

Abstract
The BMI1 oncogene promotes prostate cancer progression. High BMI1 expression predicts poor prognosis in prostate cancer (PC) patients. Recent evidence suggests that BMI1 may also play a role in Docetaxel chemoresistance. However, mechanisms and clinical significance of BMI1-related chemoresistance have not been investigated. For this purpose, BMI1 was silenced in 2 PC cell lines (LNCaP and DU 145). Cell proliferation and apoptosis after Docetaxel treatment were measured. Guanine oxidation was assessed by in-cell western. Global gene expression analysis was performed on BMI1 silenced cells. Oncomine database was employed to compare in vitro data with gene expression in PC samples. BMI1 silencing had no effect on cell proliferation, but significantly enhanced Docetaxel-induced antitumor activity. Gene expression analysis demonstrated that BMI1 silencing downregulates a set of antioxidant genes. Docetaxel treatment increased guanine oxidation, while the antioxidant N-acetyl cysteine rescued Docetaxel-induced cell death. Examination of clinical datasets revealed a positive correlation of BMI1 and antioxidant gene expression. BMI1-controlled antioxidant genes were predictive of poor prognosis in PC patients. In conclusion, BMI1 enhances antioxidant response, thereby allowing prostate cancer survival after docetaxel-based chemotherapy. BMI1-controlled antioxidant genes are overexpressed in aggressive prostate cancer, and should be tested as predictors of chemotherapy failure.
Keywords: Docetaxel, Polycomb, Prostate
Introduction
Prostate cancer is not a single disease, but an umbrella under which a plethora of heterogeneous diseases is hidden. These range from indolent localized tumors, to aggressive metastatic diseases 1. Metastatic hormone-refractory prostate cancer (MHRPC) is the most aggressive form, and is generally associated with very poor prognosis. Docetaxel is currently the only effective drug for MHRPC. Docetaxel is a microtubule-targeting drug, that suppresses spindle microtubule dynamics and, thus, blocks mitosis and induces apoptosis 2. It has been shown that Docetaxel disrupts centrosome organization in the late S, G2 and M phases of the cell cycle. This results in incomplete mitosis, accumulation of cells in G2/M phase and cell death. Two large phase III trials showed that Docetaxel treatment significantly improves survival and quality of life in MHRPC patients 3, 4. Unfortunately, the survival advantage conferred by Docetaxel is limited to 2-3 months, and life expectance for MHRPC is only 18-19 months. Indeed, after an initial response to Docetaxel, 80% of patients experience rapid disease progression 5. For this reason, insights into the mechanisms of Docetaxel chemoresistance in PC cells are warranted.
Mechanisms of resistance to Docetaxel are still not clear, although several hypotheses have been proposed. Docetaxel-induced microtubule damage inactivates the antiapoptotic protein BCL2, with subsequent triggering of the apoptotic machinery. For this reason, BCL2 overexpression in prostate cancer has been linked to chemoresistance 2. Keeping with this observation, activation of the anti-apoptotic Akt pathway leads to docetaxel resistance 6. An additional mechanism of Docetaxel resistance could be due to overexpression of efflux drug transporters. Some members of the ATP-binding cassette (ABC) transporter family are expressed by PC cells 7. ABC protein activity is associated with resistance to several chemotherapeutic drugs, including Docetaxel. Recent evidence shows that Docetaxel antitumor activity is not limited to its antimitotic effects. Docetaxel is able to trigger reactive oxygen species (ROS) formation in cancer cells, thus inducing DNA, protein and cell membrane damage 8. ROS seem to play a major role in Docetaxel antitumor activity. Indeed, head and neck carcinoma cells resistant to Docetaxel displayed an abnormal mitochondrial DNA content and an enhanced antioxidant response. Interestingly, blocking the antioxidant response was able to restore Docetaxel sensitivity 9.
A putative mediator of PC chemoresistance is the BMI1 (B-cell-specific Moloney murine leukemia virus integration site 1) oncogene. BMI1 belongs to the family of Polycomb group (PcG) genes. PcG proteins are organized in multimeric complexes, which mediate histone modifications of specific set of genes during cell development 10. BMI1 is a part of the Polycomb repressive complex 1, which triggers histone H2A ubiquitylation and gene silencing. BMI1 is responsible for p16INK4a locus silencing, thus contributing to prostate carcinogenesis 11. Although a mechanistic link has not been established, BMI1 is thought to silence many other oncosuppressors, particularly in PC cells. For example, BMI1 is essential for anchorage-independent growth and metastatic spreading of PC cells 12. This effect is likely mediated by silencing of several cell adhesion genes 13. In PC samples, BMI1 overexpression is associated with high Gleason score and increased risk of recurrence after prostatectomy 14. In addition, BMI1 is overexpressed in a subpopulation of PC cells with tumor-initiating capabilities 15. Microarray data analysis by Glinsky et al. 16 identified a BMI-1-pathway signature with concordant profiles in normal stem cells and prostate cancer metastasis. In the same study, expression of the BMI1 signature was strongly associated with poor survival and therapy failure in 5 different types of epithelial neoplasms, including PC.
Recent studies showed that BMI1 silencing enhanced 5-fluorouracyl antitumor activity in nasopharyngeal carcinoma 17. This effect seems to be dependent on the inactivation of antiapoptotic mechanisms, namely a reduced Akt phosphrylation. In addition, Hedgehog (HH) signaling activation enhanced ABC transporter expression and Docetaxel resistance in PC cells 18. BMI1 is a well known downstream effector of HH signaling 19, 20. Finally, BMI1 silencing strongly impairs antioxidant defense in different cell types 21, 22. Given its prominent role in PC carcinogenesis, progression and prognosis, we sought to investigate the role of BMI1 in PC response to Docetaxel.
Thus, we hypothesized that BMI1 silencing in PC cell could enhance Docetaxel antitumor activity by at least one of three mechanisms: (I) inactivating anti-apoptotic pathways (Akt phosophorylation); (II) downregulating ABC transporter expression, (III); impairing antioxidant defenses. For this purpose, we silenced BMI1 in 2 MHRPC cell lines: LNCaP (derived form and androgen receptor-positive tumor) and DU 145 (derived from and androgen receptor-negative tumor). We investigated putative mechanisms of BMI1-dependent chemoresistance, and we queried Oncomine database to test the clinical relevance our in vitro findings.
Our results show that BMI1 silencing impairs antioxidant defense and sensitizes PC cells to Docetaxel. Examination of clinical datasets confirmed the relationship between BMI1 expression, antioxidant response and PC aggressiveness.
Materials and Methods
Cell culture
The MHRPC cell lines LNCaP and DU 145 were obtained from American Type Culture Collection (Manassas, VA). According to ATCC, LNCaP cells are derived from a lymph node metastasis and DU 145 cells from a brain metastasis. Both cell lines are derived from androgen-independent prostate cancers, although LNCaP still expresses the androgen receptor 23. Cells were maintained in RPMI-1640 medium with 10% fetal bovine serum, glutamine (1%), and penicillin-streptomycin (1%). Docetaxel (Sigma-Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) and diluted in culture medium immediately before use. Final DMSO concentration never exceeded 0.1%. N-acetyl cysteine (NAC) (Sigma) was dissolved in sterile water and α-tocopherol (Sigma) was dissolved in ethanol and diluted in culture medium immediately before use. Final concentration for both NAC and α-tocopherol were 20 mM.
Generation of ShBMI1 LNCaP and DU 145 cells
BMI1-silenced cells were generated using the TRIPZ lentiviral doxycycline inducible Tet-On® shRNA system (Open Biosystems, Huntsville, AL), following the protocols provided by the company. They are referred as DU145ShBMI1 and LNCaPShBMI1 from therein. Non-silencing-TRIPZ lentiviral inducible ShRNAmir expressing cell lines (DU145NS and LNCaPNS) were generated and used as controls in all the experiments. Experiments were performed after at least 3 days of doxycycline (1 μg/ml) induction.
Assay of Cell Viability and Caspase Activity
Number of viable cells and caspase activity were measured though CellTiter-Glo- and CaspaseGlo luminescent assay (Promega, Madison, WL). and caspase Both assays were previously described 24. For cell viability, three kinds of experiments were performed;
To assess cell proliferation after BMI1 silencing, LNCaP and DU 145 cells (NS and ShBMI1) were plated in triplicate in 96-well plates (1000 cells/well). After 1, 3, 5 and 7 d, cell numbers were measured.
To assess cell viability after Docetaxel treatment, LNCaP and DU 145 cells (NS and ShBMI1) were plated in triplicate in 96-well plates (5000 cells/well). The following day, cells were exposed to different concentrations of Docetaxel (1, 10, 100 and 1000 nM) for 1h. After treatment, cells were allowed to grow in drug-free medium for an additional three days, as previously described 25. Cell viability was measured at the end of the three days. Docetaxel concentrations employed in this study are clinically achievable 26. The 1-h treatment has been chosen in analogy with the 1-h clinical infusion of docetaxel 26.
To asses the effect of ROS on Docetaxel antitumor activity, cells were pretreated with NAC (20 mM, as described 24), then exposed to the same Docetaxel concentration for 1h, and grown in NAC-containing medium for 3 d. For b) and c) the fraction of proliferating cells and IC50 values relative to untreated cultures were calculated by nonlinear leastsquares curve-fitting.
Western Blot
Total protein was isolated from LNCaP and DU 145 (NS and ShBMI1) cells using RIPA lysis buffer (Thermo Scientific, Waltham, MA USA) and quantified using the BCA protein assay kit (Pierce) kit. Thirty μg of protein extract was loaded per lane into a 4% to 20% Tris-glycine gel (Invitrogen, Carlsbad, CA). For the experiment testing the effects of docetaxel and antioxidants on caspase and PARP cleavage, the experiment was set up identical to the experiment testing cell viability. Proteins were transferred to a polyvinylidene fluoride membrane, blocked in 10% nonfat dry milk, 0.1% Tween-20 PBS, incubated with primary (anti-BMI1; Millipore, 1/1000 dilution; anti actin, Abcam 1/5000 dilution; anti-cleaved Caspase 3 (1:1000), anti-cleaved Caspase 8 (1:1000), anti-cleaved Caspase 9 (1:1000), anti-PARP (1:1000), Akt (1:2000), and phospho-Akt (1:1000) Cell Signaling Technology) and secondary (LI-COR Biosciences, Lincoln Nebraska USA) antibodies, and scanned by the LI-COR Odyssey IR Imaging System.
Gene Expression Analysis
Total RNA was extracted from NS and ShBMI1 (LNCaP and DU 145) cells using the TRIZOL REAGENT (Invitrogen). RNA was retrotranscribed through SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, p/n 18080-05) using deoxynucleotide triphosphates and following manufacturer's instructions.
Quantitative-RT-PCR (qRT-PCR) was performed through TaqMan gene expression assays (Applied Biosystems, Foster City, CA) and a StepOne Real time PCR machine (Applied Biosystems). Assay numbers were Hs01067802_m1, Hs00219905_m1, Hs01055362_m1, Hs00180411_m1, Hs00830226_gH and Hs99999905_m1 for ABCB1, ABCC1, ABCG2, BMI1, FTL HSP40C1 and GAPDH, respectively. Differential gene expression was calculated by the comparative Ct method.
In order to test RNA integrity for microarray analysis, we employed Agilent RNA 6000 Nano LabChip Kit (Agilent, p/n 5067-1511). All analyzed samples had RIN >9. Ten μg of total RNA from LNCaP and DU 145 (NS and ShBMI) cells were labeled by reverse transcription with Superscript II (Invitrogen) and oligo-dT in the presence of Cy3-dUTP for Universal RNA reference control (Universal Human Reference RNA Stratagene p/n 740000) or Cy5-dUTP for the samples. Whole Human Genome Oligo Microarray 4×44K format gene expression arrays (Agilent, p/n G4112F) were employed for gene expression studies. The hybridization and washes were performed using Agilent reagents and following related protocols, and the slides were scanned on a GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA, USA). Differentially expressed genes were identified by ANOVA analysis of the replicates using Partek Genomic Suite Package (Partek). Data were further analyzed through the use of Ingenuity Pathways Analysis, with statistical methods and threshold described by the manufacturer (Ingenuity® Systems, www.ingenuity.com). Unless otherwise indicated, genes that are up- or down-regulated more than 2-fold and genes modulated with a p<0.01 are referred as “significantly modulated genes”.
Quantitative Immunofluorescence
In order to measure ROS effects after Docetaxel treatment, LNCaP and DU 145 (NS and ShBMI1) cells were plated on 96-well plates. For each cell line, cells were treated with Docetaxel at IC50 concentration of NS cells, as described in “Assay of Cell Proliferation”. After treatment, cells were fixed in methanol for 15 min, washed in a solution of PBS and 1% fish oil gelatin (PBSG) and incubated in the same solution for 60 min to block. After blocking, cells were incubated overnight with primary antibodies (mouse anti-oxoguanine, Millipore, 1/200 dilution; rabbit anti-actin, Abcam, 1/500 dilution) in PBSG. After washing with PBSG + 0.1 % Tween-20, cells were incubated with IRDye 800CW labeled donkey anti-mouse and IRDye 680CW labeled donkey anti-rabbit antibody (LI-COR Biotechnology, 1/200 dilution) for 2 h. After washing with PBSG + 0.1 % Tween 20, cells were scanned with the LI-COR Odyssey IR Imaging System.
Cell Cycle Analysis
Cells were seeded at 50% confluence to ensure logarithmic growth and treated with Docetaxel and anitoxidants in the same manner as dexcribed for cell viability assays. Following treatment, one million cells were fixed in ice cold 70% ethanol overnight. Following fixation, cells were centrifuged and resuspended in PBS containing 40 μg/mL propidium iodide and 100 μg/mL RNAse A and incubated at 37°C for 1 hour.
Meta-analysis for correlation of in vitro data with clinical data
In vitro data on BMI1 silencing were compared with prostate cancer expression profiles derived from Oncomine 4.2 database analysis tool (http://www.oncomine.org). Oncomine database was also employed to investigate the correlation between antioxidant genes and prostate cancer prognosis. For this purpose, we identified 22 PC studies on Oncomine database.. We selected genes positively correlated to BMI1 based on a correlation coefficient higher than 0.5. In order to avoid a duplicated analysis on same samples, we identified studies with the same first author and analyzed the one with the highest number of BMI1-correlated genes (based on correlation coefficient). All statistical values relative to this meta-analysis were calculated as described by Chinnaiyan and colleagues 27.
Statistical Analysis
Unless otherwise specified, all experiments were done in triplicate and were repeated at least twice. Data were expressed as mean values SE or SD and were analyzed by Student's t test, ANOVA followed by Bonferroni's multiple comparison though GraphPad Prism software. The level of significance was set at P < 0.05.
Results
BMI1 silencing in PC cells
To test the effectiveness of our silencing system, we measured BMI1 mRNA and protein expression in NS and ShBMI1 cells. Both LNCaP and DU 145 ShBMI1 cells showed a more than 3-fold reduction in BMI1 mRNA levels (Fig. 1 A), and a nearly complete BMI1 protein silencing (Fig. 1 B).
Figure 1. BMI1 RNA (A) and protein (B) silencing in PC cells.
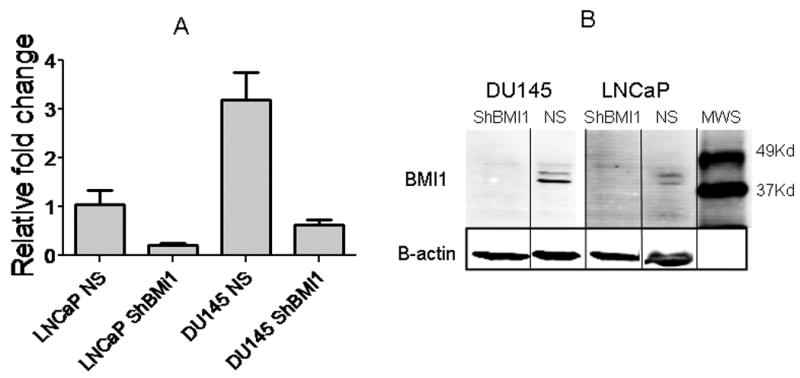
MWS, molecular weight scale.
Cell viability and caspase activity after BMI1 silencing
We first tested the hypothesis that BMI1 silencing affects PC cell proliferation. Cell growth was not significantly affected in BMI1-silenced cells (data not shown). Given these results, we investigated the effect of BMI1 silencing on Docetaxel antitumor activity. As expected, treatment with Docetaxel induced a dose-dependent growth inhibition, with IC50 concentrations of 6.53 ± 1.59 and 22.60 ± 2.64 nM (LNCaP NS and DU 145 NS, respectively). BMI1 silencing caused a significant IC50 reduction in both cell lines (2.01 ± 0.76 and 9.96 ± 1.02 nM, in LNCaP and DU 145 respectively, Fig. 2A). These results were consistent with cell cycle and apoptosis measurements. Docetaxel-treated NS and ShBMI1 LNCaP cells showed a similar cell cycle distribution (Fig. 2B), but BMI1 silencing increased both the number of apoptotic cells, as measured by flow cytometry (from 41 to 51%), and caspase 3/7 activity (Fig. 2D). To the contrary, BMI1 silencing in DU145 cells did not enhance Docetaxel-induced apoptosis (data not shown), but increased the percentage of cells arrested in G2/M phase (from 12 to 23%, Fig. 2C).
Figure 2. Effects of BMI1 silencing on Docetaxel activity.
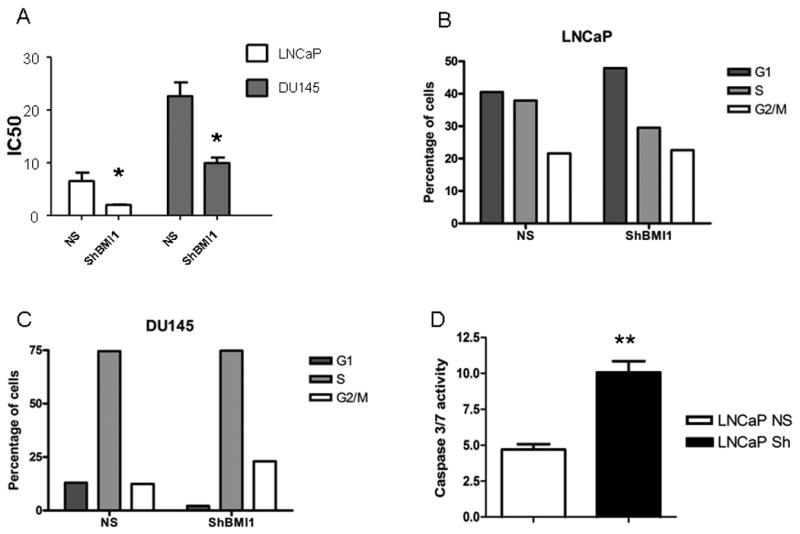
A: IC50 of cells treated at different Docetaxel concentrations * p<0.05, T test. B, C: cell cycle distribution of Docetaxel-treated cells. D: Caspase 3/7 activity in LNCaP cells treated with Docetaxel at IC50 concentration for NS cells ** p<0.01, T test.
Gene expression analysis
Since one possible explanation for BMI1-dependent chemoresistance may be drug transporter modulation, we investigated if BMI1 silencing downregulated three ABC transporters involved in Docetaxel chemoresistance 28 and expressed by PC cells 7, 29. As shown in Suppl. Fig. 1 A, LNCaP cells expressed much lower ABC transporter mRNA, compared to DU 145 cells. Interestingly, LNCaP are more sensitive to Docetaxel and express lower BMI1 than DU 145 cells (Fig 1 and 2). However, BMI1 silencing did not significantly modulate ABC transporter expression. Since Docetaxel resistance has also been linked to induction the Akt pathway, we also looked at Akt phosphorylation following BMI1 silencing. Similarly, we found that BMI1 silencing did not significantly reduce Akt phosphorylation in both cell lines (Suppl. Fig. 1 B) In order to dissect the molecular pathways involved in BMI1-dependent chemosensitivity, we compared gene expression profiles of ShBMI1 and NS cells. BMI1 silencing produced significant modulation of 564 (LNCaP) and 880 (DU 145) genes (Suppl. Tab 1 and 2). Our array data show that BMI1 silencing upregulated 280/564 (49.6%) and 492/879 (55.9%) genes in the two cell lines. In order to gain functional insights into the consequences of BMI1 silencing, we analyzed gene categories modulated by BMI1 (Suppl. Tab. 3), using Ingenuity Pathway Analysis (IPA) software. Interestingly, we found that most of the genes modulated in both cell lines were significantly correlated to Cancer (9.3e-15<p<3.9e-2). In particular, 150 and 141 genes in LNCaP and DU 145 cells respectively were correlated to tumorigenesis. We also found that “cell death” and “cell growth and proliferation” were significantly affected (Suppl. Tab. 3). Since Docetaxel sensitivity may be mediated by modulation of pro- and antiapoptotic pathways, we further assessed function and classification of these genes. In both LNCaP and DU 145 we were unable to identify a significant modulation of pro- or anti- apoptotic pathways after BMI1 silencing. These data are consistent with the similar Akt phosphorylation level in NS and Sh BMI1 cells. Since analysis of classical death pathways was not able to explain our data on Docetaxel activity, we investigated TOX pathways modulated after BMI1 silencing. This IPA analysis determines genes expression changes in response to several kinds of cell stress, including DNA damage, hypoxia and xenobiotics. Interestingly, BMI1 silencing was able to modulate ROS-related genes in both cell lines. In particular, “Oxidative Stress Response” genes ranked first among significantly affected TOX categories (Suppl. Tab 4). Keeping with our cell cycle analysis, G2/M transition genes were also significantly modulated. More interestingly, we found that BMI1 silencing was able to downregulate an antioxidant gene set in each of the cell lines (Fig. 3 A). Among these, we found classical antioxidant genes (gamma glutamyl transferase), different heat shock protein (HSP) 40 isoforms and ferritin light chain (FLT). Notably, BMI1 silencing never led to an upregulation of antioxidant genes. In DU 145 cells, this effect was coupled to upregulation of genes involved in ROS production (MAOA) 30 and ROS-dependent apoptosis (caspase 8) 31. Interestingly, BMI1 silencing also upregulated KEAP1, which is known to inhibit antioxidant response. In additon, we confirmed a significant downregulation of FTL and HSP40 genes by qRT-PCR.
Figure 3. Role of ROS in Docetaxel antitumor activity.
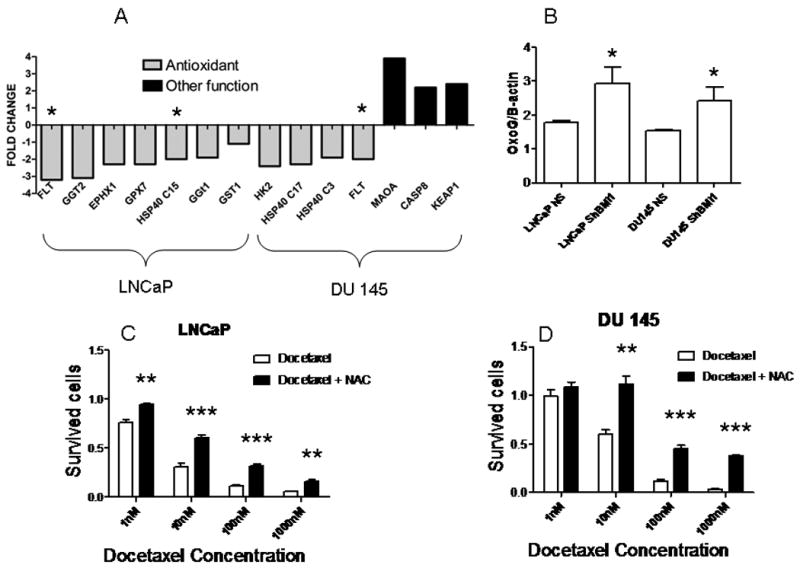
A: Oxidative stress response” genes expression profile after BMI1 silencing; *confirmed by qPCR. FTL, ferritin light chain; GGT, gamma glutamyl trasnferase; EPHX, epoxide hydrolase; GPX, glutathione peroxidase; HSP, heat shock protein; GST, gluthatione S-transferase; HK, hexokinase; MAOA, monoamine oxidase A; CASP8, caspase 8; KEAP1, Kelch-like ECH associated protein 1. B : oxoguanine levels (Docetaxel treated cells/untreated cells); *p<0.05, T test (NS vs. ShBMI1). C and D: cell survival after NAC-Docetaxel or Docetaxel only treatment in ShBMI1 cells; **p<0.01, ***p,0.001, T test (NS vs. ShBMI1)
Role of ROS in Docetaxel activity
Since our array data strongly suggested that BMI1 silencing disrupts antioxidant response in PC cells, we analyzed ROS production after Docetaxel treatment. For this purpose, we measured oxoguanine levels in NS and ShBMI1 cells. Oxoguanine is the main product of DNA oxidative damage, and is widely used to measure oxidative stress 32. As expected, Docetaxel treatment induced a 1.5- to 2-fold increase in oxoguanine levels (Fig 3 B). More importantly, BMI1 silenced cells showed a significantly higher oxoguanine concentration in both cell lines (Fig. 3 B). Finally, to show that ROS play a crucial role in Docetaxel antitumor activity, we treated BMI1-silenced cells with the antioxidant NAC. As shown in Figure 3 (C, D), NAC treatment greatly reduced Docetaxel-dependent cell death. These results show that BMI1 silencing impairs PC antioxidant response, thus enhancing Docetaxel antitumor activity.
To further corroborate our data, we measured PARP and caspase cleavage in Docetaxel treated and untreated cells. In addition, we evaluated the effect of 2 antioxidant molecules (NAC and α-tocopherol) on Docetaxel treated cells. Consistent with our previous results, LNCaP cells showed greater PARP and caspases 9 and 7 cleavage after BMI1 silencing (Figure 4). In DU 145 cells we did not detect caspase 9 cleavage but instead detected caspase 8 cleavage. However, there was no change in either caspase or PARP cleavage in the BMI1-silenced cells as compared to the NS DU 145 cells., Importantly, in both LNCaP and DU 145 cells both antioxidants were able to rescue docetaxel-treated cells form apoptosis.
Figure 4. PARP and caspase cleavage in Docetaxel-treated cells.
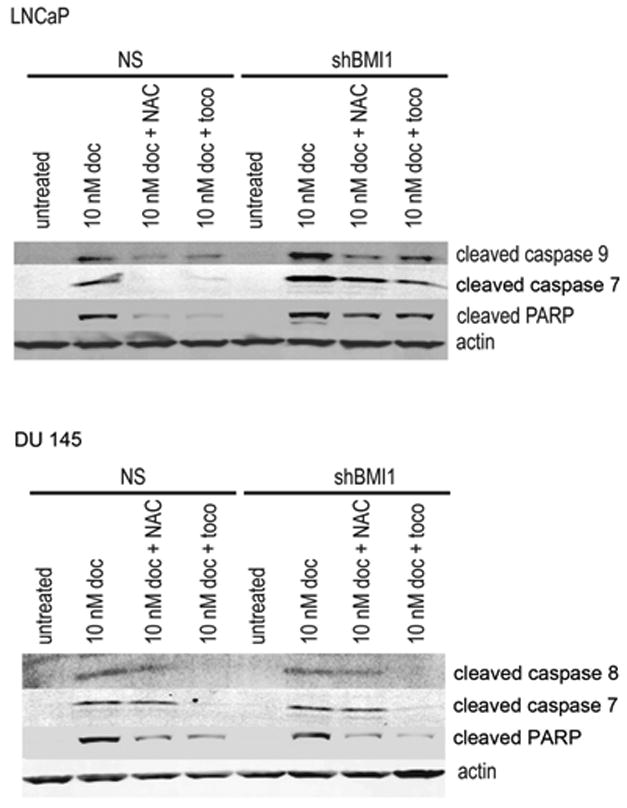
NAC, N-acetyl cysteine, toco, α-Tocopherol.
Gene expression analysis in clinical samples
Finally, we investigated the possible clinical significance of our in vitro findings. First, we tested the hypothesis that BMI1 expression is associated with ROS-related genes in PC samples. We used Oncomine database to investigate global gene expression data from 22 independent studies involving 20 to 596 prostate cancer samples of different stage and grade . We identified 1207 genes positively correlated to BMI1. . We then analyzed, through IPA, the TOX pathways associated to BMI1 in clinical samples. As shown in Tab. 1, “Oxidative Stress Response” genes were significantly correlated with BMI1 expression. Among these genes, we found 2 isoforms of HSP40, EPHX1 and GST. When we compared oxidative stress related genes modulated by BMi1 in PC cell lines (Suppl. Tab 5) and in clinical samples (Suppl. Tab. 6) we found a very significant overlap (odds ratio: 488.2, p=8.1e-8, Oncomine analysis). Moreover, in clinical samples, BMI1 expression was significantly correlated with “Hypoxia-Inducible Factor- Signalling”, which is also involved in ROS metabolism.
Tab 1. Top TOX pathways in PC samples.
Significantly modulated genes after BMI1 silencing were analyzed by IPA software. HIF, hypoxia inducible factor; RAR, retinoic acid receptor.
| Prostate cancer samples | ||
|---|---|---|
| CATEGORY | N.GENES | P VALUE |
| Oxidative Stress Response | 17 | 1.8E-2 |
| RAR Activation | 14 | 2.6E-2 |
| HIF Signaling | 11 | 4.2E-4 |
| TGF-beta Signaling | 9 | 1.0E-2 |
| G1/S transition | 8 | 2.0E-3 |
Both in vitro and clinical data indicated that BMI1 controls a set of antioxidant genes in PC cells. Our IPA analysis generated a BMI1-controlled antioxidant signature (BAS) composed of 19 genes (Suppl. Tab.5). Given the prominent role of BMI1 in PC pathogenesis and progression, we investigated if this antioxidant signature had a prognostic significance. Using the Oncomine database, we found 3 studies in which the BAS was significantly correlated with PC clinical features (Tab. 2). In these studies, the signature was highly predictive of metastasis, shorter recurrence after prostatectomy and shorter survival. Interestingly, Oncomine data showed that high BMI1 expression is predictive of shorter relapse-free survival in PC (Suppl. Fig. 2). These results show that BMI1 controls a set of antioxidant genes in PC, and that these genes are activated in poor prognosis PC.
Tab 2. Prognostic role of BAS genes in PC samples.
We generated a new oncomine concept, including 19 genes regulated by BMI1 in PC cells. This antioxidant gene set was confronted with all PC studies present in the database, to predict significant associations with clinical features. P values and Odds Ratios are calculated based on Oncomine algorithms, and represent the association between antioxidant gene expression and patient prognosis (we set significance threshold at P<0.01).
| Prediction | P Value | Odds Ratio | Patients |
|---|---|---|---|
| Metastasis (PC) | 0.004 | 7.7 | 112 |
| Dead at 5 years (PC) | 0.004 | 7.7 | 112 |
| Recurrence at 5 years (PC) | 0.006 | 7.0 | 54 |
Discussion
In this paper, we showed that BMI1 silencing enhances Docetaxel antitumor activity in PC, and that this effect is mediated by ROS production. As a part of Polycomb repressive complex 1, BMI1 can silence several genes in different cellular contexts. In PC, BMI1 is crucial for tumor progression and metastatic spreading 11, 12. In addition, BMI1 expression is higher in a small fraction of PC cells with tumor initiating abilities, which may be the seeds of chemoresistance, metastasis and recurrence 15. Clinical data strongly suggest that BMI1 expression is a poor prognostic indicator in PC patients 14. Despite all these indications, very little is known about the pathways controlled by BMI1 in PC. In addition, it has been shown that BMI1 enhances chemo- and radio-resistance in nasopharyngeal carcinoma cells 17, 33. For this reason, we investigated the relationship between BMI1 expression and PC sensitivity to Docetaxel. We also compared our in vitro findings to clinical data, in an attempt to evaluate its relevance for the development of new strategies to detect and overcome PC chemoresistance.
BMI1 silencing significantly increased Docetaxel-dependent growth arrest (Fig. 2 A). In the p53 positive LNCaP cells, this effect was due to an increased apoptotic activity. In the p53-mutated DU145 cells, BMI1 silencing enhanced G2/M phase arrest induced by Docetaxel. Interestingly, in both cell lines antioxidant drugs like NAC or tocopherol counteracted the effects of BMI1 silencing (Fig. 3 C, D).
The chemosensitivity acquired by both cell lines after BMI1 silencing was not mediated by direct apoptotic pathway modulation, as shown for nasopharyngeal carcinoma cells 33, nor by ABC transporter downregulation (Suppl. Fig. 1). Our results suggest that BMI1 antioxidant role is crucial in PC cells. Previous studies have shown that BMI1 plays a key role in maintaining the oxidative stasis of the cell 21, 22. In these models, BMI1 function has been linked to p16 and p53 suppression, with no association to antioxidant genes. In this paper, we showed that BMI1 silencing significantly increased Docetaxel-dependent oxidative stress (Fig. 3 B), and that the antioxidants NAC and α-tocopherol can completely abrogate this effect (Fig. 3 C, D). Gene expression analysis showed BMI1 silencing downregulated antioxidant genes, and upregulated pro-oxidant genes (Fig. 3 A). These results were supported by clinical samples data, showing a significant association between genes classified by IPA as playing a role in “Oxidative Stress Response” and BMI1. This set comprises genes that protect cells from oxidative stress, including epoxide hydrolase, superoxide dismutase and gamma glutamyl transferase. Interestingly, terminal caspase activity was increased after BMI1 silencing Although there is a diversity of oxidative stress-related genes regulated in these samples, HSP 40 genes are common to all datasets. HSP 40 is a family of co-chaperones comprising 41 members. Recent evidence suggests that some HSP 40 isoforms are involved in various aspects of cancer biology, and that their expression is deregulated in many neoplasms 34. In addition, HSP 40 members contribute to inhibition of apoptosis, protection from oxidative stress, and maintenance of the mitochondrial membrane potential 35. In view of our results, it would be interesting to further explore the relationship between HSP 40, BMI1 and PC progression.
The putative relationship between BMI1 antioxidant genes and PC aggressiveness is further supported by clinical data; showing a correlation between BMI1-dependent antioxidant signature (BAS) and PC aggressiveness. In particular, 3 independent studies show that BAS is predictive of metastasis, recurrence after prostatectomy and shorter survival. Interestingly, BMI1 expression is an independent predictor of shorter disease-free survival after prostatectomy 14. This relationship is confirmed by Oncomine data (Suppl Fig. 2). A recent meta-analysis on PC patients found that a shorter time of PC progression is predictive of poor response to Docetaxel 36. Our findings suggests that a subset of PC is more resistant to Docetaxel because of higher BMI1 expression and more effective antioxidant response. These tumors are also characterized by faster progression. Our in vitro results call for clinical studies on BMI1 expression in docetaxel-treated PC patients. According to our data, BMI1 should be overexpressed in chemoresistant tumors. Unfortunately, docetaxel is currently employed to treat metastatic disease, which is always unresectable1. Thus, clinical specimens from docetaxel-treated PC patients are hard to get.
These results could pave the way to new approaches to treat and detect chemoresistant PC. In particular, BMI1 expression, as well as the expression of BAS could prove to be valid predictive indicators of chemosensitivity. These genes could be employed to personalize Docetaxel regimens in MHRPC patients. Recently, some drug targeting histone modifications showed an interesting anticancer effect, and several new compounds are being tested 37. In the future, it is conceivable that specific inhibitors of Polycomb repressive complex 1 will be developed. Our results raise the possibility that Docetaxel and Polycomb-targeting drugs could be synergistic in PC cells, and that a combined regimen could improve Docetaxel activity and prevent tumor recurrence.
Supplementary Material
ABC transporter expression was measured by qRT-PCR.
Data are derived from Oncomine database.
The list represents significantly modulated genes after BMI1 silencing, as described in materials and methods.
The list represents significantly modulated genes after BMI1 silencing, as described in materials and methods.
Data were generated by IPA software.
RAR, retinoic acid receptor.
Data were generated by IPA analysis on microarray data obtained from BMI1-silenced LNCaP and DU145 cells.
Data were generated by IPA analysis on Oncomine-derived data.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Disclaimer: This publication has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract No. N01-CO-12400. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government
References
- 1.Bracarda S, de Cobelli O, Greco C, Prayer-Galetti T, Valdagni R, Gatta G, de Braud F, Bartsch G. Cancer of the prostate. Crit Rev Oncol Hematol. 2005;56:379–96. doi: 10.1016/j.critrevonc.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Mackler NJ, Pienta KJ. Drug insight: Use of docetaxel in prostate and urothelial cancers. Nature clinical practice. 2005;2:92–100. doi: 10.1038/ncpuro0099. quiz 1 p following 12. [DOI] [PubMed] [Google Scholar]
- 3.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 4.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 5.Sella A, Sternberg C, Kovel S, Yarom N, Skoneczna I. Progression after docetaxel-based chemotherapy in androgen-independent prostate cancer. BJU Int. 2007;100:533–5. doi: 10.1111/j.1464-410X.2007.07037.x. [DOI] [PubMed] [Google Scholar]
- 6.Xing H, Weng D, Chen G, Tao W, Zhu T, Yang X, Meng L, Wang S, Lu Y, Ma D. Activation of fibronectin/PI-3K/Akt2 leads to chemoresistance to docetaxel by regulating survivin protein expression in ovarian and breast cancer cells. Cancer Lett. 2008;261:108–19. doi: 10.1016/j.canlet.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 7.van Brussel JP, van Steenbrugge GJ, Romijn JC, Schroder FH, Mickisch GH. Chemosensitivity of prostate cancer cell lines and expression of multidrug resistance-related proteins. Eur J Cancer. 1999;35:664–71. doi: 10.1016/s0959-8049(98)00435-3. [DOI] [PubMed] [Google Scholar]
- 8.Rabi T, Bishayee A. d -Limonene sensitizes docetaxel-induced cytotoxicity in human prostate cancer cells: Generation of reactive oxygen species and induction of apoptosis. J Carcinog. 2009;8:9. doi: 10.4103/1477-3163.51368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizumachi T, Suzuki S, Naito A, Carcel-Trullols J, Evans TT, Spring PM, Oridate N, Furuta Y, Fukuda S, Higuchi M. Increased mitochondrial DNA induces acquired docetaxel resistance in head and neck cancer cells. Oncogene. 2008;27:831–8. doi: 10.1038/sj.onc.1210681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–18. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Fan C, He L, Kapoor A, Gillis A, Rybak AP, Cutz JC, Tang D. Bmi1 promotes prostate tumorigenesis via inhibiting p16(INK4A) and p14(ARF) expression. Biochim Biophys Acta. 2008;1782:642–8. doi: 10.1016/j.bbadis.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Berezovska OP, Glinskii AB, Yang Z, Li XM, Hoffman RM, Glinsky GV. Essential role for activation of the Polycomb group (PcG) protein chromatin silencing pathway in metastatic prostate cancer. Cell Cycle. 2006;5:1886–901. doi: 10.4161/cc.5.16.3222. [DOI] [PubMed] [Google Scholar]
- 13.Douglas D, Hsu JH, Hung L, Cooper A, Abdueva D, van Doorninck J, Peng G, Shimada H, Triche TJ, Lawlor ER. BMI-1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008;68:6507–15. doi: 10.1158/0008-5472.CAN-07-6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Leenders GJ, Dukers D, Hessels D, van den Kieboom SW, Hulsbergen CA, Witjes JA, Otte AP, Meijer CJ, Raaphorst FM. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52:455–63. doi: 10.1016/j.eururo.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Hurt EM, Farrar WL. Cancer stem cells: the seeds of metastasis? Mol Interv. 2008;8:140–2. doi: 10.1124/mi.8.3.7. [DOI] [PubMed] [Google Scholar]
- 16.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. The Journal of clinical investigation. 2005;115:1503–21. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin L, Zhang X, Zhang L, Feng Y, Weng GX, Li MZ, Kong QL, Qian CN, Zeng YX, Zeng MS, Liao DF, Song LB. Downregulation of BMI-1 enhances 5-fluorouracil-induced apoptosis in nasopharyngeal carcinoma cells. Biochemical and biophysical research communications. 2008;371:531–5. doi: 10.1016/j.bbrc.2008.04.117. [DOI] [PubMed] [Google Scholar]
- 18.Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–9. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 19.Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–41. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizo A, Olthof S, Han L, Vellenga E, de Haan G, Schuringa JJ. Repression of BMI1 in normal and leukemic human CD34(+) cells impairs self-renewal and induces apoptosis. Blood. 2009;114:1498–505. doi: 10.1182/blood-2009-03-209734. [DOI] [PubMed] [Google Scholar]
- 22.Chatoo W, Abdouh M, David J, Champagne MP, Ferreira J, Rodier F, Bernier G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–42. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. The Prostate. 2001;47:66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- 24.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fabbri F, Brigliadori G, Carloni S, Ulivi P, Vannini I, Tesei A, Silvestrini R, Amadori D, Zoli W. Zoledronic acid increases docetaxel cytotoxicity through pMEK and Mcl-1 inhibition in a hormone-sensitive prostate carcinoma cell line. Journal of translational medicine. 2008;6:43. doi: 10.1186/1479-5876-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker SD, Zhao M, Lee CK, Verweij J, Zabelina Y, Brahmer JR, Wolff AC, Sparreboom A, Carducci MA. Comparative pharmacokinetics of weekly and every-three-weeks docetaxel. Clin Cancer Res. 2004;10:1976–83. doi: 10.1158/1078-0432.ccr-0842-03. [DOI] [PubMed] [Google Scholar]
- 27.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York, NY. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat. 2003;6:71–84. doi: 10.1016/s1368-7646(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 29.Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, Bates SE. ABCG2: a perspective. Advanced drug delivery reviews. 2009;61:3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzio EA, Soliman KF. Glioma cell antioxidant capacity relative to reactive oxygen species produced by dopamine. J Appl Toxicol. 2004;24:99–106. doi: 10.1002/jat.954. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez A, Grochola LF, Bond EE, Levine AJ, Taubert H, Muller TH, Wurl P, Bond GL. Chemosensitivity profiles identify polymorphisms in the p53 network genes 14-3-3tau and CD44 that affect sarcoma incidence and survival. Cancer Res. 70:172–80. doi: 10.1158/0008-5472.CAN-09-2218. [DOI] [PubMed] [Google Scholar]
- 32.Fortini P, Pascucci B, Parlanti E, D'Errico M, Simonelli V, Dogliotti E. 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutation research. 2003;531:127–39. doi: 10.1016/j.mrfmmm.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Alajez NM, Shi W, Hui AB, Yue S, Ng R, Lo KW, Bastianutto C, O'Sullivan B, Gullane P, Liu FF. Targeted depletion of BMI1 sensitizes tumor cells to P53-mediated apoptosis in response to radiation therapy. Cell Death Differ. 2009;16:1469–79. doi: 10.1038/cdd.2009.85. [DOI] [PubMed] [Google Scholar]
- 34.Mitra A, Shevde LA, Samant RS. Multi-faceted role of HSP40 in cancer. Clinical & experimental metastasis. 2009;26:559–67. doi: 10.1007/s10585-009-9255-x. [DOI] [PubMed] [Google Scholar]
- 35.Fan GH, Qi C, Chen SD. Heat shock proteins reduce toxicity of 1-methyl-4-phenylpyridinium ion in SK-N-SH cells. Journal of neuroscience research. 2005;82:551–62. doi: 10.1002/jnr.20656. [DOI] [PubMed] [Google Scholar]
- 36.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 37.Crea F, Danesi R, Farrar WL. Epigenetics and cancer stem cell chemoresistance. Epigenomics. 2009;1(1):63–79. doi: 10.2217/epi.09.4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ABC transporter expression was measured by qRT-PCR.
Data are derived from Oncomine database.
The list represents significantly modulated genes after BMI1 silencing, as described in materials and methods.
The list represents significantly modulated genes after BMI1 silencing, as described in materials and methods.
Data were generated by IPA software.
RAR, retinoic acid receptor.
Data were generated by IPA analysis on microarray data obtained from BMI1-silenced LNCaP and DU145 cells.
Data were generated by IPA analysis on Oncomine-derived data.