

Abstract
Musashi1 (Msi1) is an evolutionarily conserved RNA-binding protein (RBP) that has profound implications in cellular processes such as stem cell maintenance, nervous system development, and tumorigenesis. Msi1 is highly expressed in many cancers, including glioblastoma, while in normal tissues, its expression is restricted to stem cells. Unfortunately, the factors that modulate Msi1 expression and trigger high levels in tumors are largely unknown. Msi1 has a long 3′ untranslated region (UTR) containing several AU- and U-rich sequences. This type of sequence motif is often targeted by HuR, another important RBP known to be highly expressed in tumor tissue such as glioblastoma, and to regulate a variety of cancer-related genes. In this report, we demonstrate an interaction between HuR and the Msi1 3′ UTR, resulting in a positive regulation of Msi1 expression. We show that HuR increased MSI1 mRNA stability and promoted its translation. We also present evidence that expression of HuR and Msi1 correlate positively in glioblastoma lines. Finally, we show that inhibition of cell proliferation, increased apoptosis, and changes in cell cycle profile as a result of silencing HuR are partially rescued when Msi1 is ectopically expressed. In sum, our results suggest that HuR is an important regulator of Msi1 in glioblastoma and that this regulation has important biological consequences during gliomagenesis.
Keywords: Musashi1, HuR, ELAV, tumorigenesis, cancer, RNA-binding protein, glioblastoma
Introduction
Cancer is a disease process ultimately triggered by aberrant gene expression. In this scenario, there is a large body of literature discussing the participation of transcription and chromatin modification factors in tumorigenesis. On the other hand, the role of post-transcriptional regulation in tumor growth is still poorly comprehended. RNA-binding proteins (RBPs) are key mediators of post-transcriptional gene regulation, mediating many aspects of RNA metabolism such as splicing, transport, localization, translational regulation, and stability (1). In addition to controlling normal processes in cellular physiology, aberrant expression of RBPs has been shown to lead to tumorigenesis when their target genes are implicated in cell proliferation, differentiation, apoptosis, invasion, and metastasis.
An RBP of particular interest in cancer is Musashi1 (Msi1), first described as a gene required for the development of the Drosophila adult external sensory organ (2). In mammals, Musashi1 is required for nervous system development during embryonic development while in adult, Musashi1 expression is mainly restricted to stem and progenitor cells of various tissues (3–6). Aberrantly high Musashi1 expression is observed in many cancers such as medulloblastoma (7), hepatocellular carcinoma (8), cervical adenocarcinoma (9), lung cancer (10), colon cancer (11), and glioblastoma multiforme (GBM). In fact, increasing expression of Musashi1 has been correlated with a poor prognosis in glioma (12), breast cancer (13), and medulloblastoma (Penalva Lab, unpublished data).
During normal development, Musashi1 maintains stem cell identity, serving as a key gene in stemness (14). However, in the tumor environment, Musashi1 enhances cancer features. High Msi1 expression is associated with increased Notch 1 expression and areas of tumor invasion/metastasis (12). Notch is a critical pathway for tumorigenesis in medulloblastoma and glioblastoma and other tumor types; Msi1 influences this pathway by repressing the regulation of NUMB mRNA, a negative regulator of Notch (15–17). In medulloblastoma, Msi1 inhibition results in increased sensitivity to the Hedgehog pathway inhibitor cyclopamine, indicating that Msi1 interfaces with the Hedgehog pathway(18). In murine in vivo xenograft experiments using breast and colon cancer cells, silencing of Msi1 via small interfering RNAs results in inhibition of tumor growth (13, 19); similar results were observed for glioblastoma and medulloblastoma cells (Penalva Lab, unpublished data). Our results indicate that Msi1 influences tumor progression in a complex manner by regulating the expression of a network of genes implicated in cancer-related processes like cell proliferation, apoptosis, cell cycle and differentiation (17).
As summarized above, a large amount of data supports the role of Msi1 as an oncogenic protein. However, the molecular determinants of increased Musashi1 expression during tumorigenesis are largely unknown. In normal stem cells, one study identified the HuD RNA-binding protein as a potential post-transcriptional regulator of Musashi1 expression, aiding neural stem cells in the transition towards differentiation (20). It has also been suggested that a potential regulatory element at the transcriptional level exist as evident by the presence of a hypoxia-responsive element which can bind the hypoxia-inducible factor 1 in times of hypoxic stress, promoting self-renewal and proliferation in neural stem cells (21). However, neither of these elements can explicate the overexpression of Musashi1 during tumorigenesis. The mRNA of Msi1 contains a long 3′ untranslated region, spanning ~1800 nucleotides, making it a likely candidate for post-transcriptional regulation. We have recently shown that Msi1 expression is regulated by several tumor suppressor microRNAs (22). Furthermore, the 3′ UTR contains several segments of AU- or U-rich cis-regulatory sequences, making it a potential target of turnover and translation regulatory RNA-binding proteins (TTR-RBPs) such as HuR (also known as ELAVL1 or HuA), a member of the Hu/ELAV (embryonic lethal abnormal vision) family (23).
HuR can enhance tumorigenesis by interacting with a subset of mRNAs that encode proteins that regulate cell proliferation, cell survival, angiogenesis, invasion, and metastasis (24). Many studies have reported elevated expression of HuR in numerous malignancies (25, 26). HuR contains three RNA recognition motifs that bind to 3′ UTRs of mRNA which bears AU- or U-rich sequences (27). In malignant tumors of the central nervous system such as glioblastoma, HuR has been linked to the augmented expression of genes such as TNF-α, IL-8, COX2, VEGF, TGF-β, and other genes involved in enhancing the tumorigenic phenotype, such as increased cell proliferation, evasion of apoptosis, angiogenesis, and invasion/metastasis (25, 26, 28). A more recent study demonstrated that HuR promotes an anti-apoptotic phenotype through the post-transcriptional control of BCL2, MCL1, and BCLXL, proteins belonging to the Bcl-2 family of antiapoptotic proteins (29).
In this study, we show that high expression of Msi1 in glioblastoma is partially triggered by HuR through its influence on Msi1 translation and mRNA stability, resulting in an increased steady-state level of MSI1 mRNA and increased Msi1 protein output. Supporting this idea, we observed that HuR and Msi1 have similar patterns of expression. Finally, we demonstrated that Msi1 transgenic expression overcomes the impact of HuR knockdown on cell proliferation, apoptosis, and cell cycle profile. In conclusion, we suggest that the HuR-Msi1 link is an important piece in gliomagenesis.
Materials and Methods
Cell culture
U251 and U343 glioblastoma cells were maintained in Dulbecco’s Modified Essential Medium (Thermo Scientific, Rockford, IL), supplemented with 10% fetal bovine serum, penicillin, and streptomycin. HeLa cervical adenocarcinoma cells were maintained in Minimum Essential Medium (Thermo Scientific, Rockford, IL) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Primary glioblastoma tumorspheres were obtained from surgically resected patient tumors and propagated in Neurobasal media containing L-glutamine, N2 supplement (Gibco, Carlsbad, CA), B27 supplement (Gibco, Carlsbad, CA), heparin (Sigma, St. Louis, MO), epidermal growth factor (EGF) (Peprotech, Inc., Rocky Hill, NJ), and basic fibroblast growth factor (bFGF) (Peprotech, Inc., Rocky Hill, NJ) (30). For growth of primary glioblastoma cells as monolayers, the cells were cultured in the presence of Dulbecco’s Modified Essential Medium with 10% fetal bovine serum, pencillin, and streptomycin. The U251 overexpression cell lines were previously established (22). U343 overexpression cell lines were created through stable G418 (Gibco, Carlsbad, CA) selection (800 μg/mL) after transfection of the pEF1/myc-His A plasmid (Invitrogen, Carlsbad, CA), which contains the expression cassette encoding for an EF-1α promoter-driven coding region (CR) for either Msi1 or GFP. After initial selection, 400 μg/mL of G418 was used for maintenance. The levels of Msi1 overexpression were monitored using quantitative PCR (qPCR) analysis.
Immunoprecipitation of HuR Ribonucleoprotein Complexes
Immunoprecipitation (IP) of HuR ribonucleoprotein (RNP) complexes (31) was performed using U251 glioblastoma cell lysates. Cell lysates were prepared and pre-cleared using 15 μg of IgG isotype control and protein A-sepharose beads, pre-swollen in NT2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM MgCl2, 5% bovine serum albumin, and 0.05% Nonidet P-40), for 30 minutes at 4°C. After pre-clearing, 100 μL of protein A-sepharose beads were incubated with 30 μg of anti-HuR antibody for 18 hours at 4°C then for 1 hour at 4°C with 3 mg of pre-cleared cell lysate. After repeated washing and centrifugation of the sepharose beads with ice cold NT2 buffer, HuR-bound RNA was released from protein using proteinase K and SDS. RNA was extracted using acid phenol-chloroform and ethanol precipitated. After reverse transcription (RT), qPCR analysis was performed using gene-specific primers for MSI1, GAPDH, and PTMA mRNAs.
In vitro transcription of biotinylated RNA and analysis of HuR bound to biotinylated RNA
A plasmid containing full length Msi1 cDNA was used as a template for PCR. The PCR products were purified and used as a template for in vitro transcription. In vitro transcription was catalyzed by T7 RNA polymerase, in the presence of biotin-CTP. Biotin pull-down assays were carried out by incubating 40 μg of cytoplasmic fractions with 1 μg of biotinylated transcripts for 1 h at room temperature. Complexes were isolated with paramagnetic streptavidin-conjugated Dynabeads (Dynal, Oslo, Norway), and bound proteins were analyzed by Western blotting by using a mouse monoclonal antibody recognizing HuR. After secondary antibody incubations, signals were visualized by chemiluminescence.
Reporter assay
For luciferase assays, HeLa cells were co-transfected with the pSGG-Msi1 reporter vector and TAP-tagged HuR expression vector with Roche Fugene 6 transfection reagent (Roche Applied Sciences, Indianapolis, IN). An empty expression vector was used as a negative control. 8 × 103 HeLa cells were plated in a 96-well culture plate 24 hours prior to transfection. Using the Fugene 6 reagent, 100 ng of the HuR expression vector (or empty vector for a negative control) and 1 ng of the UTR luciferase reporter vector were cotransfected. Forty-eight hours later, cell lysates were prepared with the Reporter Lysis Buffer (Promega, Madison, WI). Using the Luciferase Assay Reagent (Promega, Madison, WI), luminescence was read on a Berthold Technologies AutoLumat LB 953 Multi-Tube luminometer (Berthold Technologies, Oak Ridge, TN).
siRNA transfections
U251 and U343 glioblastoma cells were reverse transfected with small interfering RNAs (siRNA) with Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA). siRNAs targeting HuR were obtained from the Dharmacon RNAi Collection SMARTpool line (Thermo Scientific, Rockford, IL). Negative control siRNA reagent was obtained from Thermo Scientific Dharmacon. U251 and U343 glioblastoma cells were transfected with siRNAs in 100-mm culture plates for Musashi1 and HuR protein analysis. Seventy-two hours post-transfection, cells were scraped from the plate and harvested for analysis.
Reverse transcription – quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA). After lysis with TRIzol and separation of any RNA:protein complexes, total RNA was extracted into the aqueous phase using chloroform. Total RNA was precipitated out of the aqueous phase using isopropanol, washed with 75% ethanol, and resuspended in nuclease-free water (Ambion, Austin, TX).
Complementary DNA was synthesized using Applied Biosystems High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) using random priming. Quantitative PCR was performed using the TaqMan Gene Expression Master Mix (Applied Biosystems, Carlsbad, CA). All reactions were run on an ABI 7500 Real Time PCR machine (Applied Biosystems, Carlsbad, CA). ABI TaqMan Gene Expression Assay (Applied Biosystems, Carlsbad, CA) primer/probe sets were obtained for the ELAVL1, MSI1, and ACTB mRNAs. The data were acquired using the ABI SDS 2.0.1 software package. β-actin was used as an endogenous control and the data were analyzed using the 2−ΔΔCt method.
Western blot
After collection of cells for analysis, they were resuspended and sonicated in 2X SDS Laemmli sample buffer. A 10% SDS-PAGE gel with a 4% stacking gel was run in Tris-glycine-SDS buffer. A semi-dry transfer procedure was carried out onto a nitrocellulose membrane. After transfer, the membrane was blocked in Tris-buffered saline with Tween 20 and 5% milk. The membrane was probed with either a rabbit monoclonal anti-Musashi1 antibody (Abcam, Cambridge, MA), a mouse monoclonal anti-HuR antibody (Abcam, Cambridge, MA), mouse monoclonal anti-α-tubulin antibody (Sigma, St. Louis, MO). HRP-conjugated goat anti-rabbit antibody (Santa Cruz Biotechology, Santa Cruz, CA) was used as a secondary antibody for Musashi1 or HRP-conjugated goat anti-mouse antibody (Zymed Laboratories, Carlsbad, CA) was used as a secondary antibody for HuR and α-tubulin. Electrochemiluminescence was used to detect the Musashi1, HuR, and α-tubulin protein using the SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific, Pierce Protein Products, Rockford, IL).
mRNA stability assay
U251 glioblastoma cells were reverse transfected with either the anti-HuR siRNA or scrambled negative control siRNA, in 35-mm plates at 50% confluency in antibiotic-free media. After 12 hours, the media was switched to media containing 5 μg/mL actinomycin D (Alexis Biochemicals, Plymouth Meeting, PA) to inhibit transcription; each hour after that, total RNA was harvested using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Complementary DNA was reverse transcribed using the ABI High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). TaqMan real-time PCR was performed on the cDNA using primers (Applied Biosystems, Carlsbad, CA) for MSI1 mRNA and GAPDH ribosomal RNA. Quantitative PCR was performed on an ABI 7500 Real Time PCR system (Applied Biosystems, Carlsbad, CA), data were acquired using the ABI SDS 2.0.1 software package (Applied Biosystems, Carlsbad, CA) and analyzed using the 2−ΔΔCt method. Subsequently, the data were fitted to a linear regression, and the regression was used to calculate the half-life of the MSI1 mRNA.
Polysomal gradient preparation and analysis
HuR siRNAs, or corresponding negative control siRNA, were reverse transfected into U251 glioblastoma cells. 72 hours after transfection, translation was arrested using 0.1 mg/mL of cycloheximide. Cells were dissociated using trypsin, and cell pellets were formed. Pellets were resuspended and lysed in polysome extraction buffer (20 mM Tris-HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.3% Igepal CA-630, protease inhibitors, and 0.1 mg/mL cycloheximide. After centrifugation to remove insoluble material, the lysate was overlaid on a 10–50% sucrose gradient in a buffer solution of 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 5 mM MgCl2. After ultracentrifugation at 39000 RPM at 4°C, 1-mL fractions were obtained on a density gradient fractionation system (Brandel, Gaithersburg, MD). A254 was measured during the entire fractionation process. Total RNA was extracted from each fraction using TRIzol (Invitrogen, Carlsbad, CA) and analyzed by reverse transcription followed by quantitative PCR analysis.
Quantitative RT-PCR of clinical glioblastoma samples
Previously characterized GBM specimens were used for this study (32). Briefly, tissue samples were obtained from the solid tumor, minced and enzymatically dissociated. Primary tumor cell cultures were maintained under serum-free conditions according to Lee et al. (33). Clinical glioblastoma samples were prepared for total RNA using the RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was prepared using the ABI High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA), and real-time PCR reactions were performed with TaqMan Gene Expression assay probes (Applied Biosystems, Carlsbad, CA) for MSI1, ELAVL1, and ACTB mRNA. The data were acquired using the ABI SDS 2.0.1 software package (Applied Biosystems, Carlsbad, CA) and analyzed using the 2−ΔΔCt method. The RT-qPCR measurements of HuR/Msi1 were performed in triplicate and averaged for each tumor sample (n=20) and the control tissue sample U251. The Ct value of these genes was normalized by subtracting the Ct of the control β-actin measurement.
Tissue microarray and analysis
Immunohistochemistry was performed on the GL806 brain glioblastoma and normal tissue array, which contains 35 cases of glioblastoma, 5 normal tissue, duplicated cores per case (US Biomax Inc., Rockville, MD). Antigen retrieval was performed by heat inducation of the array slides. The primary antibody was performed with either the monoclonal anti-HuR antibody (Molecular Probes Inc., Eugene, OR) or the polyclonal anti-Msi1 antibody (R&D Systems, Inc., Minneapolis, MN).
All stained slides were scanned at 40x using an Aperio CS Scanscope (Aperio Technologies, Vista, CA) and quantified using the available Aperio algorithms. Immunodetection of Msi1 or HuR was quantified according to percent of cells stained. The percentages of stained cells were multiplied by 3 for intensively stained cells, moderately stained cells by 2, and mildly stained cell by 1, and used as a staining index for the quantitative assessment of changes in marker expression in each treated group, as previously reported (34).
Cell proliferation assay
Cell proliferation was performed on the U251 Msi1 or GFP overexpression cell lines (U251:MSI1 or U251:GFP, respectively) and U343 Msi1 or GFP overexpression cell lines (U343:MSI1 or U343:GFP, respectively). Either HuR siRNA or control siRNA was transfected into each cell line. After 48 hours after transfection, cell proliferation was measured with CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). After 1 hour, absorbance at 490nm was measured on a Synergy HT Multi-Mode Microplate Reader (Biotek, Winooski, VT).
Cell cycle analysis
Cells were prepared for cell cycle distribution using propidium iodide staining of DNA content. Cells were harvested using trypsin, pelleted via centrifugation at 1000 rpm for 3 minutes at 4°C, and resuspended in ice-cold phosphate-buffered saline (PBS). The cells were pelleted once again and resuspended in 500 μL of ice-cold PBS. The resuspended cells were then slowly dropped into a vortexing tube containing 9X volume of ice-cold 100% ethanol for fixation. The solution was incubated at 4°C for 30 minutes. The cells were then pelleted via centrifugation, washed with PBS once more, and resuspended in the propidium iodide staining column, which composed of a solution of PBS containing 0.1% (v/v) of Triton-X100 (Sigma-Aldrich, St. Louis, MO), 200 μg/mL of RNase A (Sigma-Aldrich, St. Louis, MO), and 20 ug/mL of propidium iodide (Invitrogen, Carlsbad, CA). The cells were incubated in staining solution in the dark for 30 minutes at room temperature. The cells were analyzed on the BD FACSCanto Flow Cytometry machine (BD Biosciences, San Jose, CA). Flow cytometry data was analyzed using Modfit LT (Verity Software House, Topsham, ME) to calculate cell cycle distribution.
Caspase-3/-7 activity assay
Apoptosis was assayed with the U251 Msi1 or GFP overexpression cell lines (U251:MSI1 or U251:GFP, respectively) and U343 Msi1 or GFP overexpression cell lines (U343:MSI1 or U343:GFP, respectively). Either HuR siRNA or control siRNA was transfected into each cell line. After 48 hours after transfection, apoptosis was monitored with the Caspase-Glo 3/7 Assay reagent (Promega, Madison, WI). Luciferase measurements was acquired with the SpectraMax M5 Multi-Mode Microplate Reader (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis
All statistical analysis was performed with the Student’s t-test. Data are presented as the mean ± standard error of the mean.
Results
Identification of U- and AU-rich elements in the Msi1 3′ untranslated region
The MSI1 mRNA contains a long 3′ UTR, suggestive of post-transcriptional regulation. In a previous report, our lab has identified 5 tumor suppressor miRNAs that modulate Msi1 expression (22). Upon closer observation, we also identified regions in the Msi1 3′ UTR containing potential uracil-rich elements amenable for regulation by HuR (35) (Figure 1A). In agreement with the idea that HuR functions as a positive regulator of Msi1, HuR has been shown to be upregulated in many tumors (24) including glioblastoma, where Msi1 is also highly expressed.
Figure 1.

Discovery of HuR as a trans-acting post-transcriptional regulatory factor for Msi1. (A) A schematic of the MSI1 mRNA with the 3′ untranslated region shown in the inset. The nucleotide tracts bolded, italicized, and underlined indicates the putative HuR binding sites. (B) Immunoprecipitation of RNA:HuR protein complexes identifies Musashi1 mRNA as a binding partner for HuR. HuR ribonucleoprotein complexes were pulled down with an anti-HuR specific antibody. The RNA recovered was reverse transcribed and qPCR was performed on the complementary DNA. Primers were used for GAPDH, PTMA, and MSI1 mRNAs. GAPDH mRNA is used as a negative control, and PTMA mRNA is a positive control. (C) Diagram of the mRNA fragments used in biotin pulldown experiment. Three fragments were made, one for the CR and two for each half of the 3′ UTR. (D) Biotin pulldown assay confirms HuR interaction with MSI1 mRNA. Biotinylated fragments of the MSI1 mRNA were in vitro transcribed. Three fragments of the mRNA were analyzed, one for the coding region and two for each half of the 3′ untranslated region. The GAPDH 3′ UTR was included as a negative control. After incubation of the fragments with streptavidin beads and biotinylated RNA, the protein was recovered by pulldown analysis and immunoblotted for HuR. A beads only control was included to account for any nonspecific interaction and a ‘lysate’ was included as a positive control for the immunoblot. (E) Influence of HuR on Msi1 protein expression is dependent on the 3′ untranslated region. The Msi1 3′ untranslated region was cloned downstream of a PEST-destablized luciferase gene (luc2P). The luciferase construct was cotransfected with an TAP-tagged HuR expression vector (denoted as TAP-HuR) in HeLa cervical adenocarcinoma cells. An empty vector (denoted as TAP) was utilized as a negative control. Data were analyzed with Student’s t-test and is presented as the mean ± standard error of the mean. Experiment was performed in triplicate.
Immunoprecipitation of HuR enriches for the MSI1 mRNA
First, we performed an immunoprecipitation of HuR ribonucleoprotein complexes (RNP-IP) in U251 glioblastoma cells followed by RT-qPCR to determine whether or not the MSI1 mRNA is associated with HuR protein. In three biological replicates, we observed an overall enrichment of approximately 20-fold of the MSI1 mRNA in IPs done with anti-HuR antibodies in comparison to the GAPDH mRNA, which does not bind to HuR. As a positive control for HuR binding, the mRNA encoding prothymosin α, an antiapoptotic protein, was evaluated (36). HuR binding to MSI1 mRNA seems to be even stronger than HuR binding to prothymosin α mRNA (Figure 1B).
To map the region of the MSI1 mRNA that contains the HuR binding site, we performed biotin pulldown experiments with biotinylated fragments of the MSI1 mRNA. Three fragments were made, one for the coding region (CR) of Msi1 and two for the 3′ UTR (a proximal region and a distal region) (Figure 1C). The GAPDH 3′ UTR was included as a negative control. After incubation with U251 cell lysate, the bound protein eluate was immunoblotted for HuR. The results of the pulldown-Western experiments demonstrate a strong HuR signal for the distal half of the Msi1 3′ UTR as compared to the beads only (Figure 1D). A weak but positive signal is seen for the proximal region, indicating that indeed binding exists, but most likely with low affinity. The CR fragment shows no signal, indicating a lack of a binding of HuR to this region of the MSI1 mRNA.
Luciferase reporter assay confirms interaction with Msi1 3′ UTR
To further examine the effect of HuR on Msi1 expression, a luciferase reporter carrying the Msi1 3′ UTR was constructed and tested. The luciferase CR contained a PEST degradation sequence (37) that effectively reduces the luciferase protein half-life to approximately one hour, thus reducing the chances of any ambiguous results due to protein accumulation. Compared to the empty expression vector, we detect a two-fold increase (p = 2.5 × 10−5) in luciferase activity when we cotransfect an HuR expression vector (Figure 1E) thus confirming the interaction of HuR with the Msi1 3′ UTR.
HuR silencing suggests that HuR positively regulates Msi1 expression
To further validate the regulation of Msi1 by HuR, its levels were reduced by using HuR-directed siRNA in U251 and U343 glioblastoma cells. A significant reduction in HuR (U251: p = 1.5 × 10−9; U343: p = 1.3 × 10−3) was followed by a concomitant reduction in Msi1 expression at both the mRNA (p = 3.4 × 10−7; U343: p = 5.1 × 10−3) (Figure 2A and 2B) and protein levels (Figure 2C and 2D), suggesting that HuR positively regulates Msi1 expression in glioblastoma cells.
Figure 2.

MSI1 mRNA and protein levels are dependent on HuR levels. (A and B) MSI1 mRNA levels are upregulated by HuR. Seventy-two hours after siRNA-mediated silencing of HuR (50 nM) in U251 (A) and U343 (B) glioblastoma cells, MSI1 mRNA was quantified by RT followed by qPCR analysis; β-actin mRNA was utilized as an endogenous control and the data were analyzed using the 2−ΔΔCt methodology. Data were analyzed with Student’s t-test and is presented as the mean ± standard error of the mean. Experiment was performed in triplicate. (C and D) Msi1 protein is positively dependent on HuR levels. By 72 h after siRNA-mediated silencing of HuR (50 nM) in U251 (C) and U343 (D) glioblastoma cells, Msi1 protein levels were assessed by Western blot analysis. Total RNA was extracted using Trizol reagent and cDNA was synthesized using reverse transcriptase. Western blotting of HuR and Msi1 was performed. α-tubulin served as a loading control.
HuR increases MSI1 mRNA stability
To dissect the mechanism by which HuR controls Msi1 expression, we first measured the stability of MSI1 mRNA (38). We knocked down the expression of HuR using siRNAs and 12 hours post-transfection, we treated the cells with 5 ug/mL of actinomycin D, an inhibitor of RNA polymerase II. After transcription was inhibited, mRNA decay was measured without interference. The mRNA decay rate of the MSI1 mRNA was measured in the presence and absence of the HuR RNA-binding protein. Over the course of 3 hours, we observed a faster rate of decay in HuR-silenced cells than in control cells. We fitted the data to a linear regression model and used the model to calculate the half-life of the MSI1 mRNA. The half-life of the MSI1 mRNA is 8.17 hours whereas in HuR-silenced cells, the MSI1 mRNA half-life was reduced to approximately 3.12 hours, representing a 62% increase in the rate of decay (p = 4.8 × 10−5) of the MSI1 mRNA (Figure 3).
Figure 3.
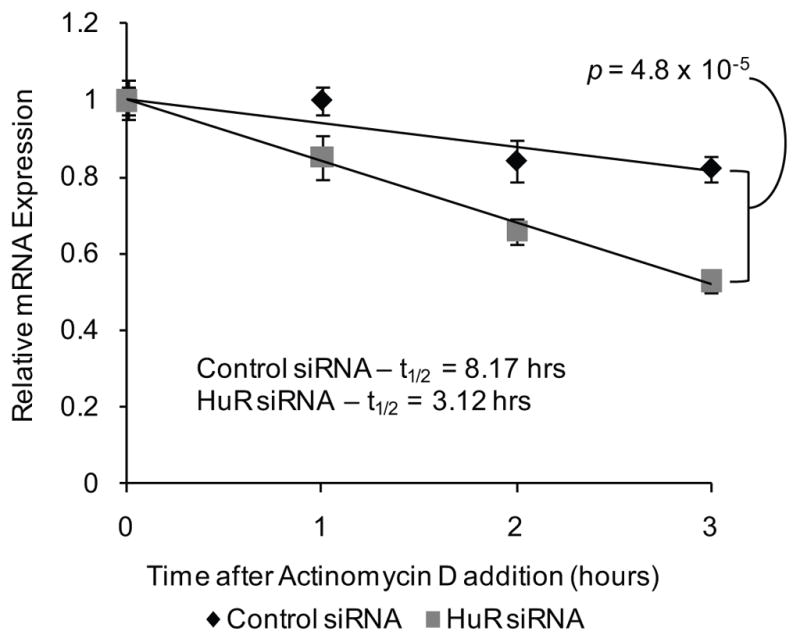
HuR stabilizes the MSI1 mRNA. The stability of the MSI1 mRNA mediated by HuR was assessed in U251 glioblastoma cells. siRNAs (50 nM) directed against HuR was transfected into U251. A scrambled siRNA was utilized as a negative control. Actinomycin D (5 ug/mL) was added to the cell media 12 hours after siRNA transfection. Total RNA samples were obtained and MSI1 mRNA levels were determined using RT-qPCR. The data were analyzed using the 2−ΔΔCt method and the GAPDH mRNA was employed as an endogenous control. Relative MSI1 mRNA levels were plotted against time after actinomycin D addition and fitted to a linear regression model. MSI1 mRNA half-life was calculated using the linear regression model. Data were analyzed with Student’s t-test and is presented as the mean ± standard error of the mean. Experiment was performed in triplicate.
HuR positively regulates Msi1 translation
In addition to mediating mRNA stability, HuR can also modulate translation (24). To determine if HuR also affects the translation of MSI1 mRNA, we performed polysome fractionation analysis using extracts from U251 control and HuR-knockdown cells (39). In this assay, polysomes, which contain actively translating mRNAs and have high sedimentation rates, are partitioned from untranslated mRNAs, which have low sedimentation rates (Figure 4A and 4B). Fine fractions are acquired from the gradients and levels of mRNAs are measured with Northern analysis or quantitative PCR. The results showed that the profile of the GAPDH mRNA, encoding a housekeeping protein, was unaffected by HuR silencing (Figure 4C) while we observed a shift in MSI1 mRNA from heavier polysomal fractions to the lighter fractions when comparing control cells to HuR knockdown cells (Figure 4D). This result indicates that HuR promotes Msi1 expression by a combination of increasing MSI1 mRNA stability and enhancing its translation.
Figure 4.

HuR promotes translation of the MSI1 mRNA. Sucrose gradients were used to fractionate lysates from control U251 cells and cells with HuR knockdown. After fractionation of the sucrose gradient post-ultracentrifugation, RNA from each fraction was extracted with TRIzol and analyzed by real-time PCR. GAPDH mRNA, a transcript that is not regulated by HuR, was included as a negative control. Panels (A) and (B) are the results of the A254 during fractionation. The first three peaks from the left are the 40S, 60S, and 80S subunits, while the peaks to the right are the heavier polysomal fractions. In panels (C) and (D), the percentage distribution of the GAPDH (C) or MSI1 (D) is shown.
HuR and Msi1 expression are positively correlated
It has been suggested that Msi1 is required to maintain the “cancer stem cell population” (18, 40). In support of this idea, previous studies done in our lab and others (30) demonstrated that tumor cells maintained as spheroids, a condition that favors the growth of cells with “stem-like” characteristics, have increased Msi1 expression compared to cells grown as adherent monolayers. We tested to see if the same applies to HuR. HuR expression was measured by qRT-PCR in two tumor lines established at UTHSCSA revealing also that as Msi1, HuR expression levels are higher in spheroid cultures relative to monolayers (Figure 5A).
Figure 5.

HuR and Msi1 expression levels correlate positively. (A) MSI1 mRNA and HuR mRNA levels are higher in tumor cells with “stem cells” characteristics. HuR and Msi1 expression was assessed in two primary glioblastoma samples grown as spheroids vs. monolayers. The ACTB mRNA was used for normalization when using the 2−ΔΔCt method for mRNA relative quantification. Data were analyzed with Student’s t-test and were presented as the mean ± standard error of the mean. Experiment was performed in triplicate. (B) HuR and MSI1 mRNA expression positively correlates in GBMs. RT-qPCR was performed on total RNA extracted from the clinical samples and the data was analyzed using the 2−ΔΔCt method. The ACTB mRNA was utilized as an endogenous control and all samples were compared to the U251 glioblastoma cell line. The ΔΔCtMsi1 was plotted on the abscissa, and the ΔΔCtHuR was plotted on the ordinate. A linear regression model was generated with a correlation score of 0.503 and p - value of 0.0201; thus, HuR and Msi1 exhibit statistically significant linear correlation in expression. (C) Representative microscopic images (40X) of immunohistochemistry of Msi1 and HuR in a glioblastoma tissue microarray. (D) Quantifying glioblastoma tissue microarray stainings of HuR and Msi1. Staining scores for HuR and Msi1 in glioblastoma (35 cores) was averaged and compared to the average score for staining in normal brain tissue (5 cores). Both HuR and Msi1 are more expressed in glioblastoma than in normal brain tissue (p – value < 0.0001). Data were analyzed by the Student’s t-test and presented as the mean ± standard error of the mean. (E) Individual scores of each glioblastoma and normal brain tissue samples for HuR and Msi1 are graphed on a vertical scatter plot. In glioblastoma, HuR had an average score of 83.48, SEM = 1.507 with a 95% confidence interval ranging from 80.47 to 86.50 while in normal brain tissue, HuR had an average score of 61.00, SEM = 9.452 with a 95% confidence interval: 39.62 to 82.38. In glioblastoma, Msi1 had an average score of 41.14, SEM = 4.017 with a 95% confidence interval ranging from 33.11 to 49.16 while in normal brain tissue, Msi1 had an average score of 0.0, SEM = 0.0 with a 95% confidence interval ranging from 0.0 to 0.0.
We then analyzed the messenger RNA expression of HuR and Msi1 in clinical samples of glioblastoma by RT-qPCR. When analyzed as on a scatter plot, a statistically significant (p = 0.0201) positive correlation of HuR and Msi1 expression is observed (Pearson correlation score = 0.503, 95% confidence interval: 0.09–0.77), indicating that glioblastoma tumors that display high Msi1 expression also express high levels of HuR (Figure 5B).
We extended our expression analysis to examine the protein expression of both HuR and Msi1. We immunostained with both anti-Msi1 and anti-HuR antibodies a tissue microarray contained 35 high-quality glioblastoma cores and 5 normal brain tissue cores in duplicate (Figure 5C). We observed that although ubiquitously expressed, HuR is more abundant in glioblastoma than in normal brain tissue (Figure 5D, tumor average score = 83.48, SEM = 1.507, 95% confidence interval: 80.47–86.50 versus normal tissue average score = 61.00, SEM = 9.452, 95% confidence interval: 39.62–82.38, p – value < 0.0001). As for Msi1, while no expression was detected in normal brain, which is consistent with the notion that Msi1 is not present in differentiated cells (3). Msi1 expression was often detected in glioblastoma samples, (Figure 5D and 5E, tumor average score = 41.14, SEM = 4.017, 95% confidence interval: 33.11–49.16 versus normal tissue average score = 0.0, SEM = 0.0, 95% confidence interval: 0.0–0.0, p – value < 0.0001). From this analysis, we also ascertain that both Msi1 and HuR expression positively correlate in glioblastoma (Pearson correlation = 0.497, p < 0.0001).
Ectopic Msi1 expression rescues cell proliferation inhibition by HuR silencing
We sought to determine if Msi1 is a main mediator of HuR function in glioblastoma cells. We silenced HuR via siRNA in control lines and in lines containing a MSI1 transgene that does not contain the 3′ UTR, rendering the ectopic Msi1 expression immune to any post-transcriptional regulation. 48 hours after transfection, we measured cell proliferation by MTS assay. While HuR knockdown caused a reduction in cell proliferation in control cells (31% reduction in U251 malignant glioma cells and 21% reduction in U343 malignant glioma cells, Figure 6A and 6B), the effect was partially ameliorated in cells with transgenic Msi1 expression (13% reduction in U251 and 2.7% in U343, Figure 6A and 6B). This result suggests that the impact of HuR on cell growth is partially mediated by Msi1.
Figure 6.

Ectopic expression of Msi1 partially reverses cell proliferation suppression induced by HuR silencing. (A) U251:GFP control and U251:MSI1 cells and (B) U343:GFP control and U343:MSI1 cells were transfected with either HuR siRNA or scrambled control siRNA. Cell growth was measured by MTS assay 48 hours post-transfection. In both cases, HuR silencing inhibited cell proliferation. However, this effect was partially recovered when Msi1 was ectopically expressed. Data were analyzed by the Student’s t-test and presented as the mean ± standard error of the mean. Experiment was performed in triplicate.
Ectopic Msi1 expression attenuates the effect of HuR silencing on cell cycle distribution
We further determined if the impact HuR has on the cell cycle is partially mediated by Msi1. Experiments were done with the U251 and U343 Msi1 and GFP control transgenic cell lines described above. Depletion of HuR by siRNAs caused an impact on cell cycle distribution in both U251 and U343 cells. In U251 cells, HuR silencing promoted a distribution of cells moving from G1 to S phase in U251:GFP cells (Figure 7A) while in U343 cells, we observed an expansion of the population in G2 phase with a concomitant decrease of cells in G1 and a slight decrease in cells found in S phase (Figure 7B). When HuR silencing was performed in U251 and U343 cells which ectopically expressing Msi1, the magnitude of the effect of HuR siRNA on the distribution of cells in each cell cycle phase is blunted (Figure 7A and 7B). In conclusion, our results indicate that HuR has a profound effect on cell cycle distribution and suggests that this effect is partially achieved via Msi1.
Figure 7.

Cell cycle distribution is altered upon HuR silencing with siRNAs and is partially attenuated with ectopic Msi1 expression. Cell cycle analysis was performed using propidium iodide staining of ethanol-fixed cells for DNA content which was analyzed using flow cytometry. (A and B) U251:GFP control and U251:MSI1 cells were transfected with either HuR siRNA or scrambled control siRNA. A similar experiment was performed in U343:GFP and U343:MSI1 cells. Cell cycle distribution was analyzed using the Modfit LT software package. Then, the percentage of cells in each phase found in the HuR siRNA experiment was subtracted from the control siRNA experiment. HuR silencing caused a large shift in cell cycle distribution in the U251:GFP (A) and U343:GFP (B) cells. However, when Msi1 is ectopically expressed, the change in cell cycle distribution induced by HuR silencing was attenuated (A and B). Data were analyzed by the Student’s t-test and presented as the mean ± standard error of the mean. Experiment was performed in triplicate.
Ectopic Msi1 expression rescues apoptosis induced by HuR silencing
Since HuR and Msi1 have been implicated in the promotion of a pro-survival/anti-apoptotic program in the cell (18, 19, 29, 36, 41–43), we sought to determine if HuR and Msi1 cooperatively to promote an anti-apoptotic phenotype. When HuR expression is depleted by siRNA transfection, caspase-3/-7 activity is increased 3.3-fold in U251:GFP control cells and 3.9-fold in U343:GFP control cells, Figure 8A and 8B, as compared to the control siRNA transfection. However, when HuR is depleted in either the U251:MSI1 or U343:MSI1 stable cell lines, caspase-3/-7 activity is only induced 2.3-fold in U251:MSI1 cells and 3.2-fold in U343:MSI1 (Figure 8A and 8B), suggesting that Msi1 transgenic expression curtails the induction of apoptosis when HuR is silenced. This result indicates that the anti-apoptotic phenotype triggered by HuR is partially mediated by Msi1.
Figure 8.

Ectopic expression of Msi1 partially recovers the effect of HuR silencing on apoptosis. (A) HuR or control siRNAs were transfected in U251:GFP and U251:MSI1 cells. (B) Similar experiments were performed in U343:GFP and U343:MSI1 cells. Apoptosis was measured 48 hours post-transfection using a luciferase-based reagent to monitor caspase-3/-7 activity. HuR silencing was able to induce apoptosis, while Msi1 ectopic expression partially inhibits this effect. Data was analyzed by the Student’s t-test and presented as the mean ± standard error of the mean. Experiment was performed in triplicate.
Discussion
In this study, we explored the post-transcriptional regulation of the Msi1 expression by HuR. We found that HuR binds to sites located in the distal portion of the Msi1 3′ UTR and controls its expression via mRNA stabilization and translational upregulation. We determined that HuR and Msi1 expression levels are positively correlated and that the identified regulation has potentially important implications for glioblastoma cell growth.
RBPs regulate mammalian gene expression at the post-transcriptional level, mediating many processes such as mRNA maturation, splicing, transport, storage, stability, and translation. Turnover and translation regulatory RNA-binding proteins (TTR-RBPs) (44) are a large family of trans-acting factors that control mRNA stability and translation through binding to cis-regulatory elements encoded by an mRNA. Many TTR-RBPs are present in the cell and likely function jointly on a particular mRNA, through additive, antagonistic, or synergistic actions. The role of pathologic and aberrant actions of RBPs in many disease processes, including cancer, has been revealed in recent years. One RBP of particular interest in cancer is HuR (HuA), a member of the Hu/ELAV family of RNA-binding proteins that also encompasses the neuronal proteins HuB, HuC, and HuD. Through interactions with specific subsets of mRNAs, HuR can potentiate biological features of cancers such as sustained proliferation, evasion of apoptosis, increased angiogenesis, and invasion/metastasis (24).
HuR is a ubiquitous, multi-faceted RBP implicated in mRNA splicing, translation regulation, and mRNA stabilization (24). In recent years, HuR has been implicated as a potent factor in tumorigenesis (24) through its ability to influences many characteristics of cancer cells (45, 46) including limitless proliferation, sustained angiogenesis, tissue invasion, and metastasis, via its interactions with cancer related genes [e.g., c-Myc (47), p27 (48), VEGF (49), and Snail (50)]. In high-grade gliomas, prominent HuR expression have been shown; specific mRNA ligands, such as VEGF, COX-2, IL6, IL8, TGFβ, and TNFα mRNAs have been identified (25, 26, 28), but the molecular characterization of the interaction of HuR with these ligands has yet to be carried out. The totality of this regulation results in the necessity of glioma cells to maintain and augment growth of glioma cells (26).
Our study explores a more global effect by HuR in glioblastoma, through HuR’s positive regulation of another oncogenic RBP, Msi1, previously implicated in controlling a wide subset of mRNAs with cancer-related functions in proliferation, differentiation, survival, and apoptosis (51). Our findings posit that HuR not only elicits direct effects on cancer cell gene expression, but it can also exert indirect gene regulatory effects by controlling expression of another key regulator of gene expression, Msi1. Our findings may be extended to other malignancies that depend on Msi1 expression such as medulloblastoma, hepatocellular carcinoma, cervical adenocarcinoma, non-small cell lung cancer, breast and colon cancer (8–10, 18, 19). High HuR expression has also been reported in other studies (25, 31, 52–54); the overlap in expression profiles of Msi1 and HuR suggests that the HuR:Msi1 relationship described in this report may exist in other tumors (Table 1). From these results, a complex post-transcriptional genetic regulatory network emerges (55) for glioblastoma that will describe the cellular and physiological functions required for gliomagenesis through the discovery of network motifs (56), modular building blocks of gene regulatory networks.
Table 1.
Summary of malignancies that exhibit increased HuR and Msi1 expression.
| Tumor Type | HuR | Msi1 | References |
|---|---|---|---|
| Glioblastoma | ↑ | ↑ | (12, 25, 64) |
| Medulloblastoma | ↑ | ↑ | (18, 25) |
| Hepatocellular Carcinoma | ↑ | ↑ | (8, 31) |
| Cervical Adenocarcinoma | ↑ | ↑ | (9, 52) |
| Non small cell lung cancer | ↑ | ↑ | (10, 53) |
| Colon cancer | ↑ | ↑ | (19, 59) |
↑ indicates increase expression. References for each study are called out in the rightmost column.
The HuR:Msi1 relationship discussed in this paper creates a positive cascade network motif (56) where the cellular logic for gliomagenesis requires HuR to essentially ‘activate’ Msi1 gene expression. Moreover, HuR also autoregulates itself bolstering its own production in a positive-feedback loop (57–59). The interaction between these two RBPs creates a two-component composite gene regulatory network. In Saccharomyces cerevisiae, 21 of the 46 studied RBPs are thought to be involved in a two- or multi-component composite gene regulatory network thus revealing the potential for a dense genetic regulatory network that has not been previously recognized (55).
However, post-transcriptional genetic regulatory network motifs are not limited by RBP nodes but extend to microRNAs. A recent study from our lab illustrates the post-transcriptional regulation of Musashi1 by tumor suppressor miRNAs (22). Moreover, we also recently showed that 25% of HuR binding sites in 3′ UTRs mapped by CLIP (crosslinking and immunoprecipitation) coincide with miRNA predicted sites, suggesting an interplay between RBPs and miRNAs (60). These complex relationships demonstrate the nonlinearity of post-transcriptional network entities, and shows that their integration creates a larger, complex network that contains the pertinent logic for the molecular pathogenesis in glioblastoma. Furthermore, post-transcriptional gene networks are intimately intertwined with the transcriptional gene network thus an inclusive analysis of the circuitry required for glioblastoma tumorigenesis surfaces, providing insights into glioblastoma cellular logic and physiology and the emergence of genetic network-directed therapies (61–63).
Acknowledgments
Grant Support
KA, JLM, KT, and MG are supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. VP is supported by the Intramural Research Program at the National Institute of Dental and Craniofacial Research, National Institutes of Health. DTV, MQ, TLB, and LOFP are supported by grants from the Children’s Brain Tumor Foundation, Association for Research of Childhood Cancer, and The Max and Minnie Tomerlin Voelcker Fund.
We thank Suzanne C. Burns for critical reading and comments on this manuscript. We are grateful to the anonymous referees for their critical reading and suggestions for this manuscript throughout the review process.
Footnotes
Conflict of interest statement: None declared.
References
- 1.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582(14):1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura M, Okano H, Blendy JA, Montell C. Musashi, a neural RNA-binding protein required for Drosophila adult external sensory organ development. Neuron. 1994;13(1):67–81. doi: 10.1016/0896-6273(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 3.Sakakibara S, Imai T, Hamaguchi K, et al. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev Biol. 1996;176(2):230–42. doi: 10.1006/dbio.1996.0130. [DOI] [PubMed] [Google Scholar]
- 4.Kayahara T, Sawada M, Takaishi S, et al. Candidate markers for stem and early progenitor cells, Musashi-1 and Hes1, are expressed in crypt base columnar cells of mouse small intestine. FEBS Lett. 2003;535(1–3):131–5. doi: 10.1016/s0014-5793(02)03896-6. [DOI] [PubMed] [Google Scholar]
- 5.Clarke RB. Isolation and characterization of human mammary stem cells. Cell Prolif. 2005;38(6):375–86. doi: 10.1111/j.1365-2184.2005.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugiyama-Nakagiri Y, Akiyama M, Shibata S, Okano H, Shimizu H. Expression of RNA-binding protein Musashi in hair follicle development and hair cycle progression. Am J Pathol. 2006;168(1):80–92. doi: 10.2353/ajpath.2006.050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokota N, Mainprize TG, Taylor MD, et al. Identification of differentially expressed and developmentally regulated genes in medulloblastoma using suppression subtraction hybridization. Oncogene. 2004;23(19):3444–53. doi: 10.1038/sj.onc.1207475. [DOI] [PubMed] [Google Scholar]
- 8.Shu HJ, Saito T, Watanabe H, et al. Expression of the Musashi1 gene encoding the RNA-binding protein in human hepatoma cell lines. Biochem Biophys Res Commun. 2002;293(1):150–4. doi: 10.1016/S0006-291X(02)00175-4. [DOI] [PubMed] [Google Scholar]
- 9.Ye F, Zhou C, Cheng Q, Shen J, Chen H. Stem-cell-abundant proteins Nanog, Nucleostemin and Musashi1 are highly expressed in malignant cervical epithelial cells. BMC Cancer. 2008;8:108. doi: 10.1186/1471-2407-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanai R, Eguchi K, Takahashi M, et al. Enhanced therapeutic efficacy of oncolytic herpes vector G207 against human non-small cell lung cancer--expression of an RNA-binding protein, Musashi1, as a marker for the tailored gene therapy. J Gene Med. 2006;8(11):1329–40. doi: 10.1002/jgm.965. [DOI] [PubMed] [Google Scholar]
- 11.Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600–8. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanemura Y, Mori K, Sakakibara S, et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation. 2001;68(2–3):141–52. doi: 10.1046/j.1432-0436.2001.680208.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang XY, Penalva LO, Yuan H, et al. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol Cancer. 9:221. doi: 10.1186/1476-4598-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glazer RI, Wang XY, Yuan H, Yin Y. Musashi1: a stem cell marker no longer in search of a function. Cell Cycle. 2008;7(17):2635–9. doi: 10.4161/cc.7.17.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guessous F, Li Y, Abounader R. Signaling pathways in medulloblastoma. J Cell Physiol. 2008;217(3):577–83. doi: 10.1002/jcp.21542. [DOI] [PubMed] [Google Scholar]
- 16.Lino MM, Merlo A, Boulay JL. Notch signaling in glioblastoma: a developmental drug target? BMC Med. 8:72. doi: 10.1186/1741-7015-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai T, Tokunaga A, Yoshida T, et al. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol Cell Biol. 2001;21(12):3888–900. doi: 10.1128/MCB.21.12.3888-3900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Diaz PC, Burton TL, Burns SC, Hung JY, Penalva LO. Musashi1 modulates cell proliferation genes in the medulloblastoma cell line Daoy. BMC Cancer. 2008;8:280. doi: 10.1186/1471-2407-8-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sureban SM, May R, George RJ, et al. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology. 2008;134(5):1448–58. doi: 10.1053/j.gastro.2008.02.057. [DOI] [PubMed] [Google Scholar]
- 20.Ratti A, Fallini C, Cova L, et al. A role for the ELAV RNA-binding proteins in neural stem cells: stabilization of Msi1 mRNA. J Cell Sci. 2006;119(Pt 7):1442–52. doi: 10.1242/jcs.02852. [DOI] [PubMed] [Google Scholar]
- 21.Okano H. Adult neural stem cells and central nervous system repair. Ernst Schering Res Found Workshop. 2006;60:215–28. doi: 10.1007/3-540-31437-7_14. [DOI] [PubMed] [Google Scholar]
- 22.Vo DT, Qiao M, Smith AD, Burns SC, Brenner AJ, Penalva LO. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 8(5) doi: 10.4161/rna.8.5.16041. [DOI] [PubMed] [Google Scholar]
- 23.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33(22):7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdisciplinary Reviews - RNA. 1(2):214–29. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61(5):2154–61. [PubMed] [Google Scholar]
- 26.Bolognani F, Gallani AI, Sokol L, Baskin DS, Meisner-Kober N. mRNA stability alterations mediated by HuR are necessary to sustain the fast growth of glioma cells. J Neurooncol. doi: 10.1007/s11060-011-0707-1. [DOI] [PubMed] [Google Scholar]
- 27.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58(2):266–77. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Tumor necrosis factor alpha induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Res. 2003;63(14):4181–7. [PubMed] [Google Scholar]
- 29.Filippova N, Yang X, Wang Y, et al. The Rna-Binding Protein Hur Promotes Glioma Growth and Treatment Resistance. Mol Cancer Res. doi: 10.1158/1541-7786.MCR-10-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23(58):9392–400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 31.Mazan-Mamczarz K, Galban S, Lopez de Silanes I, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci U S A. 2003;100(14):8354–9. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glas M, Rath BH, Simon M, et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann Neurol. 68(2):264–9. doi: 10.1002/ana.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 34.Amornphimoltham P, Patel V, Sodhi A, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65(21):9953–61. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 35.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci U S A. 2004;101(9):2987–92. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lal A, Kawai T, Yang X, Mazan-Mamczarz K, Gorospe M. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J. 2005;24(10):1852–62. doi: 10.1038/sj.emboj.7600661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belizario JE, Alves J, Garay-Malpartida M, Occhiucci JM. Coupling caspase cleavage and proteasomal degradation of proteins carrying PEST motif. Curr Protein Pept Sci. 2008;9(3):210–20. doi: 10.2174/138920308784534023. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda E, Achen MG, Breier G, Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270(34):19761–6. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- 39.Perry RP, Kelley DE. Messenger RNA-protein complexes and newly synthesized ribosomal subunits: analysis of free particles and components of polyribosomes. J Mol Biol. 1968;35(1):37–59. doi: 10.1016/s0022-2836(68)80035-x. [DOI] [PubMed] [Google Scholar]
- 40.Wang XY, Yin Y, Yuan H, Sakamaki T, Okano H, Glazer RI. Musashi1 modulates mammary progenitor cell expansion through proliferin-mediated activation of the Wnt and Notch pathways. Mol Cell Biol. 2008;28(11):3589–99. doi: 10.1128/MCB.00040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishimaru D, Ramalingam S, Sengupta TK, et al. Regulation of Bcl-2 expression by HuR in HL60 leukemia cells and A431 carcinoma cells. Mol Cancer Res. 2009;7(8):1354–66. doi: 10.1158/1541-7786.MCR-08-0476. [DOI] [PubMed] [Google Scholar]
- 42.Durie D, Lewis SM, Liwak U, Kisilewicz M, Gorospe M, Holcik M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene. 30(12):1460–9. doi: 10.1038/onc.2010.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdelmohsen K, Lal A, Kim HH, Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007;6(11):1288–92. doi: 10.4161/cc.6.11.4299. [DOI] [PubMed] [Google Scholar]
- 44.Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem. 2008;389(3):243–55. doi: 10.1515/BC.2008.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 46.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23(15):1743–8. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002;16(23):3087–99. doi: 10.1101/gad.248902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273(11):6417–23. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 50.Dong R, Lu JG, Wang Q, He XL, Chu YK, Ma QJ. Stabilization of Snail by HuR in the process of hydrogen peroxide induced cell migration. Biochem Biophys Res Commun. 2007;356(1):318–21. doi: 10.1016/j.bbrc.2007.02.145. [DOI] [PubMed] [Google Scholar]
- 51.de Sousa Abreu R, Sanchez-Diaz PC, Vogel C, et al. Genomic analyses of musashi1 downstream targets show a strong association with cancer-related processes. J Biol Chem. 2009;284(18):12125–35. doi: 10.1074/jbc.M809605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, Gorospe M. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol Cell Biol. 2006;26(8):3295–307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Zhao W, Guo Y, et al. The expression of RNA-binding protein HuR in non-small cell lung cancer correlates with vascular endothelial growth factor-C expression and lymph node metastasis. Oncology. 2009;76(6):420–9. doi: 10.1159/000216837. [DOI] [PubMed] [Google Scholar]
- 54.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23(15):3092–102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanitz A, Gerber AP. Circuitry of mRNA regulation. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2(2):245–51. doi: 10.1002/wsbm.55. [DOI] [PubMed] [Google Scholar]
- 56.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8(6):450–61. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 57.Pullmann R, Jr, Kim HH, Abdelmohsen K, et al. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol Cell Biol. 2007;27(18):6265–78. doi: 10.1128/MCB.00500-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi J, Chang N, Liu X, et al. Reduced nuclear export of HuR mRNA by HuR is linked to the loss of HuR in replicative senescence. Nucleic Acids Res. 38(5):1547–58. doi: 10.1093/nar/gkp1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Al-Ahmadi W, Al-Ghamdi M, Al-Haj L, Al-Saif M, Khabar KS. Alternative polyadenylation variants of the RNA binding protein, HuR: abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Res. 2009;37(11):3612–24. doi: 10.1093/nar/gkp223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uren PJ, Burns SC, Ruan J, Singh KK, Smith AD, Penalva LO. Genomic analyses of the RNA binding protein Hu Antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. J Biol Chem. doi: 10.1074/jbc.C111.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morrow JK, Tian L, Zhang S. Molecular networks in drug discovery. Crit Rev Biomed Eng. 38(2):143–56. doi: 10.1615/critrevbiomedeng.v38.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitano H. Computational systems biology. Nature. 2002;420(6912):206–10. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 63.Kitano H. A robustness-based approach to systems-oriented drug design. Nat Rev Drug Discov. 2007;6(3):202–10. doi: 10.1038/nrd2195. [DOI] [PubMed] [Google Scholar]
- 64.Toda M, Iizuka Y, Yu W, et al. Expression of the neural RNA-binding protein Musashi1 in human gliomas. Glia. 2001;34(1):1–7. doi: 10.1002/glia.1034. [DOI] [PubMed] [Google Scholar]