudley SC., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gibbs CL. Cardiac energetics. Physiol Rev. 1978;58:174–254. doi: 10.1152/physrev.1978.58.1.174. [DOI] [PubMed] [Google Scholar]
- [18].Suga H. Ventricular energetics. Physiol Rev. 1990;70:247–77. doi: 10.1152/physrev.1990.70.2.247. [DOI] [PubMed] [Google Scholar]
- [19].O’Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–15. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–63. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- [21].Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol. 2006;291:H2067–H2074. doi: 10.1152/ajpheart.00272.2006. [DOI] [PubMed] [Google Scholar]
- [22].Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–6. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Balaban RS. Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol. 1990;258:C377–C389. doi: 10.1152/ajpcell.1990.258.3.C377. [DOI] [PubMed] [Google Scholar]
- [24].Cortassa S, Aon MA, Marban E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84:2734–55. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Di LF, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- [26].Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- [27].Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15:241–6. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- [28].Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- [29].Schopfer P, Plachy C, Frahry G. Release of reactive oxygen intermediates (superoxide radicals, hydrogen peroxide, and hydroxyl radicals) and peroxidase in germinating radish seeds controlled by light, gibberellin, and abscisic acid. Plant Physiol. 2001;125:1591–602. doi: 10.1104/pp.125.4.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].de CJ, Wu R, Girouard H, Karas M, EL MA, Laplante MA, et al. Oxidative stress in hypertension. Clin Exp Hypertens. 2004;26:593–601. doi: 10.1081/ceh-200031904. [DOI] [PubMed] [Google Scholar]
- [31].Li N, Frigerio F, Maechler P. The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem Soc Trans. 2008;36:930–4. doi: 10.1042/BST0360930. [DOI] [PubMed] [Google Scholar]
- [32].Vassalle C, Petrozzi L, Botto N, Andreassi MG, Zucchelli GC. Oxidative stress and its association with coronary artery disease and different atherogenic risk factors. J Intern Med. 2004;256:308–15. doi: 10.1111/j.1365-2796.2004.01373.x. [DOI] [PubMed] [Google Scholar]
- [33].Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- [34].Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–7. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- [35].Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11:473–80. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- [36].Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated Levels of 8-iso-Prostaglandin F2{alpha} in Pericardial Fluid of Patients With Heart Failure: A Potential Role for In Vivo Oxidant Stress in Ventricular Dilatation and Progression to Heart Failure. Circulation. 1998;97:1536–9. doi: 10.1161/01.cir.97.16.1536. [DOI] [PubMed] [Google Scholar]
- [37].Dhalla AK, Singal PK. Antioxidant changes in hypertrophied and failing guinea pig hearts. American Journal of Physiology - Heart and Circulatory Physiology. 1994;266:H1280–H1285. doi: 10.1152/ajpheart.1994.266.4.H1280. [DOI] [PubMed] [Google Scholar]
- [38].Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96:2414–20. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- [39].Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002;8:132–40. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- [40].Vaziri ND, Lin CY, Farmand F, Sindhu RK. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003;63:186–94. doi: 10.1046/j.1523-1755.2003.00711.x. [DOI] [PubMed] [Google Scholar]
- [41].Gasparetto C, Malinverno A, Culacciati D, Gritti D, Prosperini PG, Specchia G, et al. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int J Immunopathol Pharmacol. 2005;18:487–96. doi: 10.1177/039463200501800308. [DOI] [PubMed] [Google Scholar]
- [42].Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD+ levels on sarcoplasmic reticulum Ca(2+) release in permeabilized rat ventricular myocytes. J Physiol. 2004;555:727–41. doi: 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- [44].Cortassa S, Aon MA, Winslow RL, O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J. 2004;87:2060–73. doi: 10.1529/biophysj.104.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Werns SW, Fantone JC, Ventura A, Lucchesi BR. Myocardial glutathione depletion impairs recovery of isolated blood-perfused hearts after global ischaemia. J Mol Cell Cardiol. 1992;24:1215–20. doi: 10.1016/0022-2828(92)93088-2. [DOI] [PubMed] [Google Scholar]
- [46].Ceconi C, Curello S, Cargnoni A, Ferrari R, Albertini A, Visioli O. The role of glutathione status in the protection against ischaemic and reperfusion damage: effects of N-acetyl cysteine. J Mol Cell Cardiol. 1988;20:5–13. doi: 10.1016/s0022-2828(88)80174-3. [DOI] [PubMed] [Google Scholar]
- [47].Damy T, Kirsch M, Khouzami L, Caramelle P, Le CP, Roudot-Thoraval F, et al. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS One. 2009;4:e4871. doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–8. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, et al. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. 2001;104:174–80. doi: 10.1161/01.cir.104.2.174. [DOI] [PubMed] [Google Scholar]
- [50].Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, et al. Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem. 2007;53:1652–7. doi: 10.1373/clinchem.2006.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Leftheriotis DI, Fountoulaki KT, Flevari PG, Parissis JT, Panou FK, Andreadou IT, et al. The predictive value of inflammatory and oxidative markers following the successful cardioversion of persistent lone atrial fibrillation. Int J Cardiol. 2009;135:361–9. doi: 10.1016/j.ijcard.2008.04.012. [DOI] [PubMed] [Google Scholar]
- [52].Dzhanashiya PK, Vladytskaya OV, Salibegashvili NV. Efficiency and mechanisms of the antioxidant effect of standard therapy and refracterin in the treatment of chronic heart failure in elderly patients with postinfarction cardiosclerosis. Bull Exp Biol Med. 2004;138:412–4. doi: 10.1007/s10517-005-0056-1. [DOI] [PubMed] [Google Scholar]
- [53].Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–30. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- [54].Choudhary G, Dudley SC., Jr. Heart failure, oxidative stress, and ion channel modulation. Congest Heart Fail. 2002;8:148–55. doi: 10.1111/j.1527-5299.2002.00716.x. [DOI] [PubMed] [Google Scholar]
- [55].Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002;295:1895–7. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- [56].Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–27. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sies H. Metabolic Compartmentation. Academic press; London: 1982. [Google Scholar]
- [58].Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Blinova K, Carroll S, Bose S, Smirnov AV, Harvey JJ, Knutson JR, et al. Distribution of mitochondrial NADH fluorescence lifetimes: steady-state kinetics of matrix NADH interactions. Biochemistry. 2005;44:2585–94. doi: 10.1021/bi0485124. [DOI] [PubMed] [Google Scholar]
- [60].Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, et al. Cardiac Na+ current regulation by pyridine nucleotides. Circ Res. 2009;105:737–45. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–30. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- [62].Dzhanashiya PK, Vladytskaya OV, Salibegashvili NV. Efficiency and mechanisms of the antioxidant effect of standard therapy and refracterin in the treatment of chronic heart failure in elderly patients with postinfarction cardiosclerosis. Bull Exp Biol Med. 2004;138:412–4. doi: 10.1007/s10517-005-0056-1. [DOI] [PubMed] [Google Scholar]
- [63].O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–6. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- [64].Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–13. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–77. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, et al. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- [68].Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519–28. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- [70].Cai H, Li Z, Goette A, Mera F, Honeycutt C, Feterik K, et al. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. 2002;106:2854–8. doi: 10.1161/01.cir.0000039327.11661.16. [DOI] [PubMed] [Google Scholar]
- [71].Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, et al. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002;277:48311–7. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- [72].Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89:E32–E38. doi: 10.1161/hh1801.097644. [DOI] [PubMed] [Google Scholar]
- [73].Shiroshita-Takeshita A, Schram G, Lavoie J, Nattel S. Effect of simvastatin and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia remodeling in dogs. Circulation. 2004;110:2313–9. doi: 10.1161/01.CIR.0000145163.56529.D1. [DOI] [PubMed] [Google Scholar]
- [74].Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Q, Taylor WR, et al. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- [75].Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- [76].Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, et al. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension. 2008;51:319–25. doi: 10.1161/HYPERTENSIONAHA.107.101980. [DOI] [PubMed] [Google Scholar]
- [77].Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200–6. doi: 10.1006/bbrc.2001.4493. [DOI] [PubMed] [Google Scholar]
- [78].Dudley SC, Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, et al. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation. 2005;112:1266–73. doi: 10.1161/CIRCULATIONAHA.105.538108. [DOI] [PubMed] [Google Scholar]
- [79].Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, et al. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–36. doi: 10.1161/01.RES.0000183735.09871.61. [DOI] [PubMed] [Google Scholar]
- [80].Shang LL, Gao G, Dudley SC., Jr. The tail of the cardiac sodium channel. Channels (Austin) 2008:2. doi: 10.4161/chan.2.3.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu H, Clancy C, Cormier J, Kass R. Mutations in cardiac sodium channels: clinical implications. Am J Pharmacogenomics. 2003;3:173–9. doi: 10.2165/00129785-200303030-00003. [DOI] [PubMed] [Google Scholar]
- [82].Letsas KP, Sideris A, Efremidis M, Pappas LK, Gavrielatos G, Filippatos GS, et al. Prevalence of paroxysmal atrial fibrillation in Brugada syndrome: a case series and a review of the literature. J Cardiovasc Med (Hagerstown) 2007;8:803–6. doi: 10.2459/JCM.0b013e3280112b21. [DOI] [PubMed] [Google Scholar]
- [83].Charital YM, Haasteren GV, Massiha A, Schlegel W, Fujita T. A functional NF-kappaB enhancer element in the first intron contributes to the control of c-fos transcription. Gene. 2008 doi: 10.1016/j.gene.2008.10.014. [DOI] [PubMed] [Google Scholar]
- [84].Kim YH, Lim DS, Lee JH, Shim WJ, Ro YM, Park GH, et al. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp Mol Med. 2003;35:336–49. doi: 10.1038/emm.2003.45. [DOI] [PubMed] [Google Scholar]
- [85].Genolet R, Wahli W, Michalik L. PPARs as drug targets to modulate inflammatory responses? Curr Drug Targets Inflamm Allergy. 2004;3:361–75. doi: 10.2174/1568010042634578. [DOI] [PubMed] [Google Scholar]
- [86].Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–8. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, et al. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–C379. doi: 10.1152/ajpcell.00186.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. 2006;6:111–30. doi: 10.1385/ct:6:2:111. [DOI] [PubMed] [Google Scholar]
- [89].Bowie A, O’Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- [90].Shang LL, Dudley SC., Jr. Tandem promoters and developmentally regulated 5′- and 3′-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J Biol Chem. 2005;280:933–40. doi: 10.1074/jbc.M409977200. [DOI] [PubMed] [Google Scholar]
- [91].Lebeche D, Kaprielian R, Hajjar R. Modulation of action potential duration on myocyte hypertrophic pathways. J Mol Cell Cardiol. 2006;40:725–35. doi: 10.1016/j.yjmcc.2006.01.018. [DOI] [PubMed] [Google Scholar]
- [92].Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, et al. Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol. 2007;293:H182–H192. doi: 10.1152/ajpheart.00684.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gassanov N, Brandt MC, Michels G, Lindner M, Er F, Hoppe UC. Angiotensin II-induced changes of calcium sparks and ionic currents in human atrial myocytes: potential role for early remodeling in atrial fibrillation. Cell Calcium. 2006;39:175–86. doi: 10.1016/j.ceca.2005.10.008. [DOI] [PubMed] [Google Scholar]
- [94].Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, et al. Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–54. doi: 10.1161/CIRCRESAHA.107.152918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zhou A, Ou AC, Cho A, Benz EJ, Jr., Huang SC. Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5′ splice site selection. Mol Cell Biol. 2008;28:5924–36. doi: 10.1128/MCB.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Gao G, Shang L, Zhou A, Zhe J, Gaconnet G, Dudley S. The possible mechanism of acquired sodium channel mRNA splicing variants with human heart failure. Circulation. 2008 [Google Scholar]
- [97].London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and Functional Characterization of Novel Glycerol-3-Phosphate Dehydrogenase 1-Like Gene (GPD1-L) Mutations in Sudden Infant Death Syndrome. Circulation. 2007;116:2253–9. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Papadatos GA, Wallerstein PMR, Head CEG, Ratcliff R, Brady PA, Benndorf K, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene SCN5a. Proc Natl Acad Sci U S A. 2002;99:6210–5. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Bruzzone S, Moreschi I, Guida L, Usai C, Zocchi E, De-aflora A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem J. 2006;393:697–704. doi: 10.1042/BJ20051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Romanello M, Padoan M, Franco L, Veronesi V, Moro L, D’Andrea P. Extracellular NAD+ induces calcium signaling and apoptosis in human osteoblastic cells. Biochem Biophys Res Commun. 2001;285:1226–31. doi: 10.1006/bbrc.2001.5325. [DOI] [PubMed] [Google Scholar]
- [102].Zhou J, Yi J, Hu N, George AL, Jr., Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000;87:33–8. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- [103].Shin HG, Murray KT. Conventional protein kinase C isoforms and cross-activation of protein kinase A regulate cardiac Na+ current. FEBS Lett. 2001;495:154–8. doi: 10.1016/s0014-5793(01)02380-8. [DOI] [PubMed] [Google Scholar]
- [104].Tipparaju SM, Saxena N, Liu SQ, Kumar R, Bhatnagar A. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am J Physiol Cell Physiol. 2005;288:C366–C376. doi: 10.1152/ajpcell.00354.2004. [DOI] [PubMed] [Google Scholar]
- [105].Tipparaju SM, Liu SQ, Barski OA, Bhatnagar A. NADPH binding to beta-subunit regulates inactivation of voltage-gated K+ channels. Biochem Biophys Res Commun. 2007;359:269–76. doi: 10.1016/j.bbrc.2007.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jungling E, Zitt C, et al. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J. 2003;371:1045–53. doi: 10.1042/BJ20021975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Herson PS, Dulock KA, Ashford ML. Characterization of a nicotinamideadenine dinucleotide-dependent cation channel in the CRI-G1 rat insulinoma cell line. J Physiol. 1997;505:65–76. doi: 10.1111/j.1469-7793.1997.065bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sasaki N, Sato T, Marban E, O’Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H1882–H1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- [110].Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–35. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O’Rourke B. From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol. 2009;41:1940–8. doi: 10.1016/j.biocel.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Brown DA, Aon MA, Akar FG, Liu T, Sorarrain N, O’Rourke B. Effects of 4′-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovasc Res. 2008;79:141–9. doi: 10.1093/cvr/cvn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Brown DA, O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88:241–9. doi: 10.1093/cvr/cvq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Beavis AD, Davatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J Bioenerg Biomembr. 1996;28:207–14. doi: 10.1007/BF02110652. [DOI] [PubMed] [Google Scholar]
- [115].Faivre JF, Findlay I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim Biophys Acta. 1990;1029:167–72. doi: 10.1016/0005-2736(90)90450-3. [DOI] [PubMed] [Google Scholar]
- [116].Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004;101:4447–52. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, et al. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–9. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].O’Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–15. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–24. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- [120].Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–15. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- [121].Laurita KR, Rosenbaum DS. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J Mol Cell Cardiol. 2008;44:31–43. doi: 10.1016/j.yjmcc.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Bers DM. Excitation-contraction (E-C) coupling and cardiac contractile force. Kluwer Academic Press; 2001. [Google Scholar]
- [123].Jin H, Lyon A, Akar FG. Arrhythmia mechanisms in the failing heart. Pacing and Clinical Electrophysiology. 2008;31:1048–56. doi: 10.1111/j.1540-8159.2008.01134.x. [DOI] [PubMed] [Google Scholar]
- [124].Weiss JN, Nivala M, Garfinkel A, Qu Z. Alternans and arrhythmias: from cell to heart. Circ Res. 2011;108:98–112. doi: 10.1161/CIRCRESAHA.110.223586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Overend CL, Eisner DA, O’Neill SC. Altered cardiac sarcoplasmic reticulum function of intact myocytes of rat ventricle during metabolic inhibition. Circ Res. 2001;88:181–7. doi: 10.1161/01.res.88.2.181. [DOI] [PubMed] [Google Scholar]
- [126].Fearon IM, Palmer ACV, Balmforth AJ, Ball SG, Varadi G, Peers C. Modulation of recombinant human cardiac L-type Ca2+ channel +|1C subunits by redox agents and hypoxia. The Journal of Physiology. 1999;514:629–37. doi: 10.1111/j.1469-7793.1999.629ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–93. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hool LC. Hypoxia increases the sensitivity of the L-type Ca(2+) current to beta-adrenergic receptor stimulation via a C2 region-containing protein kinase C isoform. Circ Res. 2000;87:1164–71. doi: 10.1161/01.res.87.12.1164. [DOI] [PubMed] [Google Scholar]
- [129].Lederer WJ, Nichols CG, Smith GL. The mechanism of early contractile failure of isolated rat ventricular myocytes subjected to complete metabolic inhibition. J Physiol. 1989;413:329–49. doi: 10.1113/jphysiol.1989.sp017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Losito VA, Tsushima RG, Diaz RJ, Wilson GJ, Backx PH. Preferential regulation of rabbit cardiac L-type Ca2+ current by glycolytic derived ATP via a direct allosteric pathway. J Physiol. 1998;511(Pt 1):67–78. doi: 10.1111/j.1469-7793.1998.067bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Chantawansri C, Huynh N, Yamanaka J, Garfinkel A, Lamp ST, Inoue M, et al. Effect of metabolic inhibition on couplon behavior in rabbit ventricular myocytes. Biophys J. 2008;94:1656–66. doi: 10.1529/biophysj.107.114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Goldhaber JI, Parker JM, Weiss JN. Mechanisms of excitation-contraction coupling failure during metabolic inhibition in guinea-pig ventricular myocytes. J Physiol. 1991;443:371–86. doi: 10.1113/jphysiol.1991.sp018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Xu KY, Zweier JL, Becker LC. Hydroxyl Radical Inhibits Sarcoplasmic Reticulum Ca2+-ATPase Function by Direct Attack on the ATP Binding Site. Circ Res. 1997;80:76–81. doi: 10.1161/01.res.80.1.76. [DOI] [PubMed] [Google Scholar]
- [134].Scherer N, Deamer D. Oxidation of thiols in the Ca2+-ATPase of sarcoplasmic reticulum microsomes. Biochim Biophys Acta. 1986;862:309–17. doi: 10.1016/0005-2736(86)90233-6. [DOI] [PubMed] [Google Scholar]
- [135].Gao Z, Barth AS, DiSilvestre D, Akar FG, Tian Y, Tanskanen A, et al. Key pathways associated with heart failure development revealed by gene networks correlated with cardiac remodeling. Physiol Genomics. 2008;35:222–30. doi: 10.1152/physiolgenomics.00100.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- [137].Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovascular Research. 2006;71:310–21. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- [138].Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–72. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Anzai K, Ogawa K, Kuniyasu A, Ozawa T, Yamamoto H, Nakayama H. Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem Biophys Res Commun. 1998;249:938–42. doi: 10.1006/bbrc.1998.9244. [DOI] [PubMed] [Google Scholar]
- [140].Liu G, Abramson JJ, Zable AC, Pessah IN. Direct evidence for the existence and functional role of hyperreactive sulfhydryls on the ryanodine receptor-triadin complex selectively labeled by the coumarin maleimide 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin. Molecular Pharmacology. 1994;45:189–200. [PubMed] [Google Scholar]
- [141].Zable AC, Favero TG, Abramson JJ. Glutathione Modulates Ryanodine Receptor from Skeletal Muscle Sarcoplasmic Reticulum. Journal of Biological Chemistry. 1997;272:7069–77. doi: 10.1074/jbc.272.11.7069. [DOI] [PubMed] [Google Scholar]
- [142].Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin Binding and Inhibition of Cardiac Muscle Calcium Release Channel (Ryanodine Receptor) Journal of Biological Chemistry. 2001;276:20144–53. doi: 10.1074/jbc.M010771200. [DOI] [PubMed] [Google Scholar]
- [143].Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD+ levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J Physiol. 2004;555:727–41. doi: 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- [145].Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218–25. doi: 10.1161/01.res.0000028455.36940.5e. [DOI] [PubMed] [Google Scholar]
- [146].Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- [147].Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–92. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- [148].Hinata M, Matsuoka I, Iwamoto T, Watanabe Y, Kimura J. Mechanism of Na+/Ca2+ exchanger activation by hydrogen peroxide in guinea-pig ventricular myocytes. J Pharmacol Sci. 2007;103:283–92. doi: 10.1254/jphs.fp0060015. [DOI] [PubMed] [Google Scholar]
- [149].Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–65. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–93. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- [152].Wang Y, Joyner RW, Wagner MB, Cheng J, Lai D, Crawford BH. Stretch-activated channel activation promotes early afterdepolarizations in rat ventricular myocytes under oxidative stress. Am J Physiol Heart Circ Physiol. 2009;296:H1227–H1235. doi: 10.1152/ajpheart.00808.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Fotopoulos GD, Mason MJ, Walker S, Jepson NS, Patel DJ, Mitchell AG, et al. Stabilisation of medically refractory ventricular arrhythmia by intra-aortic balloon counterpulsation. Heart. 1999;82:96–100. doi: 10.1136/hrt.82.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kamkin A, Kiseleva I, Isenberg G. Stretch-activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res. 2000;48:409–20. doi: 10.1016/s0008-6363(00)00208-x. [DOI] [PubMed] [Google Scholar]
- [155].Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden Death Associated With Short-QT Syndrome Linked to Mutations in HERG. Circulation. 2004;109:30–5. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- [156].Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, et al. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1446–H1455. doi: 10.1152/ajpheart.01356.2005. [DOI] [PubMed] [Google Scholar]
- [157].Berube J, Caouette D, Daleau P. Hydrogen peroxide modifies the kinetics of HERG channel expressed in a mammalian cell line. J Pharmacol Exp Ther. 2001;297:96–102. [PubMed] [Google Scholar]
- [158].Kolbe K, Schonherr R, Gessner G, Sahoo N, Hoshi T, Heinemann SH. Cysteine 723 in the C-linker segment confers oxidative inhibition of hERG1 potassium channels. J Physiol. 2010;588:2999–3009. doi: 10.1113/jphysiol.2010.192468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Su Z, Limberis J, Martin RL, Xu R, Kolbe K, Heinemann SH, et al. Functional consequences of methionine oxidation of hERG potassium channels. Biochem Pharmacol. 2007;74:702–11. doi: 10.1016/j.bcp.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Xu Z, Patel KP, Lou MF, Rozanski GJ. Up-regulation of K+ channels in diabetic rat ventricular myocytes by insulin and glutathione. Cardiovascular Research. 2002;53:80–8. doi: 10.1016/s0008-6363(01)00446-1. [DOI] [PubMed] [Google Scholar]
- [161].Marionneau C, Aimond F, Brunet S, Niwa N, Finck B, Kelly DP, et al. PPAR[alpha]-mediated remodeling of repolarizing voltage-gated K+ (Kv) channels in a mouse model of metabolic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2008;44:1002–15. doi: 10.1016/j.yjmcc.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Rozanski GJ, Xu Z. Glutathione and K+ channel remodeling in postinfarction rat heart. American Journal of Physiology - Heart and Circulatory Physiology. 2002;282:H2346–H2355. doi: 10.1152/ajpheart.00894.2001. [DOI] [PubMed] [Google Scholar]
- [163].Rozanski GJ, Xu Z, Zhang K, Patel KP. Altered K+ current of ventricular myocytes in rats with chronic myocardial infarction. American Journal of Physiology - Heart and Circulatory Physiology. 1998;274:H259–H265. doi: 10.1152/ajpheart.1998.274.1.H259. [DOI] [PubMed] [Google Scholar]
- [164].Gomez R, Caballero R, Barana A, Amoros I, Calvo E, Lopez JA, et al. Nitric Oxide Increases Cardiac IK1 by Nitrosylation of Cysteine 76 of Kir2.1 Channels. Circ Res. 2009;105:383–92. doi: 10.1161/CIRCRESAHA.109.197558. [DOI] [PubMed] [Google Scholar]
- [165].Spach MS, Kootsey JM. The nature of electrical propagation in cardiac muscle. Am J Physiol. 1983;244:H3–22. doi: 10.1152/ajpheart.1983.244.1.H3. [DOI] [PubMed] [Google Scholar]
- [166].Unger VM, Kumar NM, Gilula NB, Yeager M. Expression, two-dimensional crystallization, and electron cryo-crystallography of recombinant gap junction membrane channels. J Struct Biol. 1999;128:98–105. doi: 10.1006/jsbi.1999.4184. [DOI] [PubMed] [Google Scholar]
- [167].Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover: implications for cardiac excitability. Circ Res. 2000;86:723–8. doi: 10.1161/01.res.86.7.723. [DOI] [PubMed] [Google Scholar]
- [168].Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431–88. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- [169].Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–12. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Duffy HS, Ashton AW, O’Donnell P, Coombs W, Taffet SM, Delmar M, et al. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–22. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- [171].Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–62. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- [172].Jain SK, Schuessler RB, Saffitz JE. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ Res. 2003;92:1138–44. doi: 10.1161/01.RES.0000074883.66422.C5. [DOI] [PubMed] [Google Scholar]
- [173].Miura T, Ohnuma Y, Kuno A, Tanno M, Ichikawa Y, Nakamura Y, et al. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;286:H214–H221. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- [174].Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–21. [PMC free article] [PubMed] [Google Scholar]
- [175].Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49:6819–32. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- [176].Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- [177].Pham NA, Magalhaes JM, Do T, Schwock J, Dhani N, Cao PJ, et al. Activation of Src and Src-associated signaling pathways in relation to hypoxia in human cancer xenograft models. Int J Cancer. 2009;124:280–6. doi: 10.1002/ijc.23912. [DOI] [PubMed] [Google Scholar]
- [178].Trevino JG, Summy JM, Lesslie DP, Parikh NU, Hong DS, Lee FY, et al. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am J Pathol. 2006;168:962–72. doi: 10.2353/ajpath.2006.050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Wojcik EJ, Sharifpoor S, Miller NA, Wright TG, Watering R, Tremblay EA, et al. A novel activating function of c-Src and Stat3 on HGF transcription in mammary carcinoma cells. Oncogene. 2006;25:2773–84. doi: 10.1038/sj.onc.1209306. [DOI] [PubMed] [Google Scholar]
- [180].Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23:4598–610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Khadaroo RG, He R, Parodo J, Powers KA, Marshall JC, Kapus A, et al. The role of the Src family of tyrosine kinases after oxidant-induced lung injury in vivo. Surgery. 2004;136:483–8. doi: 10.1016/j.surg.2004.05.029. [DOI] [PubMed] [Google Scholar]
- [183].Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- [184].Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- [185].Sovari AA, Jiao Z, Dolmatova E, Kumar V, Wang K, Bonini MG, et al. Abstract 17524: c-Src Tyrosine Kinase Is a Potential Therapeutic Target to Prevent Angiotensin-II Mediated Connexin43 Remodeling and Ventricular Arrhythmia. Circulation. 2010;122:A17524. [Google Scholar]
- [186].Sochman J, Kolc J, Vrana M, Fabian J. Cardioprotective effects of N-acetylcysteine: the reduction in the extent of infarction and occurrence of reperfusion arrhythmias in the dog. Int J Cardiol. 1990;28:191–6. doi: 10.1016/0167-5273(90)90060-i. [DOI] [PubMed] [Google Scholar]
- [187].Ganitkevich V, Reil S, Schwethelm B, Schroeter T, Benndorf K. Dynamic responses of single cardiomyocytes to graded ischemia studied by oxygen clamp in on-chip picochambers. Circ Res. 2006;99:165–71. doi: 10.1161/01.RES.0000232321.89714.0e. [DOI] [PubMed] [Google Scholar]
- [188].Walker MK, Vergely C, Lecour S, Abadie C, Maupoil V, Rochette L. Vitamin E analogues reduce the incidence of ventricular fibrillations and scavenge free radicals. Fundam Clin Pharmacol. 1998;12:164–72. doi: 10.1111/j.1472-8206.1998.tb00937.x. [DOI] [PubMed] [Google Scholar]
- [189].Torok B, Roth E, Mezey B, Temes G, Toth K, Pollak Z. Promising reduction of ventricular fibrillation in experimentally induced heart infarction by antioxidant therapy. Basic Res Cardiol. 1987;82(Suppl 2):347–53. doi: 10.1007/978-3-662-11289-2_34. [DOI] [PubMed] [Google Scholar]
- [190].Neuzil J, Widen C, Gellert N, Swettenham E, Zobalova R, Dong LF, et al. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 2007;12:148–62. doi: 10.1179/135100007X200227. [DOI] [PubMed] [Google Scholar]
- [191].Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. The FASEB Journal. 2005;19:1088–95. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- [192].Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–8. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- [193].Beavis AD. Properties of the inner membrane anion channel in intact mitochondria. J Bioenerg Biomembr. 1992;24:77–90. doi: 10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- [194].Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–20. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- [195].Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006;8:E277–E283. doi: 10.1007/BF02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–90. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- [197].Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–19. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]