

Abstract
Rad9 plays a crucial role in maintaining genomic stability by regulating cell cycle checkpoints, DNA repair, telomere stability and apoptosis. Rad9 controls these processes mainly as part of the heterotrimeric 9-1-1 (Rad9-Hus1-Rad1) complex. However, in recent years it has been demonstrated that Rad9 can also act independently of the 9-1-1 complex as a transcriptional factor, participate in immunoglobulin class switch recombination and show 3’–5’ exonuclease activity. Aberrant Rad9 expression has been associated with prostate, breast, lung, skin, thyroid and gastric cancers. High expression of Rad9 is causally related to, at least, human prostate cancer growth. On the other hand, deletion of Mrad9, the mouse homolog, is responsible for increased skin cancer incidence. These results reveal that Rad9 can act as an oncogene or tumor suppressor. Which of the many functions of Rad9 are causally related to initiation and progression of tumorigenesis and the mechanistic details by which Rad9 induces or suppresses tumorigenesis are presently not known, but are crucial for the development of targeted therapeutic interventions.
Keywords: Rad9, oncogene, tumor suppressor, cell cycle checkpoint, DNA repair
Environmental stress and normal metabolic events like cell metabolism and DNA replication pose a constant threat to genomic stability of all living organisms. To maintain genomic stability cells have developed evolutionarily conserved signal transduction pathways called DNA damage response (DDR) pathways that sense the damage, transiently arrest the cell cycle and recruit specialized proteins that repair the lesions or when damage is severe activate an apoptotic program. The main DDR pathways that respond to different kinds of DNA damage are controlled by ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad53-related) kinases. ATM and ATR phosphorylate numerous downstream proteins that culminate in the activation of CHK2 and CHK1 checkpoint kinases. Malfunctioning of DNA damage/repair pathways results in cell death or various health related issues, including developmental defects, neurodegenerative diseases and cancer.
Rad9 is an evolutionarily conserved gene that regulates cell cycle checkpoints and promotes resistance to DNA damage [Lieberman, 2006]. Rad9 forms a complex with Hus1 and Rad1 [Volkmer and Karnitz, 1999] and that complex serves as an early damage sensor in the cell checkpoint control pathway [Ciccia and Elledge, 2010]. Inactivation of Rad9 promotes spontaneous chromosome aberrations and embryonic lethality in mice, thus emphasizing its importance in the DNA damage response pathway and perhaps other critical functions [Hopkins et al., 2004].
Besides its role in cell cycle checkpoint control, human Rad9 has many additional functions, including DNA repair, radioresistance [Lieberman, 2006], transactivation of downstream genes [Yin et al., 2004], apoptosis [Komatsu et al., 2000a], 3’–5’ exonuclease activity [Bessho and Sancar, 2000], androgen receptor signaling repression [Wang et al., 2004], ribonucleotide metabolism (in conjunction with CAD, carbamoyl phosphate synthetase/aspartate transcarbamoylase/dihydroorotase multienzyme protein complex), immunoglobulin class switching [An et al., 2010] and telomere maintenance [Francia et al., 2006]. A multitude of proteins interact with Rad9 either directly or indirectly through the Rad9-Hus1-Rad1 complex and mediate diverse physiological outcomes (Table 1).
TABLE I. Human Rad9 interacting proteins.
The list of Rad9-interacting partners includes proteins that have been shown experimentally to associate directly with Rad9 and proteins that associate with at least the 9-1-1 complex, which may or may not interact directly with Rad9.
| Protein | Rad9/9-1-1 | Function | References |
|---|---|---|---|
| Cell cycle checkpoint control | |||
| RPA70 | Rad9 | DNA damage checkpoint | Wu et al., 2005; Xu et al., 2008 |
| RPA32 | Rad9 | DNA damage checkpoint | Wu et al., 2005 |
| Hus1 | Rad9 | DNA damage checkpoint | Volkmer & Karnitz, 1999 |
| Rad1 | Rad9 | DNA damage checkpoint | Volkmer & Karnitz, 1999 |
| TopBP1 | Rad9 | DNA damage checkpoint | Delacroix et al, 2007 |
| RHINO | 9-1-1 | DNA damage checkpoint, HR | Cotta-Ramusino et al., 2011 |
| Casein kinase II | Rad9 | Promotes interaction of 9-1-1 with TopBP1 | Takeishi et al., 2010 |
| p53 | Rad9 | p21 modulation | Ishikawa et al., 2007 |
| PRMT5 | Rad9 | S/M and G2/M checkpoint | He et al., 2011 |
| HDAC1 | Hus1, maybe Rad9 | G2/M checkpoint control | Cai et al., 2000 |
| DNA repair | |||
| NEIL1 | Rad9 | BER | Guan et al., 2007a |
| TDG | Rad9 | BER | Guan et al., 2007b |
| OGG1 | Rad9 | BER | Park et al., 2009 |
| DNA ligase I | 9-1-1 | BER | Wang et al., 2006 |
| DNA polymerase β | 9-1-1 | BER | Toueille et al., 2004 |
| APE1 | 9-1-1 | BER | Gembka et al., 2007 |
| FEN1 | 9-1-1 | BER | Wang et al., 2004 |
| MLH1 | Rad9 | MMR | He et al., 2008 |
| hMSH2 | Rad9 | MMR | Bai et al., 2010 |
| hMSH3 | Rad9 | MMR | Bai et al., 2010 |
| hMSH6 | Rad9 | MMR | Bai et al., 2010 |
| TLK1B | Rad9 | DSBR | Sunavala-Dossabhoy & De Benedetti, 2009 |
| Rad51 | Rad9 | HR | Pandita, et al. 2006 |
| TRF2 | Rad9 | Telomere integrity | Pandita, et al. 2006 |
| CAD | Rad9 | Ribonucleotide synthesis | Lindsey-Boltz et al., 2004 |
| Apoptosis | |||
| Frag1 | Rad9 | Apoptosis | Ishii et al., 2005 |
| Bcl-2 | Rad9 | Apoptosis | Komatsu et al., 2000 |
| Bcl-XL | Rad9 | Apoptosis | Komatsu et al., 2000 |
| c-Abl | Rad9 | Apoptosis | Yoshida et al., 2002 |
| PKC delta | Rad9 | Assembly of 9-1-1; apoptosis | Yoshida et al., 2003 |
| Other functions | |||
| AR | Rad9 | Inhibition of AR | Wang et al., 2004 |
RPA70: replication protein A 70kDa subunit; RPA32: replication protein A 32kDa subunit; TopBP1: topoisomerase IIβ binding protein 1; RHINO: Rad9, Rad1, Hus1 interacting nuclear orphan; PRMT5: protein methyltransferase 5; HDCA1: histone deacetylase 1; BER: base excision repair; MMR: mismatch repair; DSBR: double-strand break repair; HR: homologous recombination; OGG1: 8-oxoguanine DNA glycosylase; NEIL1: NEI-like DNA glycosylase 1; TDG: thymine DNA glycosylase; APE1: apurinic/apyrimidinic endonuclease 1; FEN1: flap endonuclease 1; MLH1: MutL homolog 1; hMSH2, 3, 6: human mutS homolog 2, 3, 6; CAD: carbamoyl phosphate synthetase/aspartate transcarbamoylase/dihydroorotase; TLK1B: tousled-like kinase 1B; TRF2: Telomeric repeat-binding factor 2; FRAG1: fragility-associated gene 1; bcl-2: B-cell CLL/lymphoma 2; bcl-XL: apoptosis regulator Bcl-X large isoform; AR: androgen receptor.
In recent years it has become apparent that Rad9 is a multifunctional protein and plays a key role in several types of cancer. However, it is not clear at this time which functions impact on tumorigenesis. Below is a review of the many activities assigned to Rad9, and speculation as to which influence its function in tumor development.
STRUCTURE OF RAD9 PROTEIN
Human Rad9 protein is a highly complex molecule, with multiple protein interaction sites and functional domains (Figure 1). The protein can be broadly divided into two functional regions, the N-terminal part (aa1-270) that contains two proliferating cell nuclear antigen (PCNA)-like domains, responsible for binding Hus1 and Rad1, and the C-terminal tail (aa271-391) that does not participate in the assembly of the 9-1-1 complex. The latter is, however, important for full activation of the DNA damage response. In the N-terminal portion there is a functional BH3 (bcl-2 homology 3) motif and 3’–5’ exonuclease activity. The C-terminal tail contains a proline-rich region, as well as a nuclear localization signal, and is constitutively phosphorylated at a number of serines and threonines (i.e., S277, S328, S336, S341, T355 and S387; St Onge et al, 2001). Cell cycle dependent phosphorylation of Rad9 at T292 occurs during mitosis in a cdc2-dependent manner [St Onge et al, 2003]. Moreover, additional phosphorylation in the Rad9 C-terminal tail is observed in response to DNA damage [Roos-Mattjus et al, 2003]. Neither constitutive nor damage-induced phosphorylation influences interaction of Rad9 with its partners Rad1 and Hus1 [Volkmer and Karnitz, 1999]. The nuclear localization signal (NLS) is functional and it is needed not only for the translocation of Rad9 to the nucleus, but also for Rad1 and Hus1. Overexpression of a deletion mutant of Rad9 lacking the NLS-containing C-terminal region can bypass the G2 checkpoint and result in cell death after ionizing radiation or hydroxyurea treatment [Hirai and Wang, 2002].
Figure 1. Human Rad9 structural and functional domains.
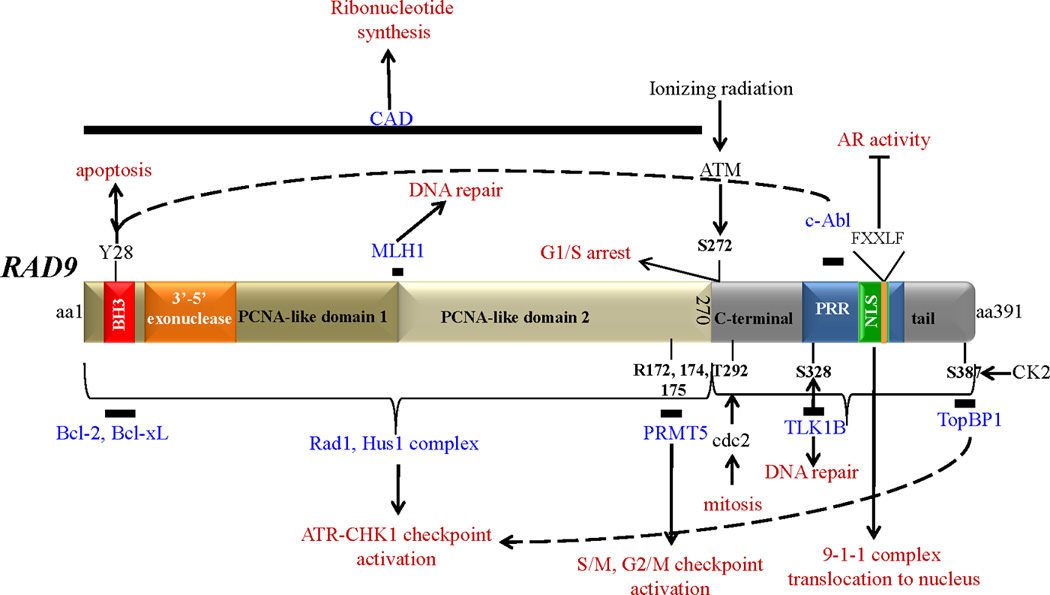
Rad9 is a 391 aa protein, roughly divided into two parts; the N-terminal aa1-270 contains two PCNA-like domains, primarily involved in assembling the 9-1-1 complex, and the C-terminal tail domain aa271-391 that does not participate in the 9-1-1 complex, but binds a number of proteins crucial for its function, is highly phosphorylated, and contains a proline-rich domain (PRR) and a nuclear localization signal (NLS). Solid bars denote the position of Rad9 that interacts with the depicted proteins (shown in blue). Proteins that Rad9 interacts with, but their exact binding position is not known, are not shown. FXXLF is the Rad9 sequence that binds to AR. The functional outcome of Rad9-protein association is shown in red. Phosphorylation sites: Y28, S272, T292, S328, S387.
Methylation sites: R172, R174, R175. Abbreviations are as follows: BH3: bcl-2 homology motif 3; PCNA: proliferating cell nuclear antigen; PRR: proline-rich domain; NLS: nuclear localization signal; PRMT5: protein methyltransferase 5; TLK-1B: tousled-like kinase 1B; TopBP1: topoisomerase IIb binding protein; MLH1: mutL homolog 1; CAD: carbamoyl phosphate synthetase/aspartate transcarbamoylase/dihydroorotase; AR: androgen receptor; CK2: casein kinase 2; ATM: Ataxia telangiectasia mutated; ATR: ATM and Rad3-related; CHK1: checkpoint kinase 1.
RAD9 AND CELL CYCLE CHECKPOINTS
Certain DNA lesions caused either by endogenous factors (e.g. reactive oxygen species produced by normal cellular metabolism), exogenous stress (e.g. UV light) or replication stress, can activate the ATR DNA damage pathway. These lesions create single stranded DNA (ssDNA) around the damaged site, which is then coated by replication protein A (RPA), thus generating a platform for binding of other proteins. RPA binds and recruits ATR/ATRIP through its direct interaction with ATRIP, and independently RPA recruits the Rad17-RFC (replication factor C) clamp loader. The Rad17-RFC complex then assists in the loading of Rad9-Hus1-Rad1 onto DNA. The 9-1-1 heterotrimeric sliding clamp shows structural similarity to the PCNA clamp, which serves as a processivity factor for DNA polymerases during normal replication. Subsequently, the 9-1-1 complex binds the recently discovered RHINO (Rad9, Rad1, Hus1 interacting nuclear orphan) [Cotta-Ramusino, 2011] and TopBP1 [Lee et al., 2007a], and localizes them to the damage site. TopBP1 and RHINO participate in the amplification of the ATR signal, which is absolutely necessary for full activation of Chk1, as well as the multiple substrates downstream that culminate in cell cycle arrest /delay. TopBP1 is recruited to the damage site exclusively through its interaction with Rad9 and requires that S-387 of Rad9 be phosphorylated [Delacroix et al., 2007]. Serine-387 (along with serine-341) is phosphorylated by casein kinase 2, and when both serines are mutated cells become sensitive to UV irradiation and methyl methane sulfonate (MMS) treatment [Takeishi et al., 2010].
Double-strand breaks (DSB) activate another branch of the DDR pathway that is controlled by ATM kinase [Ciccia and Elledge, 2010], and Rad9 has a role in it as well. Mrad9−/− mouse ES cells are sensitive not only to hydroxyurea and UV light (which activate ATR kinase), but also to ionizing radiation (IR; activates the ATM pathway) [Roos-Mattju et al, 2003]. Rad9 is known to associate with chromatin at sites of DSBs in human cells [Greer et al., 2003; Warmerdam et al., 2009] and human Rad18, an E3 ubiquitin ligase involved in replicative damage bypass and DNA double strand break repair, recruits Rad9 to DSB sites [Inagaki et al., 2011]. Furthermore, ATM kinase phosphorylates Rad9 at S-272 in response to IR. ATM-mediated phosphorylation of Rad9 is required for IR-induced G1 checkpoint activation [Chen et al., 2001]. However, depletion of Rad9 does not cause CHK2 phosphorylation and does not impair G2/M checkpoint activation in response to IR [Bao et al., 2004; Pandita et al., 2006], implying that Rad9 has functions beyond checkpoint activation, such as activities related to the repair of these lesions.
Phosphorylation is not the only post-translational modification important for the role of Rad9 in checkpoint control. Arginine methylation has an impact on Rad9 function as well [He et al., 2011]. Rad9 interacts with protein arginine methyltransferase 5 (PRMT5), resulting in the methylation of Rad9 arginines 172, 174, 175 in the glycine-arginine rich sequence commonly found in proteins methylated by PRMTs. Arginine methylation of Rad9 is required for genotoxin-induced CHK1 activation, as well as S/M and G2/M cell cycle checkpoints.
RAD9 AND DNA REPAIR
The function of Rad9 is not limited to checkpoint activation. It plays a critical role in DNA repair and is involved in almost every aspect of this process. In fact, it has been proposed that the main role of Rad9 is in DNA repair [Pandita et al., 2006].
Reactive oxygen species are commonly generated as by-products of normal cellular metabolism, when cells are challenged with ionizing radiation or are exposed to various chemicals. This causes the induction of oxidative base lesions, which are repaired by the dedicated base excision repair (BER) mechanism. Rad9 and the 9-1-1 complex are intimately involved in this process and influence every step of the pathway. Rad9 interacts with a number of DNA glycosylases that function in the first step of BER. Human NEIL1 DNA glycosylase, involved in repairing oxidatively damaged DNA bases, thymine DNA glycosylase that repairs deaminated cytosines and 5-methyl-cytosines caused by agents such as N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), and 8-oxoguanine DNA glycosylase (OGG1) that repairs the specific oxidized nucleotide caused by hydrogen peroxide, [Guan et al., 2007a; Guan et al., 2007b; Park et al., 2009] physically interact with and are stimulated by Rad9, either as free protein or as part of the 9-1-1 complex. Furthermore, in S. pombe, 9-1-1 physically interacts with the MYH (MutY homolog) DNA glycosylase [Chang and Lu, 2005].
Rad9 as part of the 9-1-1 complex also interacts with and stimulates the activity of apurinic/apyrimidinic endonuclease 1 (APE1), an enzyme that follows the action of DNA glycosylases on the damaged site [Gembka et al., 2007; Balakrishnan et al., 2009]. The next step in the BER process is mediated by DNA polymerase β (Polβ), which is the major repair polymerase in human cells and is critical for BER. The 9-1-1 complex physically interacts with Polβ and stimulates its activity [Toueille et al., 2004]. Likewise, Flap endonuclease 1 (FEN1), which is a key nuclease in BER following the action of Polβ, interacts with the 9-1-1 complex resulting in stimulated activity [Wang et al., 2004]. Finally, DNA ligase I is the enzyme at the last step of BER, and also associates with as well as is stimulated by the 9-1-1 complex [Smirnova et al, 2005; Wang et al., 2006]. Many of the BER proteins have been linked to cancer as tumor suppressors. Rad9 could impact on tumorigenesis by controlling the activation of these proteins (see below).
Mismatch repair (MMR) is an important mechanism that corrects base-base mismatches and deletion/insertion mispairs accidentally generated during DNA replication or in cells treated with alkylating agents. Central to this pathway are two protein complexes, MutLα (MLH1-PMS2) and MutSα (MSH2-MSH6). MMR proteins interact with members of the ATR-Chk1 pathway [Liu et al., 2010]. Human and mouse Rad9 can interact with MLH1 [He et al., 2008]. When the interaction is disrupted by a single point mutation in Rad9, MMR activity is greatly reduced, thus indicating that Rad9 is important for the function of this pathway. Rad9 and 9-1-1 also interact with human MSH2, MSH3, and MSH6. Rad9 and MSH6 co-localize to nuclear foci in cells exposed to MNNG [Bai et al., 2010].
Rad9 is also involved in nucleotide excision repair (NER). DDC1, the Rad9 homolog in fission yeast S. pombe, interacts with Rad14, which is a homolog of human XPA, and participates in lesion recognition during NER [Giannattasio et al., 2004]. In mammalian cells, it has been postulated that NER triggers ATR checkpoint signaling, and downregulation of early stage NER proteins XPA and XPC (xeroderma pigmentosum proteins A and C) impairs localization of Rad9 to sites of DNA damage after exposure to UV light [Warmerdam et al., 2009].
DNA interstrand cross-links (ICL) are toxic lesions induced by anti-proliferative agents such as mitomycin C, cisplatin and photoactivated psoralens that inhibit DNA synthesis. Among the multiple factors required to repair ICLs are those from the Fanconi Anemia pathway. Rad9 and Rad17 have recently been implicated in ICL resistance and optimal activation of FANCD2. Depletion of either Rad9 or Rad17 results in lower mono-ubiquitylation and thus reduced activation of FANCD2 through ATR activation [Guervilly et al., 2008].
Two main pathways of DSB repair exist in mammalian cells: error-free homologous recombination (HR) that is active during S and G2 phases of the cell cycle, and error-prone non-homologous end joining (NHEJ) that predominates at the G1 phase. Human Rad9 interacts with hRad51 and influences HR repair of DSBs [Pandita et al., 2006]. On the other hand, in the same report it was concluded that Rad9 expression has no impact on NHEJ.
Rad9 as part of the 9-1-1 clamp has a role in post-replication repair [Jansen et al., 2007]. Translesion synthesis (TLS) is catalyzed by specialized DNA polymerases (e.g. polη and polκ) and is used by the cell to bypass unrepaired DNA lesions. TLS proceeds through two pathways, the error-prone mono-ubiquitylation and the error-free poly-ubiquitylation of PCNA. 9-1-1 favors the latter pathway. In S. pombe, damage-induced phosphorylation of Rad9 at T-227 interacts with the Mms2-Ubc13-Rad5 complex that poly-ubiquitylates PCNA, thus favoring the relatively error-free branch of TLS [Lazzaro et al., 2009].
Transcriptional induction of ribonucleotide reductase and the subsequent increase in dNTP pools follows the acquisition of DNA damage. Rad9 binds and stimulates the carbamoyl phosphate synthetase II (CPSase II) activity of CAD, which is the rate-limiting step for the de novo synthesis of pyrimidine nucleotides that leads to cell growth. Only free Rad9, not Rad9 as part of the 9-1-1 complex, binds and activates CPSase II [Lindsey-Boltz et al., 2004]. CPSase II activity is also regulated transcriptionally by c-myc and post-translationally by mitogen-activated protein kinase and protein kinase A phosphorylation [Huang and Graves, 2003], and it is increased in tumor cells [Sigoillot et al., 2004]. It is therefore conceivable that Rad9 may promote tumorigenesis through the regulation of CPSase II activity.
RAD9 AND APOPTOSIS
Rad9 has a function as a death mediator, and numerous reports support this role. Human and yeast Rad9 contain a BH3 motif that is common in all BH3-only pro-apoptotic members of the Bcl-2 family proteins [Komatsu et al., 2000a; Komatsu et al., 2000b]. When Rad9 is overexpressed, it translocates to mitochondria and, much like BH3-only proteins, it binds and neutralizes the anti-apoptotic activity of Bcl-2 and Bcl-xL proteins, thus promoting cell death. Moreover, a tyrosine residue within the BH3 motif is subject to phosphorylation by c-Abl [Yoshida et al., 2002]. In response to DNA damaging agents, c-Abl phosphorylates Rad9 at Y-28, inducing the binding of Rad9 to Bcl-xL and promoting apoptosis. PKC delta is also able to phosphorylate and activate Rad9 pro-apoptotic function in response to genotoxin exposure [Yoshida et al., 2003].
Fragility-associated gene 1 (FRAG1; official symbol ATAD5), a homolog of alternative replication factor C subunits, is a caretaker gene that ensures genomic stability under conditions of replication stress and plays a role in the decision of the cell to arrest the cell cycle or undergo apoptosis in response to DNA damage. Activation of ATR after replication stress results in downregulation of FRAG1 expression and induction of apoptosis through the release of Rad9 from damaged chromatin. Free Rad9 binds and inhibits Bcl-2, which in turn allows activation of Bax and cell death [Ishii et al., 2005].
Lastly, p63 induces apoptosis by activating signaling via death receptors and mitochondria [Gressner et al., 2005]. Specifically, TP63α upregulates expression of pro-apoptotic Bcl-2 family members like Bax and BCL2L11 (BIM), and expression of Rad9, DAP3 (death associated protein 3) and APAF1 (apoptotic protease-activating factor 1).
Although Rad9 overexpression induces apoptosis, its absence can also result in apoptosis. Replication stress in Rad9−/− DT40 cells has been found to promote cell death, because there is no proper CHK1 activation, which is important for cell survival after replication fork stalling [Zachos et al., 2003]. In addition, Mrad9−/− mouse ES cells demonstrate high spontaneous levels of apoptosis [Zhu et al., 2005]. Apparently, cancer cells that overexpress Rad9 have developed the mechanisms to neutralize the pro-apoptotic function of Rad9 while retaining its tumor promoting activity. Other proteins involved in DNA repair display the same dual mode of action. Deletion of Rad51, that codes for a recombinase that participates in homologous recombination in mice, is embryonic lethal, whereas overexpression of Rad51 protein causes aberrant homologous recombination, increased genomic instability and cell death [Flygare et al., 2001]. Nevertheless, many tumors upregulate Rad51 protein levels, and high levels of Rad51 confer resistance to drug therapies [Klein, 2008].
RAD9 AS A TRANSCRIPTION FACTOR
Human Rad9 can function as a sequence specific transcription factor. Specifically, Rad9 can bind to p53 DNA-binding consensus sequences in the promoter region of p21Waf1/Cip1, even in the absence of p53, and enhance transcription of the gene [Yin et al., 2004]. Overexpression of Rad9 increases expression of p21 at both mRNA and protein levels, controlling G1/S cell cycle transition [Yin et al., 2004]. In contrast, human Rad1 or Hus1 cannot interact with the p53 DNA binding consensus sequence within the p21 promoter [Yin et al., 2004]. Co-expression of Rad9 and p53 induces p21 transcription, but the level is comparable to that observed when either protein alone is overproduced [Yin et al., 2004]. Furthermore it has been shown that Rad9 physically associates with p53 and that the phosphorylation status of Rad9 may influence the binding affinity of p53 to the p21 promoter [Ishikawa et al., 2007]. In the future, it would be interesting to examine the effect of Rad9 on other p53 target genes, especially those involved in apoptosis.
In addition to p21, Rad9 is able to transactivate other target genes, a subset of which are also regulated by p53 [Yin et al., 2004]. Thus, Rad9 might bind as of yet unidentified DNA sequences or it can participate as a co-factor of transcriptional complexes.
The finding that Rad9 can transactivate p21 and other target genes has added Rad9 to the growing number of DNA repair proteins like MDC1 (mediator of DNA damage checkpoint protein 1), BRCA1 (breast cancer 1), and PARP-1 (poly [ADP-ribose] polymerase 1) that have canonical functions in DNA damage response and repair, but also display transcriptional activity either directly as transcription factors or indirectly by regulating the function of other transcription factors. BRCA1, for example, is known to affect transcription either by binding the promoters of genes (e.g. amphiregulin, psoriasin) [Lamber et al., 2010; Kennedy et al., 2005] or by acting as a co-repressor (e.g. Myc, ERα) or co-activator (STAT1, p53) of other transcriptional factors [Mullan et al., 2006]. DNA microarray analysis showed that MDC1 affects transcription independently of DNA damage or p53, although it was not assessed whether directly or indirectly. One such gene was caveolin-1 that affects cell adhesion and migration [Wilson et al., 2011]. PARP-1 attenuates TGFβ-induced epithelial to mesenchymal transition by ADP-ribosylating the SMAD3 and SMAD4 transcriptional complex and dissociating them from DNA [Lonn et al., 2010].
THE ROLE OF RAD9 IN TUMORIGENESIS
Genomic instability is a hallmark of cancer. A function for Rad9 in tumorigenesis can be inferred by the fact that Rad9 is important in maintaining genomic stability. Mrad9−/− cells demonstrate a marked increase in spontaneous chromosomal aberrations, indicating responsibility for genomic integrity. These cells are extremely sensitive to UV light, gamma rays and hydroxyurea [Hopkins et al., 2004]. Genomic instability usually emerges from enhanced DNA damage and/or unfaithful DNA repair, and therefore DNA repair is generally considered tumor suppressive (Hoeijmakers, 2001). In line with a tumor suppressive role, Rad9 influences the activity of base excision enzymes like FEN1, which is considered a tumor suppressor [Zheng et al., 2010]. Likewise, OGG1 glycosylase in the BER pathway is considered a tumor suppressor and single nucleotide polymorphisms are associated with various types of cancer [Nohmi et al, 2005]. Furthermore, in fission yeast, Rad9 promotes translesion synthesis through the more faithful recombination-like pathway that employs the undamaged sister chromatid, and not the error-prone branch of TLS that uses translesion DNA polymerases to copy the strand containing damage [Kai et al., 2007]. However, accumulating evidence both in vitro and in vivo indicate that Rad9 also displays a role as tumor promoter, and this oncogenic role is more difficult to reconcile based on its established function in DNA checkpoint/control of repair.
The Rad9 gene is located at chromosome 11q13, which is amplified in several types of malignant tumors. The 11q13 locus harbors a number of oncogenes, including cyclin D1, which is amplified in several types of cancer [Sherr and Roberts, 2004], multiple endocrine neoplasia 1 (MEN1) amplified in metastatic prostate cancer [Paris et al., 2004; Paris et al., 2009], EMS1 (cortactin) in advanced breast cancer [Ormandy et al., 2003], and NF-kappa B/p65 (RELA), which as a complex with p50 is necessary for the development of numerous cancers [Chaturvedi et al., 2011]. Furthermore, HER2 amplification is associated with genetic alterations in the 11q13 locus that promote a more aggressive tumor phenotype [Ellsworth et al., 2008].
Immunohistochemical analysis of surgically resected nonsmall cell lung carcinomas revealed that 33% of specimens displayed elevated levels of Rad9 expression and accumulation of the protein in the nucleus [Maniwa et al., 2005]. Western blot analysis on frozen specimens showed that Rad9 was highly phosphorylated in the malignant specimens compared with benign tissue. Furthermore, expression of Rad9 was correlated with expression of tumor proliferation marker Ki-67. Reducing levels of Rad9 by RNA interference in lung adenocarcinoma cell lines suppressed cell growth, thus confirming the relationship between Rad9 expression and tumor growth [Yuki et al., 2008]. Finally, examination of the nucleotide sequence of Rad9 detected a non-synonymous His239Arg single nucleotide polymorphism that might be associated with the development of lung adenocarcinoma [Maniwa et al., 2006].
A role for Rad9 in thyroid cancer cell growth has also been suggested [Kebebew et al., 2006]. Specifically, Rad9 mRNA was shown to be overexpressed in malignant thyroid cancer of follicular cell origin, compared with benign thyroid neoplasms. The authors suggested that Rad9 along with minichromosome maintenance proteins MCM5 and MCM7, which were overexpressed in thyroid cancer as well, could serve as markers of the disease in conjunction with adjunct fine-needle aspiration biopsy.
The abundance of Rad9 has been correlated with breast cancer cell proliferation and local invasion. Semi-quantitative reverse transcription PCR showed that Rad9 mRNA levels were elevated in 52.1% of breast tumors, and this up-regulation was correlated positively with tumor size and local recurrence [Cheng et al., 2005]. Increased levels of Rad9 mRNA were due to either an increase in Rad9 gene number or differential methylation of two putative Sp1/3 binding sites within the first and second intron of the Rad9 gene. Rad9 was necessary for cell proliferation in vitro, as reduction of the protein level by RNA interference inhibited growth of the MCF-7 breast cancer cell line. Immunohistochemical analysis further revealed that, as in the case of lung carcinomas, overexpressed Rad9 was localized to breast cancer cell nuclei, and the protein was highly phosphorylated compared with benign breast tissue [Chan et al., 2008].
A functional relationship between Rad9 expression and prostate cancer development and progression has been shown [Zhu et al., 2008]. In a large study including 339 human prostate adenocarcinoma specimens, it was shown that Rad9 is overexpressed in 153 (45.1%) of them. On the other hand, only 2 out of 52 benign prostate specimens showed somewhat elevated Rad9 levels. Likewise, four prostate cancer cell lines showed a 7.8 to 15.5-fold increase in Rad9 protein compared with a non-cancer prostate epithelial cell line. Most importantly, silencing Rad9 in human prostate cancer cell lines reduced tumorigenicity in vivo, as assessed by injection into nude mice as a xenograft. In addition to tumor growth, there was a strong correlation between Rad9 protein levels and cancer stage. Immunohistochemical staining for Rad9 protein in benign and tumor prostate tissue revealed that Rad9 was more abundant in stage III and IV prostate adenocarcinomas. Two mechanisms responsible for the high levels of Rad9 observed in prostate cancer cells were identified, namely, gene amplification and aberrant DNA hypermethylation in intron 2. This second intron contains sequences that inhibit Rad9 gene expression, but are suppressed by DNA methylation. As mentioned above, the same mode of Rad9 overexpression was detected in breast cancer specimens as well.
Although higher expression of Rad9 in a variety of cancers points to an oncogenic role for Rad9, this role is not universal for all cancers. In contrast to high expression of Rad9 in lung, thyroid, breast and prostate cancer, other types of tumors like skin and gastric carcinoma show diminished levels of Rad9 expression. In mouse skin keratinocytes, targeted deletion of Rad9 actually enhances tumor development after 7, 12-dimethylbenzathracene (DMBA) carcinogen application to the skin of the animals [Hu et al., 2008]. In this report, DMBA treatment caused earlier onset and more frequent formation of tumors in Mrad9−/− mice, compared with Mrad9+/− and Mrad9+/+ littermates. Moreover, immunostaining of 659 gastric cancers with antibodies against several proteins, including Rad9, and comparing them with benign tissue, revealed that Rad9 expression along with a number of known tumor suppressor proteins like PTEN, Retinoblastoma, SMAD4 and E-cadherin, was lost in the tumor specimens [Lee et al., 2007b]. These findings strongly suggest that Rad9 might act as a tumor suppressor in some cancers.
RAD9 AND OTHER DUAL FUNCTION GENES IN TUMORIGENESIS
Cancer arises from a series of genetic and epigenetic changes that result in the inactivation of tumor suppressing genes and the activation of proto-oncogenes. As described above, Rad9 has emerged as a dual action gene that can function as an oncogene to promote tumor growth or a tumor suppressor gene where loss is associated with tumorigenesis and enhanced sensitivity to genotoxin-induced tumorigenesis. The functional interaction of Rad9 with CAD and Rad51, for example, supports a role for Rad9 as an oncogene. In addition, Rad9 is able to transactivate a number of oncogenes [Yin et al., 2004]. In contrast, a tumor suppressive role for Rad9 can be inferred by the fact that it associates with and stimulates the activity of many enzymes involved in DNA repair, some of which have been established as tumor suppressors. Likewise, the upregulation of p21 transcription by Rad9 can generally be viewed as tumor suppressive.
To elucidate the function of Rad9, much has to be gained by studying other genes that function in a similar fashion. Examples of such genes include E2F1, transforming growth factor-beta (TGFβ), Notch, cyclin D1, EZH2 and parafibromin all of which can act as tumor suppressors or oncogenes depending on the type of cancer or the stage of the tumor.
E2F1 is a member of the E2F family of transcription factors that control a wide array of biological processes, including cell cycle progression, DNA replication, mitosis, DNA repair, differentiation, development and apoptosis [Polager and Ginsberg, 2008]. Deregulated E2F activity is seen in almost all human tumors. E2F family members can either induce (activator E2F) or repress (repressor E2F) gene expression. Activator E2Fs and specifically E2F1 are able to induce either cell proliferation or apoptosis. E2F1 promotes cell cycle progression and proliferation through upregulation of a number of genes involved in DNA replication, such as DNA polymerases, and minichromosome maintenance complex (MCM) components and cell cycle progression, such as cyclin E. Ectopic expression of E2F1 leads to p53-dependent and - independent apoptosis in cell cultures and transgenic mice. E2F1 can induce phosphorylation and activation of p53 in response to DNA damage, or it can up-regulate p53 pro-apoptotic co-factors, such as ASPP1 and 2 (apoptosis stimulating protein of p53). In addition, it has been demonstrated that E2F1 binds directly to p53 and, independently of its transcriptional activity, stimulates pro-apoptotic action of p53. E2F1 can also promote apoptosis in a p53-independent manner by transactivating pro-apoptotic genes, such as Apaf-1 (apoptotic protease activating factor 1), caspases, and bcl-2 homology (BH3)-only proteins. Akt inhibits E2F1-induced apoptosis through the phosphorylation of TopBP1 (topoisomerase IIβ-binding protein 1), which binds E2F1 and represses its pro-apoptotic function [Liu et al., 2006].
Transforming growth factor-beta is possibly the best described dual function gene. In early stage tumors it acts as a tumor suppressor by inhibiting cell cycle progression and proliferation by up-regulating p21Waf1/Cip1 via the RUNX3 transcription factor, as well as by inhibiting expression of c-myc, a transcription factor that promotes cell proliferation. TGFβ can also induce apoptosis by up-regulating pro-apoptotic genes, including DAPK (death-associated protein kinase) and BH3-only BIM (Bcl-2 interacting mediator of cell death) [Ikushima and Miyazono, 2010].
By contrast, TGFβ functions as a metastasis promoting gene at later stages in tumor development. TGFβ can induce the expression, secretion and activation of matrix metalloproteases (MMP) 2 and 9, and down-regulate expression of tissue inhibitor of metalloproteinase (TIMP) in tumor and endothelial cells [Ikushima and Miyazono, 2010]. It plays a pivotal role in the execution of the epithelial to mesenchymal transition, thought to be important in the metastatic process [Thiery, 2002]. Finally, TGFβ signaling can promote angiogenesis by inducing, among other genes, expression of the vascular remodeling gene ANGPTL4 (angiopoietin-like 4) [Padua et al., 2008] and suppresses inflammation, thereby inhibiting immune system cells from recognizing and destroying cancer cells. Increased TGFβ1 expression correlates with colorectal and prostate cancer progression. High levels of TGFβ are positively correlated with breast, colorectal and prostate metastases [Ikushima and Miyazono, 2010].
NOTCH is a transmembrane receptor that regulates differentiation, proliferation, stem cell maintenance, angiogenesis, and apoptosis. In tumorigenesis NOTCH has been identified as either oncogenic or tumor suppressive. It is an oncogene in prostate cancer by promoting tumor invasion and metastasis [Shou et al., 2001; Bin Hafeez et al., 2009; Wang et al., 2010], and its ligand Jagged1 correlates with a more aggressive disease course in prostate cancer [Santagata et al., 2004]. In contrast, NOTCH acts as a tumor suppressor in mouse skin cancer. NOTCH1 expression in keratinocytes leads to upregulation of p21Waf1/Cip1, which promotes cell cycle exit and downregulates Wnt signaling downstream of Notch1 [Devgan et al., 2005]. Loss of NOTCH1 in skin results in activation of β-catenin-mediated signaling [Nicolas et al., 2003] and impairs the skin barrier integrity, thus creating a chronic injury/wound-like microenvironment [Demehri et al., 2009]. These oncogene-prostate and tumor suppressor-skin relationships in particular are reminiscent of Rad9.
Cyclin D1, a gene with established oncogenic activity, binds and activates cyclin-dependent kinases CDK2 and CDK4, which in turn phosphorylate and inactivate Rb allowing entry into S phase of the cell cycle and proliferation. In the absence of Rb, cyclin D1 activity becomes dispensable for cell cycle progression. However, it still plays an important role in DNA repair. In response to DNA damage, cyclin D1 is recruited to DNA damage sites in a BRCA2-dependent fashion, physically interacts with Rad51 and facilitates homologous recombination [Jirawatnotai et al., 2011]. So it appears that cyclin D1 has a tumor suppressive function (DNA repair) or it acts as an oncogene (activation of CDKs and cell proliferation), depending on the cellular context, which in this particular case is defined by Rb status.
EZH2, polycomb histone methyltransferase enhancer of zeste 2, is generally considered an oncogene in various types of cancers. In one downstream oncogenic pathway, EZH2 activates NF-κB. In breast cancer, however, EZH2 can either be an activator or inhibitor of NF-κB, depending on the estrogen receptor (ER) status of the tumor. Thus, in ER-negative, basal-like, breast cancer cells, it activates NF-κB, but in ER-positive, luminal-like cells it represses NF-κB [Lee et al., 2011], thus acting like an oncogene in the first case or a tumor suppressor in the second.
Post-translational modifications are also involved in the decision as to whether a dual function gene acts as oncogene or tumor suppressor. In the case of the parafibromin/cdc73 gene, a single dephosphorylation event suffices to convert it from a tumor suppressor to oncogene. Parafibromin is normally a tumor suppressor that binds to promoters and strongly inhibits cyclin D1 and c-myc transcription. At the same time, it binds and drives transcription of the β-catenin gene, albeit at very low levels. However, dephosphorylation of parafibromin by SHP2 tyrosine phosphatase coverts parafibromin to a strong inducer of β-catenin and activator of the Wnt oncogenic pathway, thereby acting as a tumor promoter [Takahashi et al., 2011]. Rad9 phosphorylation and other post-translational modifications (e.g. methylation) are very important in the context of cell cycle checkpoints. However, the effect these modifications have on the transcriptional activity of Rad9 toward its target genes has not been studied.
FUTURE DIRECTIONS
Rad9 controls a multitude of physiological functions and has been associated with the development of tumorigenesis. Which of these biological functions have a causal relationship to cancer initiation and progression is presently not known and will be the focus of future studies. The differential phosphorylation of Rad9 and other post-translational modifications, protein-protein interactions and subcellular localization will impact on the final physiological outcome. The abundance of the protein will also prove to be important as too much Rad9 or too little induces cell death in some cell types. Another critical factor for the elucidation of the role of Rad9 in tumorigenesis is the cellular context in which the protein functions. Evidence is growing that Rad9 acts as a context-specific tumor promoter or tumor suppressor gene.
The capability of Rad9 as a DNA repair gene to preserve genomic integrity could explain its role as a tumor suppressor in skin cancer [Lieberman et al., 2011]. However, Rad9-mediated oncogenic action in prostate (and other) cancer cannot be attributed merely to the ability to repair DNA as overexpression of Rad9 in a prostate immortalized cell line causes the ability to form abnormal growths when subcutaneously injected into nude mice [Zhu et al., 2008]. Furthermore, high levels of expression do not necessarily translate into more efficient DNA repair. For example, malignant prostate cancer cells display defective repair of DNA breaks, alkali-labile sites and oxidative base damage, despite increased expression of DNA repair genes [Fan et al., 2004].
Another area of investigation will be the functional interaction between Rad9 and p53. Rad9 induces p21 expression in the absence of p53, whereas it presumably competes with p53 for binding to the promoter of p21 when both are present. It will be interesting to examine the effect of Rad9 on other p53-induced genes involved in regulating the cell cycle or apoptosis in both a p53-null or p53 wild type background, as well as examine expression of p53-dependent downstream target genes in the presence of Rad9. Besides p21, Rad9 is able to transactivate a number of other genes that are not known to participate in or be related to cell cycle checkpoint and DNA repair pathways, thus pointing to additional, novel functions for Rad9.
There are genes critical for cancer etiology that do not fit the oncogene-tumor suppressor dichotomy and display context-dependent opposing roles in cancer. Rad9 is emerging as a dual function gene in this regard. Because of such complexity, it is paramount to first elucidate the function of each of these kinds of genes in relation to the genetic background and the tissue-specific origin of the tumor, as a prelude to the development of beneficial therapeutic strategies.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant numbers: 5R01CA130536-17, 5R01GM079107-10, 5P01CA49062-20.
REFERENCES
- An L, Wang Y, Liu Y, Yang X, Liu C, Hu Z, He W, Song W, Hang H. Rad9 is required for B cell proliferation and immunoglobulin class switch recombination. J Biol Chem. 2010;285:35267–35273. doi: 10.1074/jbc.M110.161208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Madabushi A, Guan X, Lu AL. Interaction between human mismatch repair recognition proteins and checkpoint sensor Rad9-Rad1-Hus1. DNA Repair (Amst) 2010:9478–9487. doi: 10.1016/j.dnarep.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Brandt PD, Lindsey-Boltz LA, Sancar A, Bambara RA. Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex. J Biol Chem. 2009;284:15158–15172. doi: 10.1074/jbc.M109.000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Lu T, Wang X, Zheng H, Wang LE, Wei Q, Hittelman WN, Li L. Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene. 2004;23:5586–5593. doi: 10.1038/sj.onc.1207753. [DOI] [PubMed] [Google Scholar]
- Bessho T, Sancar A. Human DNA damage checkpoint protein hRAD9 is a 3' to 5' exonuclease. J Biol Chem. 2000;275:7451–7454. doi: 10.1074/jbc.275.11.7451. [DOI] [PubMed] [Google Scholar]
- Cai RL, Yan-Neale Y, Cueto MA, Xu H, Cohen D. HDAC1, a histone deacetylase, forms a complex with Hus1 and Rad9, two G2/M checkpoint Rad proteins. J Biol Chem. 2000;275:27909–27916. doi: 10.1074/jbc.M000168200. [DOI] [PubMed] [Google Scholar]
- Chan V, Khoo US, Wong MS, Lau K, Suen D, Li G, Kwong A, Chan TK. Localization of hRad9 in breast cancer. BMC Cancer. 2008;8:196. doi: 10.1186/1471-2407-8-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DY, Lu AL. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J Biol Chem. 2005;280:408–417. doi: 10.1074/jbc.M406800200. [DOI] [PubMed] [Google Scholar]
- Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: 'one size does not fit all'. Oncogene. 2011;30:1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY. ATM-dependent phosphorylation of human Rad9 is required for ionizing radiation-induced checkpoint activation. J Biol Chem. 2001;276:16580–16586. doi: 10.1074/jbc.M008871200. [DOI] [PubMed] [Google Scholar]
- Cheng CK, Chow LW, Loo WT, Chan TK, Chan V. The cell cycle checkpoint gene Rad9 is a novel oncogene activated by 11q13 amplification and DNA methylation in breast cancer. Cancer Res. 2005;65:8646–8654. doi: 10.1158/0008-5472.CAN-04-4243. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta-Ramusino C, McDonald ER, 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–1317. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005;19:1485–1495. doi: 10.1101/gad.341405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth RE, Ellsworth DL, Patney HL, Deyarmin B, Love B, Hooke JA, Shriver CD. Amplification of HER2 is a marker for global genomic instability. BMC Cancer. 2008;8:297. doi: 10.1186/1471-2407-8-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R, Kumaravel TS, Jalali F, Marrano P, Squire JA, Bristow RG. Defective DNA strand break repair after DNA damage in prostate cancer cells: implications for genetic instability and prostate cancer progression. Cancer Res. 2004;64:8526–8533. doi: 10.1158/0008-5472.CAN-04-1601. [DOI] [PubMed] [Google Scholar]
- Flygare J, Fält S, Ottervald J, Castro J, Dackland AL, Hellgren D, Wennborg A. Effects of HsRad51 overexpression on cell proliferation, cell cycle progression, and apoptosis. Exp Cell Res. 2001;268:61–69. doi: 10.1006/excr.2001.5265. [DOI] [PubMed] [Google Scholar]
- Francia S, Weiss RS, Hande MP, Freire R, d'Adda di Fagagna F. Telomere and telomerase modulation by the mammalian Rad9/Rad1/Hus1 DNA-damage-checkpoint complex. Curr Biol. 2006;16:1551–1558. doi: 10.1016/j.cub.2006.06.066. [DOI] [PubMed] [Google Scholar]
- Furuya K, Miyabe I, Tsutsui Y, Paderi F, Kakusho N, Masai H, Niki H, Carr AM. DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol Cell. 2010;40:606–618. doi: 10.1016/j.molcel.2010.10.026. [DOI] [PubMed] [Google Scholar]
- Gembka A, Toueille M, Smirnova E, Poltz R, Ferrari E, Villani G, Hübscher U. The checkpoint clamp, Rad9-Rad1-Hus1 complex, preferentially stimulates the activity of apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta in long patch base excision repair. Nucleic Acids Res. 2007;35:2596–2608. doi: 10.1093/nar/gkl1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J. 2004;23:429–438. doi: 10.1038/sj.emboj.7600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, Davey S. hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res. 2003;63:4829–4835. [PubMed] [Google Scholar]
- Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, Lena AM, Candi E, Terrinoni A, Catani MV, Oren M, Melino G, Krammer PH, Stremmel W, Müller M. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 2005;24:2458–2471. doi: 10.1038/sj.emboj.7600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A. BID as a double agent in cell life and death. Cell Cycle. 2006;5:582–584. doi: 10.4161/cc.5.6.2575. [DOI] [PubMed] [Google Scholar]
- Guan X, Bai H, Shi G, Theriot CA, Hazra TK, Mitra S, Lu AL. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007a;35:2463–2472. doi: 10.1093/nar/gkm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Madabushi A, Chang DY, Fitzgerald ME, Shi G, Drohat AC, Lu AL. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007b;35:6207–6218. doi: 10.1093/nar/gkm678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guervilly JH, Macé-Aimé G, Rosselli F. Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum Mol Genet. 2008;17:679–689. doi: 10.1093/hmg/ddm340. [DOI] [PubMed] [Google Scholar]
- He W, Zhao Y, Zhang C, An L, Hu Z, Liu Y, Han L, Bi L, Xie Z, Xue P, Yang F, Hang H. Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1. Nucleic Acids Res. 2008;36:6406–6417. doi: 10.1093/nar/gkn686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Ma X, Yang X, Zhao Y, Qiu J, Hang H. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 2011;39:4719–4727. doi: 10.1093/nar/gkq1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai I, Wang HG. A role of the C-terminal region of human Rad9 (hRad9) in nuclear transport of the hRad9 checkpoint complex. J Biol Chem. 2002;277:25722–25727. doi: 10.1074/jbc.M203079200. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Hopkins KM, Auerbach W, Wang XY, Hande MP, Hang H, Wolgemuth DJ, Joyner AL, Lieberman HB. Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol Cell Biol. 2004;24:7235–7248. doi: 10.1128/MCB.24.16.7235-7248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Liu Y, Zhang C, Zhao Y, He W, Han L, Yang L, Hopkins KM, Yang X, Lieberman HB, Hang H. Targeted deletion of Rad9 in mouse skin keratinocytes enhances genotoxin-induced tumor development. Cancer Res. 2008;68:5552–5561. doi: 10.1158/0008-5472.CAN-07-5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Graves LM. De novo synthesis of pyrimidine nucleotides; emerging interfaces with signal transduction pathways. Cell Mol Life Sci. 2003;60:321–336. doi: 10.1007/s000180300027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- Inagaki A, Sleddens-Linkels E, van Cappellen WA, Hibbert RG, Sixma TK, Hoeijmakers JH, Grootegoed JA, Baarends WM. Human RAD18 Interacts with Ubiquitylated Chromatin Components and Facilitates RAD9 Recruitment to DNA Double Strand Breaks. PLoS One. 2011;6:e23155. doi: 10.1371/journal.pone.0023155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Ishii H, Murakumo Y, Mimori K, Kobayashi M, Yamamoto K, Mori M, Nishino H, Furukawa Y, Ichimura K. Rad9 modulates the P21WAF1 pathway by direct association with p53. BMC Mol Biol. 2007;8:37. doi: 10.1186/1471-2199-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Inageta T, Mimori K, Saito T, Sasaki H, Isobe M, Mori M, Croce CM, Huebner K, Ozawa K, Furukawa Y. Frag1, a homolog of alternative replication factor C subunits, links replication stress surveillance with apoptosis. Proc Natl Acad Sci U S A. 2005;102:9655–9660. doi: 10.1073/pnas.0504222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JG, Fousteri MI, de Wind N. Send in the clamps: control of DNA translesion synthesis in eukaryotes. Mol Cell. 2007;28:522–529. doi: 10.1016/j.molcel.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, Correll M, Quackenbush J, Livingston DM, Gygi SP, Sicinski P. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474:230–234. doi: 10.1038/nature10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Furuya K, Paderi F, Carr AM, Wang TS. Rad3-dependent phosphorylation of the checkpoint clamp regulates repair-pathway choice. Nat Cell Biol. 2007;9:691–697. doi: 10.1038/ncb1600. [DOI] [PubMed] [Google Scholar]
- Kennedy RD, Gorski JJ, Quinn JE, Stewart GE, James CR, Moore S, Mulligan K, Emberley ED, Lioe TF, Morrison PJ, Mullan PB, Reid G, Johnston PG, Watson PH, Harkin DP. BRCA1 and c-Myc associate to transcriptionally repress psoriasin, a DNA damage-inducible gene. Cancer Res. 2005;65:10265–10272. doi: 10.1158/0008-5472.CAN-05-1841. [DOI] [PubMed] [Google Scholar]
- Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu K, Miyashita T, Hang H, Hopkins KM, Zheng W, Cuddeback S, Yamada M, Lieberman HB, Wang HG. Human homologue of S. pombe Rad9 interacts with BCL-2/BCL-xL and promotes apoptosis. Nat Cell Biol. 2000a;2:1–6. doi: 10.1038/71316. [DOI] [PubMed] [Google Scholar]
- Komatsu K, Hopkins KM, Lieberman HB, Wang H. Schizosaccharomyces pombe Rad9 contains a BH3-like region and interacts with the anti-apoptotic protein Bcl-2. FEBS Lett. 2000b;481:122–126. doi: 10.1016/s0014-5793(00)01975-x. [DOI] [PubMed] [Google Scholar]
- Lamber EP, Horwitz AA, Parvin JD. BRCA1 represses amphiregulin gene expression. Cancer Res. 2010;70:996–1005. doi: 10.1158/0008-5472.CAN-09-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Giannattasio M, Puddu F, Granata M, Pellicioli A, Plevani P, Muzi-Falconi M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst) 2009;8:1055–1067. doi: 10.1016/j.dnarep.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007a;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- Lee HS, Cho SB, Lee HE, Kim MA, Kim JH, Park do J, Kim JH, Yang HK, Lee BL, Kim WH. Protein expression profiling and molecular classification of gastric cancer by the tissue array method. Clin Cancer Res. 2007b;13:4154–4163. doi: 10.1158/1078-0432.CCR-07-0173. [DOI] [PubMed] [Google Scholar]
- Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-Specific Regulation of NF-κB Target Gene Expression by EZH2 in Breast Cancers. Mol Cell. 2011;43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Lieberman HB. Rad9, an evolutionarily conserved gene with multiple functions for preserving genomic integrity. J Cell Biochem. 2006;97:690–697. doi: 10.1002/jcb.20759. [DOI] [PubMed] [Google Scholar]
- Lieberman HB, Bernstock JD, Broustas CG, Hopkins KM, Leloup C, Zhu A. The role of RAD9 in tumorigenesis. J Mol Cell Biol. 2011;3:39–43. doi: 10.1093/jmcb/mjq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey-Boltz LA, Wauson EM, Graves LM, Sancar A. The human Rad9 checkpoint protein stimulates the carbamoyl phosphate synthetase activity of the multifunctional protein CAD. Nucleic Acids Res. 2004;32:4524–4530. doi: 10.1093/nar/gkh789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Paik JC, Wang B, Lin FT, Lin WC. Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. EMBO J. 2006;25:4795–4807. doi: 10.1038/sj.emboj.7601355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fang Y, Shao H, Lindsey-Boltz L, Sancar A, Modrich P. Interactions of human mismatch repair proteins MutSalpha and MutLalpha with proteins of the ATR-Chk1 pathway. J Biol Chem. 2010;285:5974–5982. doi: 10.1074/jbc.M109.076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönn P, van der Heide LP, Dahl M, Hellman U, Heldin CH, Moustakas A. PARP-1 attenuates Smad-mediated transcription. Mol Cell. 2010;40:521–532. doi: 10.1016/j.molcel.2010.10.029. [DOI] [PubMed] [Google Scholar]
- Maniwa Y, Yoshimura M, Bermudez VP, Yuki T, Okada K, Kanomata N, Ohbayashi C, Hayashi Y, Hurwitz J, Okita Y. Accumulation of hRad9 protein in the nuclei of nonsmall cell lung carcinoma cells. Cancer. 2005;103:126–132. doi: 10.1002/cncr.20740. [DOI] [PubMed] [Google Scholar]
- Maniwa Y, Yoshimura M, Bermudez VP, Okada K, Kanomata N, Ohbayashi C, Nishimura Y, Hayashi Y, Hurwitz J, Okita Y. His239Arg SNP of HRAD9 is associated with lung adenocarcinoma. Cancer. 2006;106:1117–1122. doi: 10.1002/cncr.21705. [DOI] [PubMed] [Google Scholar]
- Mullan PB, Quinn JE, Harkin DP. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 2006;25:5854–5863. doi: 10.1038/sj.onc.1209872. [DOI] [PubMed] [Google Scholar]
- Nohmi T, Kim SR, Yamada M. Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes. Mutat Res. 2005;591:60–73. doi: 10.1016/j.mrfmmm.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Musgrove EA, Hui R, Daly RJ, Sutherland RL. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003;78:323–335. doi: 10.1023/a:1023033708204. [DOI] [PubMed] [Google Scholar]
- Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, MassaguÈ J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang J, Powell SN, Roti Roti JL, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–1864. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris PL, Andaya A, Fridlyand J, Jain AN, Weinberg V, Kowbel D, Brebner JH, Simko J, Watson JE, Volik S, Albertson DG, Pinkel D, Alers JC, van der Kwast TH, Vissers KJ, Schroder FH, Wildhagen MF, Febbo PG, Chinnaiyan AM, Pienta KJ, Carroll PR, Rubin MA, Collins C, van Dekken H. Whole genome scanning identifies genotypes associated with recurrence and metastasis in prostate tumors. Hum Mol Genet. 2004;13:1303–1313. doi: 10.1093/hmg/ddh155. [DOI] [PubMed] [Google Scholar]
- Paris PL, Sridharan S, Hittelman AB, Kobayashi Y, Perner S, Huang G, Simko J, Carroll P, Rubin MA, Collins C. An oncogenic role for the multiple endocrine neoplasia type 1 gene in prostate cancer. Prostate Cancer Prostatic Dis. 2009;12:184–191. doi: 10.1038/pcan.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MJ, Park JH, Hahm SH, Ko SI, Lee YR, Chung JH, Sohn SY, Cho Y, Kang LW, Han YS. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair (Amst) 2009;8:1190–1200. doi: 10.1016/j.dnarep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18:528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Roos-Mattjus P, Hopkins KM, Oestreich AJ, Vroman BT, Johnson KL, Naylor S, Lieberman HB, Karnitz LM. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J Biol Chem. 2003;278:24428–24437. doi: 10.1074/jbc.M301544200. [DOI] [PubMed] [Google Scholar]
- Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, Kim R, Tang J, Montie JE, Chinnaiyan AM, Rubin MA, Aster JC. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004;64:6854–6857. doi: 10.1158/0008-5472.CAN-04-2500. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Sigoillot FD, Sigoillot SM, Guy HI. Breakdown of the regulatory control of pyrimidine biosynthesis in human breast cancer cells. Int. J. Cancer. 2004;109:491–498. doi: 10.1002/ijc.11717. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Toueille M, Markkanen E, Hübscher U. The human checkpoint sensor and alternative DNA clamp Rad9-Rad1-Hus1 modulates the activity of DNA ligase I, a component of the long-patch base excision repair machinery. Biochem J. 2005;389:13–17. doi: 10.1042/BJ20050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Onge RP, Besley BD, Park M, Casselman R, Davey S. DNA damage-dependent and -independent phosphorylation of the hRad9 checkpoint protein. J Biol Chem. 2001;276:41898–41905. doi: 10.1074/jbc.M105152200. [DOI] [PubMed] [Google Scholar]
- St Onge RP, Besley BD, Pelley JL, Davey S. A role for the phosphorylation of hRad9 in checkpoint signaling. J Biol Chem. 2003;278:26620–26628. doi: 10.1074/jbc.M303134200. [DOI] [PubMed] [Google Scholar]
- Sunavala-Dossabhoy G, De Benedetti A. Tousled homolog, TLK1, binds and phosphorylates Rad9; TLK1 acts as a molecular chaperone in DNA repair. DNA Repair (Amst) 2009;8:87–102. doi: 10.1016/j.dnarep.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Tsutsumi R, Kikuchi I, Obuse C, Saito Y, Seidi A, Karisch R, Fernandez M, Cho T, Ohnishi N, Rozenblatt-Rosen O, Meyerson M, Neel BG, Hatakeyama M. SHP2 tyrosine phosphatase converts parafibromin/Cdc73 from a tumor suppressor to an oncogenic driver. Mol Cell. 2011;43:45–56. doi: 10.1016/j.molcel.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi Y, Ohashi E, Ogawa K, Masai H, Obuse C, Tsurimoto T. Casein kinase 2-dependent phosphorylation of human Rad9 mediates the interaction between human Rad9-Hus1-Rad1 complex and TopBP1. Genes Cells. 2010;15:761–771. doi: 10.1111/j.1365-2443.2010.01418.x. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, Friedrich-Heineken E, Villani G, Hottiger MO, Hübscher U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase beta and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 2004;32:3316–3324. doi: 10.1093/nar/gkh652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmer E, Karnitz LM. Human homologs of Schizosaccharomyces pombe rad1, hus1, and rad9 form a DNA damage-responsive protein complex. J Biol Chem. 1999;274:567–570. doi: 10.1074/jbc.274.2.567. [DOI] [PubMed] [Google Scholar]
- Wang W, Brandt P, Rossi ML, Lindsey-Boltz L, Podust V, Fanning E, Sancar A, Bambara RA. The human Rad9-Rad1-Hus1 checkpoint complex stimulates flap endonuclease 1. Proc Natl Acad Sci U S A. 2004;101:16762–16777. doi: 10.1073/pnas.0407686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lindsey-Boltz LA, Sancar A, Bambara RA. Mechanism of stimulation of human DNA ligase I by the Rad9-rad1-Hus1 checkpoint complex. J Biol Chem. 2006;281:20865–20872. doi: 10.1074/jbc.M602289200. [DOI] [PubMed] [Google Scholar]
- Warmerdam DO, Freire R, Kanaar R, Smits VA. Cell cycle-dependent processing of DNA lesions controls localization of Rad9 to sites of genotoxic stress. Cell Cycle. 2009;8:1765–1774. doi: 10.4161/cc.8.11.8721. [DOI] [PubMed] [Google Scholar]
- Wilson KA, Colavito SA, Schulz V, Wakefield PH, Sessa W, Tuck D, Stern DF. NFBD1/MDC1 regulates Cav1 and Cav2 independently of DNA damage and p53. Mol Cancer Res. 2011;9:766–781. doi: 10.1158/1541-7786.MCR-10-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Shell SM, Zou Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005;24:4728–4735. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol Cell Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Zhu A, Jin YJ, Liu YX, Zhang X, Hopkins KM, Lieberman HB. Human RAD9 checkpoint control/proapoptotic protein can activate transcription of p21. Proc Natl Acad Sci U S A. 2004;101:8864–8869. doi: 10.1073/pnas.0403130101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Komatsu K, Wang HG, Kufe D. c-Abl tyrosine kinase regulates the human Rad9 checkpoint protein in response to DNA damage. Mol Cell Biol. 2002;22:3292–3300. doi: 10.1128/MCB.22.10.3292-3300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Wang HG, Miki Y, Kufe D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki T, Maniwa Y, Doi T, Okada K, Nishio W, Hayashi Y, Okita Y. DNA damage sensor protein hRad9, a novel molecular target for lung cancer treatment. Oncol Rep. 2008;20:1047–1052. [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003;22:713–723. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Jia J, Finger LD, Guo Z, Zer C, Shen B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011;39:781–794. doi: 10.1093/nar/gkq884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Zhou H, Leloup C, Marino SA, Geard CR, Hei TK, Lieberman HB. Differential impact of mouse Rad9 deletion on ionizing radiation-induced bystander effects. Radiat Res. 2005;164:655–661. doi: 10.1667/rr3458.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Zhang CX, Lieberman HB. Rad9 has a functional role in human prostate carcinogenesis. Cancer Res. 2008;68:1267–1274. doi: 10.1158/0008-5472.CAN-07-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]