

Abstract
The Group IVA cytosolic phospholipase A2 (GIVA cPLA2) plays a central role in inflammation. Long chain 2-oxoamides constitute a class of potent GIVA cPLA2 inhibitors that exhibit potent in vivo anti-inflammatory and analgesic activity. We have now gained insight into the binding of 2-oxoamide inhibitors in the GIVA cPLA2 active site through a combination of molecular docking calculations and molecular dynamics simulations. Recently, the location of the 2-oxoamide inhibitor AX007 within the active site of the GIVA cPLA2 was determined using a combination of deuterium exchange mass spectrometry followed by molecular dynamics simulations. After the optimization of the AX007-GIVA cPLA2 complex using the docking algorithm Surflex-Dock, a series of additional 2-oxoamide inhibitors have been docked in the enzyme active site. The calculated binding affinity presents a good statistical correlation with the experimental inhibitory activity (r2 = 0.76, N = 11). A molecular dynamics simulation of the docking complex of the most active compound has revealed persistent interactions of the inhibitor with the enzyme active site and proves the stability of the docking complex and the validity of the binding suggested by the docking calculations. The combination of molecular docking calculations and molecular dynamics simulations is useful in defining the binding of small-molecule inhibitors and provides a valuable tool for the design of new compounds with improved inhibitory activity against GIVA cPLA2.
Introduction
Phospholipase A2 (PLA2) enzymes are characterized by their ability to catalyze the hydrolysis of the ester bond at the sn-2 position of membrane phospholipids.1, 2 The products of this enzymatic activity are free fatty acids and lysophospholipids, which both play important roles as lipid second messengers. Among the various groups of PLA2s, the Group IVA cytosolic PLA2 (GIVA cPLA2) catalyzes the preferential release of arachidonic acid from the sn-2 position of intracellular membrane phospholipids.3 Arachidonic acid is subsequently metabolized by downstream enzymes, such as cyclooxygenase or lipoxygenase to form eicosanoids, while lysophospholipids may be either converted to lysophosphatidic acid or acetylated to produce platelet activating factor. Results from experiments performed using knockout mice have demonstrated a critical role for the GIVA cPLA2 in many inflammatory diseases.4, 5 In addition, the elucidation of GIVA cPLA2 function, using synthetic inhibitors or antisense techniques that reduce the enzyme activity in vitro, ex vivo, and in vivo has revealed confirmatory findings about the role of the enzyme in pathophysiology.2, 6 Thus, GIVA cPLA2 is an attractive target for the development of new anti-inflammatory agents.
The human GIVA cPLA2 enzyme was purified in 1991 from the cytosol of mammalian macrophages and was cloned.7, 8 Its structure was discovered to be composed of a C2 domain, which is responsible for the calcium-dependent membrane translocation, and an α/β hydrolase domain containing the active site. It was discovered through site-directed mutagenesis that GIVA cPLA2 utilizes an unusual catalytic dyad Ser228/Asp549,9 and this was later confirmed by X-ray crystallography of the enzyme.10 The Asp549 residue activates Ser228 by abstracting a proton form the hydroxyl group during its nucleophilic attack at the sn-2 ester bond to form an acyl-enzyme intermediate on the serine.9–12 The backbone amide groups of residues Gly197/Gly198 form the “oxyanion hole”, which is located close to the catalytic Ser228 and stabilizes the transition state of the tetrahedral intermediate through hydrogen bonding. In the same loop with the “oxyanion hole” Arg200 is located, which contributes to the stabilization of the tetrahedral intermediate interacting with the phosphate moiety of the substrate.10 The crystal structure of GIVA cPLA2 reveals an amphipathic “lid region”, which consists of residues 413–457, and blocks access of the phospholipid substrate into the active site.10 Residues 408–412 which lead into the “lid region” and residues 433–457, are highly flexible regions and may act as hinges that allow the “lid region” to open. Given this flexibility, the lid is in the open form in the presence of substrate (lipid vesicles, which are the natural substrate of the enzyme) or when an inhibitor is bound in the active site.13
Since GIVA cPLA2 is associated with various inflammatory diseases, the development of selective inhibitors of the enzyme has attracted a lot of interest.14, 15 Potent and selective GIVA cPLA2 inhibitors include indole derivatives developed by Wyeth,16–19 pyrrolidine-based inhibitors by Shionogi Pharmaceuticals20–22 and substituted propan-2-ones by Astra Zeneca and the Lehr group.23–26 The 2-oxoamides are another important class of GIVA cPLA2 inhibitors, which were developed by the Kokotos and Dennis groups.27–32 It has been shown that 2-oxoamides based on γ- or δ-amino acids containing a free carboxyl group are selective inhibitors of GIVA cPLA2 not affecting the other major intracellular Ca2+-independent GVIA iPLA2, and show potent in vivo activity.27 The corresponding esters inhibit both GIVA cPLA2 and GVIA iPLA2.28, 29
The molecular modelling studies reported to date for GIVA cPLA2 are very limited contrary to those for secreted sPLA2 enzymes, which have been studied extensively using molecular modelling techniques.33–37 Two inhibitors docked in the enzyme active site have been reported, but the docking complexes have not given insight into the binding interactions between the inhibitor and the active site of the enzyme.19, 38 Recently, the location of two inhibitors bound in the GIVA cPLA2 active site has been determined using a combination of Molecular Dynamics (MD) simulations and Deuterium Exchange Mass Spectrometry (DXMS).39 The two inhibitors are the pyrrolidine-derived inhibitor pyrrophenone and the 2-oxoamide inhibitor AX007.
Using rational drug design approaches to develop new 2-oxoamide inhibitors with improved activity against GIVA cPLA2 has been a challenge. In the present study, molecular docking calculations were performed in an effort to better understand the binding mode of 2-oxoamide inhibitors in the GIVA cPLA2 active site. For the docking calculations, the previously reported39 complex of GIVA cPLA2 with the 2-oxoamide inhibitor AX007, resulted from the MD simulation, was used. The aforementioned GIVA cPLA2-AX007 complex has been optimized using the docking algorithm Surflex-Dock. Then, a series of 2-oxoamide inhibitors was docked in the enzyme active site and the calculated binding affinity was correlated with the experimental in vitro inhibitory activity. The aim was to reveal the contribution of the pharmacophore segments of each ligand to the binding. The docking complex of the most active compound was subjected to molecular dynamics simulations using the MacroModel 9.740 to identify persistent interactions of the inhibitor with the enzyme active site. The resultant understanding of the mechanism of action of the 2-oxoamide inhibitors should guide the rational design of new GIVA cPLA2 inhibitors with improved inhibitory activity against the enzyme.
Results and Discussion
Design of 2-oxoamide inhibitors
2-Oxoamides are potent GIVA cPLA2 inhibitors that were originally designed through a substrate-based approach.32 The design was based on the principle that the inhibitors should consist of several segments that target particular residues in the GIVA cPLA2 active site (Figure 1). The 2-oxoamide functionality (an electrophilic functionality, which contains the activated 2-carbonyl group) is a replacement of the sn-2 ester group of the phospholipid substrate and has been reported to interact with the catalytic serine (Ser228) of lipolytic serine hydrolases.41 The residue Arg200 has been proposed to interact with the negative charged phosphate moiety of the substrate head group,10 thus a bioisosteric carboxylic acid moiety may ensure such an interaction. The lipophilic segments are replacements of the sn-1 and sn-2 esterified fatty acids and they are necessary for hydrophobic interactions with the hydrophobic region of the GIVA cPLA2 active site. Structure-activity relationship studies have revealed the optimum structural features of 2-oxoamide inhibitors.27, 28, 31 However, we do not know the precise interactions of each pharmacophore with particular residues of the enzyme and a model of the 2-oxoamide-GIVA cPLA2 complex would be of great value for designing inhibitors with improved properties.
Figure 1.

Substrate-based design of 2-oxoamide inhibitors.
Optimization of the GIVA cPLA2-AX007 complex
The complex of AX007-GIVA cPLA2 arising from the combination of MD simulations with DXMS39 (Figure 2) showed a hydrogen bond between the carboxylic acid moiety and the Arg200 residue (O…H 1.90 Å, O…N 2.90 Å). The long aliphatic 2-oxoacyl chain was placed in the hydrophobic region of the active site and interacted with several residues, such as Phe199, Pro263, Leu264 and Phe683. The short aliphatic chain of the γ-amino acid interacted with residues, such as Asn555, Gly551 and Leu552. The 2-oxoamide functionality did not seem to participate into any hydrogen bonds with the catalytic Ser228 or with the “oxyanion hole” (residues Gly197/Gly198). The two carbonyl groups of the oxoamide functionality adopted an almost S-cis configuration (dihedral angle 51.7°, atoms O=C-C=O) and the two carbonyl oxygen atoms were located about 7.5 Å and 7.0 Å away from the oxygen atom of the hydroxyl group of Ser228 and the nitrogen atom of Gly197, respectively.
Figure 2.

AX007-GIVA cPLA2 complex generated by a combination of molecular dynamics simulations and DXMS.39
The first step in our study was to examine if the complex of GIVA cPLA2 with AX007 bound in the active site was suitable for docking calculations. The complex was prepared and structurally optimized using the “Protein Preparation Wizard” in Schrödinger suite 2009.42 Inhibitor AX007 was docked in the enzyme active site using the prepared structure of the enzyme and the Surflex-Dock algorithm. The ligand was sketched using the molecular sketcher of SYBYL 8.0 molecular modelling package43 and was subjected in full energy minimization using the Powell energy minimization algorithm with gradient 0.01 kcal mol−1 Å−1. Thereafter, the conformational space of AX007 was investigated using the simulated annealing method.44 The goal was to generate several low-energy conformations with a view to using them as initial structures for the docking calculations in order to avoid “false positives” given the high flexibility of the AX007. The 100 annealed structures were docked in the enzyme active site using the Surflex-Dock algorithm. The scoring function of the Surflex-Dock calculates the -logKd using the hydrophobic contact and polar contact terms which are the dominant ones as well as the repulsive, entropic and solvation terms which possess lower contributions.45, 46 The -logKd is an indicator of the binding affinity of a ligand-receptor complex, and in the present manuscript is referred to as the calculated binding affinity. The docking complex assumed to represent the ligand-receptor interactions was selected based on the following three criteria: (i) the docking score of the pose possessed the highest calculated binding affinity; (ii) its orientation of the long and short aliphatic chains of the ligand oriented in the hydrophobic region of the active site in a similar with the initial complex orientation; and (iii) two key interactions are preserved, namely the interaction of the 2-oxoamide functionality with the catalytic Ser228 and the interaction of the carboxylic acid moiety with Arg200, since these two groups were designed to target Ser228 and Arg200, respectively.
The docking complex that was generated using the Surflex-Dock is presented in Figure 3. The calculated binding affinity (−logKd) is 9.26. The particular complex reveals two hydrogen bonds of the 2-oxoamide carbonyl group with Ser228 (O…H 1.80 Å, O…O 2.70 Å) and Gly197 (O…H 2.30 Å, O…N 3.00 Å). The 2-oxoamide N-H group participates into a hydrogen bond with Asn555 (H…O 2.20 Å, N…O 3.10 Å). The carboxylic acid moiety participates into two hydrogen bonds, one with Arg200 (O…H 2.00 Å, O…N 2.90 Å) and the other with Ser577 (O…H 2.30 Å, 3.20 Å). The long aliphatic 2-oxoacyl chain is placed in the hydrophobic region and interacts with several residues, such as Phe199, Pro263, Leu264 and Phe683, while the short aliphatic chain interacts with residues Asn555, Leu421, Gly551 and Leu552.
Figure 3.

AX007-GIVA cPLA2 complex generated by the Surflex-Dock algorithm.
In comparison with the initial complex (Figure 2), the binding based on the docking complex (Figure 3) shows that the 2-oxoamide functionality is involved in three hydrogen bonds with the residues Ser228, Gly197 and Asn555. The carboxylic acid moiety participates into one additional hydrogen bond with Ser577 and the two aliphatic chains are placed in the hydrophobic region of the active site in a similar with the initial complex orientation. The superimposition of the AX007 pose (pose is the conformation and the orientation of the ligand in the active site) (Figure 4) in the initial complex and in the docking complex (RMSD 1.45 Å for 25 atom pairs) indicates that the main conformational differences are detected in the configuration of the 2-oxoamide functionality and the orientation of the γ-amino acid, which contains the carboxylic acid moiety. As was noted, the configuration of the 2-oxoamide functionality in the initial complex is almost S-cis (dihedral angle 51.7°, atoms O=C-C=O) and is placed in a long distance from Ser228 and Gly197. In contrast, in the Surflex-Dock complex the configuration of the 2-oxoamide functionality is almost S-trans (dihedral angle 149.0°, atoms O=C-C=O) and it is in spatial proximity with Ser228 and Gly197. The optimization of the initial GIVA cPLA2-AX007 complex using the Surflex-Dock suggests that the algorithm should be useful for modelling complexes of the enzyme with other 2-oxoamide inhibitors. Thus, the next goal was to ascertain whether the calculated binding affinity is correlated with the experimental inhibitory activity (XI(50)).
Figure 4.

Superimposition of the AX007 pose in the initial complex (blue colour) and the pose generated by the Surflex-Dock algorithm (magenta colour).
In vitro inhibitory data and calculated binding affinities for the 2-oxoamide inhibitors
The inhibitory potency of various 2-oxoamides has been previously reported in a series of articles.27, 28, 31, 32 The inhibitory activity was reported as XI(50) value, which is the mole fraction of the inhibitor in the total substrate interface required to inhibit the enzyme activity by 50%. The experimental in vitro inhibitory activity was compared with the calculated binding affinity (Table 1).
Table 1.
Structures, XI(50) values, calculated binding affinity (−logKd), polar score that represents the contribution of hydrogen bonding to the calculated binding affinity, and the hydrophobic score that represents the contribution of the hydrophobic interactions to the calculated binding affinity for the 2-oxoamide inhibitors are shown.
| Code | Molecular Structure | XI(50) | −logKd | Polar | Hydrophobic |
|---|---|---|---|---|---|
| AX074 |
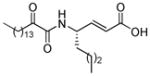
|
0.003a | 11.94 | 4.87 | 7.07 |
| AX109 |
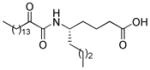
|
0.005b | 10.11 | 2.98 | 7.13 |
| AX007 |
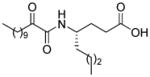
|
0.009c | 9.26 | 3.49 | 5.77 |
| AX073 |
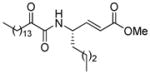
|
0.018a | 8.24 | 3.17 | 5.07 |
| AX063 |
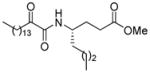
|
0.019b | 8.20 | 2.57 | 5.63 |
| AX013 |
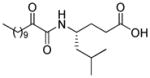
|
0.021d | 7.84 | 3.17 | 4.67 |
| GK165 |
|
0.023e | 7.53 | 3.79 | 3.74 |
| AX006 |
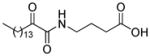
|
0.024f | 7.21 | 4.08 | 3.13 |
| AX021 |
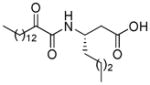
|
0.027b | 7.10 | 2.77 | 4.33 |
| AX016 |
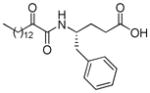
|
0.035d | 7.19 | 2.76 | 4.43 |
| AX008 |
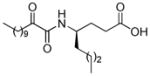
|
0.068c | 5.25 | 2.90 | 2.35 |
The correlation between the calculated binding affinities of the 2-oxoamide inhibitors with the experimental XI(50) values was studied. Even though the size of the dataset is small to be statistically relevant, the aim of the linear regression statistics applied was to detect possible correlation between the calculated binding affinity and experimental XI(50) values. The linearity of the plot (r2 = 0.76, N = 11, Figure 5) demonstrates a good correlation between the calculated binding affinity (−logKd) and the experimental inhibitory activity (XI(50)). Based on the linear regression plot, inhibitor AX074 which possesses the highest calculated binding affinity deviates in comparison with the rest of the inhibitors. Nevertheless, there is a tendency for the most active compound to possess the highest calculated binding affinity and for the least active compound to possess the lowest calculated binding affinity.
Figure 5.

Plot of the calculated binding affinity (−logKd) versus the experimental inhibitory activity (XI(50)) for the 2-oxoamide inhibitors.
Insight into the binding interactions of 2-oxoamide inhibitors with GIVA cPLA2
Using the docking complex of each inhibitor, it was possible to understand the main interactions with the GIVA cPLA2 active site. The interactions of selected inhibitors (AX074 presenting the highest score and inhibitory activity in comparison to AX007, AX013 and AX016 presenting a low score and activity, and AX008 which is the enantiomer of AX007) will be discussed here in detail. Data and discussion for the other 2-oxoamide inhibitors are included in the supporting information (see discussion and SF1-SF4).
The 2-oxoamide inhibitor AX074 exhibits the highest experimental inhibitory activity and also the highest calculated binding affinity. The hydrogen bonding network of the AX074-GIVA cPLA2 complex presented high similarities with that of the AX007-GIVA cPLA2 complex (Figure 3). The 2-oxoamide functionality participates into three hydrogen bonds with residues Ser228 (O…H 1.80 Å, O…O 2.70 Å), Gly197 (O…H 2.30 Å, O…N 3.00 Å) and Asn555 (H…O 2.10 Å, N…O 3.00 Å) (Figure 6). The carboxylic acid moiety interacts with the residues Arg200 (O…H 1.90 Å, O…N 2.90 Å) and Ser577 (O…H 2.30 Å, 3.10 Å). Despite the fact that the two inhibitors (AX074 and AX007) show the same hydrogen bonding network, the polar score for AX074 is higher than that of AX007 (Table 1). The distances of the hydrogen bonds of AX074 are smaller, which means that the hydrogen bonding is optimized. The long aliphatic 2-oxoacyl chain of AX074 has four carbon atoms more than the one of the AX007. Thus, the hydrophobic score of AX074 is higher than that of AX007 due to the optimization of the hydrophobic interactions with the hydrophobic region of the enzyme active site. The short aliphatic chain interacts with the residues Asn555, Leu421, Gly551 and Leu552 in a similar mode like the one of AX007.
Figure 6.

AX074-GIVA cPLA2 complex generated by the Surflex-Dock algorithm.
The leucine variant (AX013) and the phenylalanine variant (AX016) are 7-fold and 12-fold less potent than AX074, respectively. The calculated binding affinity of these compounds is also significantly lower than that of AX074 (Table 1). The leucine and phenylalanine chains are bulkier than the short linear aliphatic chain of AX074, and the docking complexes (Figure 7A and 7B) demonstrate that it is difficult to be accommodated in the hydrophobic pocket. They may clash with residues Asn555, Leu421, Gly551 and Leu552. The carboxylic acid moiety of AX013 participates into two hydrogen bonds with residues Arg200 (O…H 1.70 Å, O…N 2.30 Å) and Ser577 (O…H 1.80 Å, 2.60 Å), but only one of the oxygen atoms participates into hydrogen bonding (Figure 7A). In contrast, the carboxylic acid moiety of AX016 participates into a hydrogen bond with Arg200 (O…H 1.90 Å, O…N 2.80 Å), but does not interact with Ser577 (Figure 7B). The hydrogen bond of AX016 with residue Asn555 is also not observed. The polar score of AX016 is lower than that of AX013, but the hydrophobic score is almost the same (Table 1). The lack of the aforementioned hydrogen bonds may be the reason that AX016 possesses lower experimental inhibitory activity than AX013.
Figure 7.

(A) AX013-GIVA cPLA2 complex generated by the Surflex-Dock algorithm; (B) AX016-GIVA cPLA2 complex generated by the Surflex-Dock algorithm.
The previous studies on 2-oxoamide inhibitors suggested the presence of a binding pocket that interacts with the short linear aliphatic chain adjacent to the amide.31 This binding pocket exhibits enantioselectivity and accommodates a short, preferably linear, carbon chain. This part of the inhibitor may correspond to the sn-1 acyl chain of the natural substrate phospholipid. Alternatively, because the inhibition is higher with shorter chains than with a typical sn-1 acyl chain, it is possible that the short linear chain is fitting into a small binding pocket of the GIVA cPLA2, which is not exploited in substrate binding. Based on previous studies, it is believed that the small binding pocket is constructed by residues Leu421, Gly551 and Leu552.39 The common feature of the inhibitors GK165 and AX006 is the lack of the short linear aliphatic chain of the γ-amino acid (Table 1). These compounds exhibit almost the same experimental inhibitory activity and almost the same calculated binding affinity and they are about 8-fold less potent than AX074.
The chirality of the γ-amino acid influences the inhibition of GIVA cPLA2. Inhibitor AX008 ((R)-enantiomer of AX007) is 8-fold less potent than AX007 ((S)-enantiomer) (Table 1). It is clear that the stereogenic center provides stereoselective enhancement of the binding interactions. The docking complex of inhibitor AX008 (Figure 8) in the GIVA cPLA2 active site suggests a completely different binding of the (R)-enantiomer in comparison with the (S)-enantiomer. The 2-oxoamide functionality is positioned about 11.0 Å away from the catalytic Ser228 and participates into two hydrogen bonds with Met417 (O…H 1.90 Å, O…N 2.90 Å) and Glu418 (O…H 2.70 Å, O…N 3.50 Å). The carboxylic acid moiety interacts with residue Arg200 (O…H 1.80 Å, O…N 2.70 Å) and with residue Gly198 (O…H 1.60 Å, O…N 2.50 Å). The short aliphatic chain is not accommodated inside the small binding pocket (residues Leu421, Gly551 and Leu552). The low hydrophobic score of inhibitor AX008 indicates that the small binding pocket lacks interactions.
Figure 8.

AX008-GIVA cPLA2 complex generated by the Surflex-Dock algorithm.
Molecular dynamics simulation on the AX074-GIVA cPLA2 docking complex
Molecular docking techniques can be combined with the molecular dynamics (MD) simulations to predict more reliable receptor-ligand complexes. Each technique has strengths and weaknesses and the combination can be complementary. The docking technique can be used for the investigation of the ligand conformational space and the binding in the receptor active site, but allows poor or no flexibility for the receptor, which is not permitted to adjust its conformation upon ligand binding. On the other hand, MD simulations allow flexibility both for the ligand and the receptor and permit an induced fit of the receptor active site around the introduced ligand. However, the main problems with the MD simulations are that they are time-consuming and that the system can be trapped in local minima. Therefore, the molecular docking procedure can be used for introducing the ligand in the receptor active site and the MD simulations are then applied to explore the conformational space, to relax and optimize the structure and to check the stability of the docking complex (the term stability of the docking complex means that the ligand remains in the active site of the receptor during the MD simulation even if can adopt different low energy conformers).
An MD simulation after the minimization of the AX074-GIVA cPLA2 docking complex (Figure 6) was performed using the MacroModel 9.7,40 with the aim of identifying persistent interactions between the inhibitor and its binding site. For this purpose the distances and the population of the hydrogen bonds, which were observed in the docking complex have been analysed. In principle, a significant increase in the hydrogen bond distance is assumed to describe a system where the ligand moves apart from the protein, while a relatively fixed distance can be interpreted as a close and profitable interaction between the ligand and the receptor. The hydrogen bond population describes the ratio between the number of samples (snapshots of the AX074-GIVA cPLA2 complex, which were collected during the MD simulation) where a hydrogen bond is observed and the total number of samples that were studied. Three requirements for an interaction to be considered as a hydrogen bond should be filled: (i) the maximum H…Y distance should be 2.5 Å; (ii) the minimum X-H…Y angle should be 120°; and (iii) the minimum H…Y-Z angle should be 90°, where H is the hydrogen atom, X is the donor atom, Y is the acceptor atom, and Z is the atom covalently bonded to the Y atom. If the distance is greater than the maximum or if the angles are smaller than the minimum, the H-bond is not counted in the current population survey.
The analysis of the MD simulation was initiated by plotting the temperature and the potential energy versus the time of the simulation in order to confirm successful equilibration of the system. The temperature of the system was equilibrated around 310 K as was expected and it was retained during the simulation (SF 5a). The potential energy plot (SF 5b) after an initial decrease was stabilized.
Hydrogen bonding of the 2-oxoamide functionality
The docking complex of AX074 with GIVA cPLA2 reveals three hydrogen bonds of the 2-oxoamide functionality with residues Ser228, Gly197 and Asn555. Based on the results of the MD simulation the distance between the hydrogen and oxygen atom, for the hydrogen bond of the 2-oxoamide functionality with the catalytic Ser228, is smaller than 2.5 Å (Figure 9a) and the angle of the atoms H…O=C is greater than 90° (Figure 9c) during the simulation. On the other hand, the angle of the atoms O-H…O for the first 315 ps is lower than 120° (Figure 9b) and the population of the hydrogen bond is 40% (Figure 9d), because one of the requirements is not observed. After the first 315 ps, the angle becomes higher than 120° and the population of hydrogen bond starts an upswing, and at the end of the simulation becomes 90% indicating that the hydrogen bond is persistent.
Figure 9.

Plots for the hydrogen bond between the 2-oxoamide functionality and the catalytic Ser228: a. the H…O distance; b. the O-H…O angle; c. the H…O=C angle; and d. the population of the hydrogen bond.
Although the angle of the atoms H…O=C is higher than 90° (SF 6c), the distance between the hydrogen and the oxygen atom, for the hydrogen bond of the 2-oxoamide functionality with Gly197, is greater than 2.5 Å (SF 6a) suggesting the absence of one of the basic requirements. Besides the absence of the distance requirement, the angle of the atoms N-H…O is smaller than 120° (SF 6b) and consequently the population of the hydrogen bond is about 1.2% at the end of the simulation (SF 6d) indicating that the hydrogen bond is not retained. The third hydrogen bond, in which the 2-oxoamide functionality is involved, is with Asn555. Although the angle requirements are filled (SF 7b and 7c), the distance of the hydrogen and the oxygen atom is greater than 2.5 Å (SF 7a) during the simulation. The population of the hydrogen bond is almost 0%, which declares the thorough absence of this interaction (SF 7d).
Hydrogen bonding of the carboxylic acid moiety
According to the docking complex generated for AX074 with the enzyme, the carboxylic acid moiety participates into two hydrogen bonds with residues Arg200 and Ser577. The hydrogen bond with residue Arg200 fills all the requirements (SF 8a, 8b, and 8c). The population of this interaction is almost 100% during the simulation (SF 8d). This hydrogen bond, along with the hydrogen bond with Ser288, is of paramount importance for the binding since it is believed that these residues play an important role in the catalytic mechanism of the GIVA cPLA2. In particular, the catalytic Ser228 attacks the sn-2 ester bond of a phospholipid molecule whereas the Arg200 stabilizes the head group of a phospholipid molecule. Therefore, it is important for an inhibitor to interact with these particular residues. The MD simulation reveals that these two hydrogen bonds (with Ser228 and Arg200) are persistent during the simulation.
The hydrogen bond of the carboxylic acid moiety with Ser577 is also retained during the simulation despite the flexibility of the linker, which contains the carboxylic acid moiety and of the side chain of Ser577. The hydrogen bond requirements are filled (SF 9a, 9b, and 9c) and the population (SF 9d) is almost 100% during the simulation and that demonstrates the persistency of this interaction.
Based on the above analysis, the two hydrogen bonds of the 2-oxoamide functionality with residues Gly197 and Asn555 are not retained during the simulation. The superposition of the two complexes of AX074 with GIVA cPLA2 before (docking complex) and after (MD complex) the simulation is presented in Figure 10 (RMSD 5.96 Å). The position of the short aliphatic chain in the MD complex differs appreciably in comparison with the position in the docking complex. Consequently, it does not interact with Gly551 and Asn555, but is still in spatial vicinity with Leu421 and Leu552. The long aliphatic 2-oxoacyl chain after the MD simulation adopts an extended conformation instead of the coiled one that it had adopted in the docking complex. It is obvious that the eclipsed and gauche conformations between the carbon atoms in the docking complex relax and finally at the end of the simulation all the carbon atoms adopt anti (staggered) conformations, which correspond to the lower energy ones. Therefore, the relaxation of the long aliphatic 2-oxoacyl chain obligates the side chains of Leu264, Phe295 and Phe397 to change their conformation appreciably in comparison with the docking complex. In addition, the extended long aliphatic 2-oxoacyl chain interacts with Phe401, an interaction that was not observed in the docking complex. In summary, the MD simulation has revealed three persistent hydrogen bonds of AX074 with Ser228, Arg200 and Ser577, the displacement of the short aliphatic chain, which interacts with Leu421 and Leu552 and the relaxation of the long aliphatic 2-oxoacyl chain, which interacts with Leu264, Phe295, Phe397 and Phe401. Above all the MD simulations proves the stability of the docking complex and the validity of the binding mode, which was suggested by the docking calculations.
Figure 10.

Superimposition of the two complexes of AX074 with GIVA cPLA2. The docking complex, which is the initial complex for the MD simulation, is highlighted in magenta and the MD complex, which is the complex after the simulation, is highlighted in green.
Computational methods
Preparation of the GIVA cPLA2 structure
For the docking calculations the complex of GIVA cPLA2 with AX007, which was derived by applying a combination of MD simulations and DXMS was used.39 The structure of the complex was prepared using the “Protein Preparation Wizard” in Schrödinger Suite 2009.42 All the hydrogen atoms were added, and the bond orders and the disulfide bonds were assigned. The prediction of the het groups ionization and tautomeric states was achieved using Epik 2.0.47, 48 To refine the structure, a restrained minimization was performed using the Impref utility, which is available in the “Protein Preparation Wizard”. The specified RMSD of the atoms displacement for terminating the minimizations was 0.30 Å. The refined structure was used for the docking calculations in PDB format.
Preparation of the 2-oxoamide structures
The 2-oxoamide ligands were sketched using the SYBYL 8.0 molecular sketcher.43 All the hydrogen atoms were added to define the correct ionization and tautomeric states, and the oxygen atoms of the carboxylic acid moiety were considered as equivalent with atom type for the oxygen atoms of O.co2. The molecules were subjected to energy minimization using the standard Tripos49 molecular mechanics force field of SYBYL 8.0 molecular modelling package. The Powell50 energy minimization algorithm with gradient 0.01 kcal mol−1 Å−1 was used for the minimization procedure.
Simulated annealing method
The investigation of the conformational space of each inhibitor, using the method of simulated annealing44 was performed. Each inhibitor was heated at a temperature of 500 K for 2000 fs and was cooled at a temperature of 0 K for 10000 fs for 100 cycles. For the simulation, the Tripos49 standard molecular mechanic force field was used. The dielectric function was selected to be distance dependent with value of 1 for the distance-dependent dielectric constant and nonbonded cutoffs 8.0 Å. The annealed structures were minimized using the Powell energy minimization algorithm with gradient 0.01 kcal mol−1 Å−1. For the simulated annealing experiments the SYBYL 8.043 was used.
Molecular docking procedure
Molecular docking calculations were performed using the Surflex-Dock algorithm, which is available in SYBYL 8.0 molecular modelling package.51 Surflex-Dock is a fast, flexible docking method that employs an idealized active site ligand called protomol, as a target to generate putative poses of molecules. These putative poses are scored using the Hammerhead scoring function.46, 52 The scoring function contains the dominant hydrophobic contact and polar contact terms as well as a repulsive, an entropic, and a solvation term. In this study, the ligand mode was adopted to generate the protomol, leaving the threshold and bloat parameters at their default values of 0.50 and 0 Å, respectively. During this procedure, the conformation of the receptor is rigid.
Molecular dynamics simulation
The MD simulation was performed using the MacroModel 9.7, which is available in Schrödinger Suite 2009.40 The docking complex of AX074 with GIVA cPLA2 was first subjected in energy minimization for 25 000 steps using the Polak-Ribiere Conjugate Gradient (PRCG) algorithm,53 to relieve unfavorable contacts between the inhibitor and the enzyme. For the MD simulation the OPLS_2005 force flied has been used.54–56 Solvation effects were simulated using the implicit GB/SA water model.57 The GB/SA continuum solvation model requires the constant-dielectric electrostatic treatment with dielectric constant of 1. The minimized structure was relaxed by performing 500 ps of MD simulation (equilibration phase) at a temperature of 310 K. The system was subsequently submitted in 2 ns MD simulation at a temperature of 310 K, and snapshot structures were extracted every 1 ps. Amino acid atoms at a distance greater than a 20 Å shell from the atoms of the inhibitor were kept fixed during the MD simulation. The hydrogen bonds, which were identified in the complex obtained from the docking calculations, were monitored to check their persistency during the MD simulations. MacroModel periodically examines the geometry around each monitored hydrogen bond. If the bond meets the three hydrogen bond criteria, it is counted in the hydrogen bond population survey.
Conclusion
Recently, a combination of deuterium exchange mass spectrometry (DXMS) and molecular dynamics (MD) has been applied to analyze the complexes between GIVA cPLA2 and two structurally different inhibitors, pyrrophenone and the 2-oxoamide AX007. The resulting models showed that both inhibitors interact with key-enzyme residues, however in entirely different manners. Pyrrophenone is bound to the protein through numerous hydrophobic residues located distal from the active site, while the 2-oxoamide is bound mainly through contacts near the active site. The present molecular docking study analyzed in detail the interactions of eleven 2-oxoamide inhibitors with key-residues of the enzyme. The present study extends the previous model to other related 2-oxoamide inhibitors and suggests an approach for docking studies on other inhibitors targeted to the active site of GIVA cPLA2.
From the present study, it became clear that the interactions of the oxoamide functionality with the active site Ser228 and the “oxyanion hole” residues as well as the carboxyl group with Arg200 are of paramount importance for the stabilization of the inhibitor-GIVA cPLA2 complex. Most recently, we have reported that 2-oxoamides based on α-amino acids may inhibit secreted GIIA sPLA2.34 In that case, docking calculations indicated that the 2-oxoamide functionality interacts with the calcium ion and participates into two hydrogen bonds with Gly29 of GIIA sPLA2.34 The free carboxyl group of the inhibitor interacts with the calcium ion and is involved in a hydrogen bond with Lys62 through a water molecule placed near the hydrophilic region of the GIIA sPLA2 active site. It seems that both the 2-oxoamide functionality and the free carboxyl group of synthetic inhibitors play different roles and bind to different enzyme residues in the active site of each particular class of PLA2. The distance between the 2-oxoamide functionality and the carboxyl group is a critical factor determining the selectivity between the GIVA cPLA2 and the GIIA sPLA2.
In conclusion, the present molecular docking and molecular dynamics studies have provided insight into the mode of binding of 2-oxoamide inhibitors in the GIVA cPLA2 active site. The optimization of the initial AX007-GIVA cPLA2 complex and the good statistical correlation of the experimental inhibitory activities with the calculated binding affinities, suggest that the Surflex-Dock algorithm is useful for predicting the calculated binding affinity of new compounds. The information derived from the docking complexes of the 2-oxoamide inhibitors with the GIVA cPLA2 improves our understanding of the structural features that affect the inhibitory activity of these compounds. The MD simulation on the docking complex of the most active compound reveals persistent interactions of the inhibitor and proves the stability of the complex, validating also the binding suggested by the docking calculations. This molecular modelling study on GIVA cPLA2 is the first that gives information on the binding interactions of small-molecule inhibitors with GIVA cPLA2. Thus, we have developed a model which should be a useful tool for the design of new inhibitors with improved inhibitory properties toward GIVA cPLA2.
Supplementary Material
Acknowledgments
The project is co-funded by the European Social Fund and National Resources (EPEAEK II) (G.K.) and by a grant from the National Institutes of Health (GM 20501) (EAD).
Abbreviations
- GIVA cPLA2
group IVA cytosolic phospholipase A2
- PLA2
phospholipase A2
- MD
Molecular Dynamics
- DXMS
deuterium exchange mass spectrometry
- RMSD
root mean square deviation
- PRCG
polak-ribiere conjugate gradient
- OPLS
optimized potentials for liquid simulations
- GB/SA
generalized born/surface area
Footnotes
Supporting Information Available: Data and discussion for the interactions of 2-oxoamide inhibitors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–85. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Prog Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 6.Magrioti V, Kokotos G. Phospholipase A2 inhibitors as potential therapeutic agents for the treatment of inflammatory diseases. Expert Opin Ther Pat. 2010;20:1–18. doi: 10.1517/13543770903463905. [DOI] [PubMed] [Google Scholar]
- 7.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–51. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 8.Kramer RM, Roberts EF, Manetta J, Putnam JE. The Ca2+-sensitive cytosolic phospholipase A2 is a 100-kDa protein in human monoblast U937 cells. J Biol Chem. 1991;266:5268–72. [PubMed] [Google Scholar]
- 9.Pickard RT, Chiou XG, Strifler BA, DeFelippis MR, Hyslop PA, Tebbe AL, Yee YK, Reynolds LJ, Dennis EA, Kramer RM, Sharp JD. Identification of essential residues for the catalytic function of 85-kDa cytosolic phospholipase A2. Probing the role of histidine, aspartic acid, cysteine, and arginine. J Biol Chem. 1996;271:19225–31. doi: 10.1074/jbc.271.32.19225. [DOI] [PubMed] [Google Scholar]
- 10.Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97:349–360. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- 11.Sharp JD, Pickard RT, Chiou XG, Manetta JV, Kovacevic S, Miller JR, Varshavsky AD, Roberts EF, Strifler BA, Brems DN, et al. Serine 228 is essential for catalytic activities of 85-kDa cytosolic phospholipase A2. J Biol Chem. 1994;269:23250–23254. [PubMed] [Google Scholar]
- 12.Huang Z, Payette P, Abdullah K, Cromlish WA, Kennedy BP. Functional identification of the active-site nucleophile of the human 85-kDa cytosolic phospholipase A2. Biochemistry. 1996;35:3712–3721. doi: 10.1021/bi952541k. [DOI] [PubMed] [Google Scholar]
- 13.Burke JE, Hsu YH, Deems RA, Li S, Woods VL, Jr, Dennis EA. A phospholipid substrate molecule residing in the membrane surface mediates opening of the lid region in group IVA cytosolic phospholipase A2. J Biol Chem. 2008;283:31227–31236. doi: 10.1074/jbc.M804492200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magrioti V, Kokotos G. Synthetic Inhibitors of Group IVA and Group VIA Phospholipase A2. Anti-Inflammatory Anti-Allergy Agents Med Chem. 2006;5:189–203. [Google Scholar]
- 15.Lehr M. Inhibitors of Cytosolic Phospholipase A2α as Potential Anti-Inflammatory Drugs. Anti-Inflammatory & Anti-Allergy Agents Med Chem. 2006;5:149–161. [Google Scholar]
- 16.McKew JC, Lee KL, Shen MW, Thakker P, Foley MA, Behnke ML, Hu B, Sum FW, Tam S, Hu Y, Chen L, Kirincich SJ, Michalak R, Thomason J, Ipek M, Wu K, Wooder L, Ramarao MK, Murphy EA, Goodwin DG, Albert L, Xu X, Donahue F, Ku MS, Keith J, Nickerson-Nutter CL, Abraham WM, Williams C, Hegen M, Clark JD. Indole cytosolic phospholipase A2α inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(dipheny lmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J Med Chem. 2008;51:3388–3413. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 17.Lee KL, Foley MA, Chen L, Behnke ML, Lovering FE, Kirincich SJ, Wang W, Shim J, Tam S, Shen MW, Khor S, Xu X, Goodwin DG, Ramarao MK, Nickerson-Nutter C, Donahue F, Ku MS, Clark JD, McKew JC. Discovery of Ecopladib, an indole inhibitor of cytosolic phospholipase A2α. J Med Chem. 2007;50:1380–1400. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]
- 18.McKew JC, Foley MA, Thakker P, Behnke ML, Lovering FE, Sum FW, Tam S, Wu K, Shen MW, Zhang W, Gonzalez M, Liu S, Mahadevan A, Sard H, Khor SP, Clark JD. Inhibition of cytosolic phospholipase A2α: hit to lead optimization. J Med Chem. 2006;49:135–158. doi: 10.1021/jm0507882. [DOI] [PubMed] [Google Scholar]
- 19.McKew JC, Lovering F, Clark JD, Bemis J, Xiang Y, Shen M, Zhang W, Alvarez JC, Joseph-McCarthy D. Structure-activity relationships of indole cytosolic phospholipase A2α inhibitors: substrate mimetics. Bioorg Med Chem Lett. 2003;13:4501–4504. doi: 10.1016/j.bmcl.2003.08.070. [DOI] [PubMed] [Google Scholar]
- 20.Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, Seno K. Characterization of a novel inhibitor of cytosolic phospholipase A2α pyrrophenone. Biochem J. 2002;363:727–735. doi: 10.1042/0264-6021:3630727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T. Pyrrolidine inhibitors of human cytosolic phospholipase A2. Part 2: synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘pyrrophenone’. Bioorg Med Chem Lett. 2001;11:587–590. doi: 10.1016/s0960-894x(01)00003-8. [DOI] [PubMed] [Google Scholar]
- 22.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. Pyrrolidine inhibitors of human cytosolic phospholipase A2. J Med Chem. 2000;43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 23.Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, Hartopp P, Jackson CG, King SJ, Lawrence L, Mete A, Murray D, Robinson DH, Smith GM, Stein L, Walters I, Wells E, Withnall WJ. Design and synthesis of a novel and potent series of inhibitors of cytosolic phospholipase A2 based on a 1,3-disubstituted propan-2-one skeleton. J Med Chem. 2002;45:1348–1362. doi: 10.1021/jm011050x. [DOI] [PubMed] [Google Scholar]
- 24.Fritsche A, Elfringhoff AS, Fabian J, Lehr M. 1-(2-Carboxyindol-5-yloxy)propan-2-ones as inhibitors of human cytosolic phospholipase A2α: synthesis, biological activity, metabolic stability, and solubility. Bioorg Med Chem. 2008;16:3489–3500. doi: 10.1016/j.bmc.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 25.Hess M, Schulze Elfringhoff A, Lehr M. 1-(5-Carboxy- and 5-carbamoylindol-1-yl)propan-2-ones as inhibitors of human cytosolic phospholipase A2α: bioisosteric replacement of the carboxylic acid and carboxamide moiety. Bioorg Med Chem. 2007;15:2883–2891. doi: 10.1016/j.bmc.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Ludwig J, Bovens S, Brauch C, Elfringhoff AS, Lehr M. Design and synthesis of 1-indol-1-yl- propan-2-ones as inhibitors of human cytosolic phospholipase A2α. J Med Chem. 2006;49:2611–2620. doi: 10.1021/jm051243a. [DOI] [PubMed] [Google Scholar]
- 27.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker SF, Dennis EA, Kokotos G. Structure-activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phospholipase A2. J Med Chem. 2007;50:4222–4235. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 28.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. Differential inhibition of group IVA and group VIA phospholipases A2 by 2-oxoamides. J Med Chem. 2006;49:2821–2828. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. Systemic and intrathecal effects of a novel series of phospholipase A2 inhibitors on hyperalgesia and spinal prostaglandin E2 release. J Pharmacol Exp Ther. 2006;316:466–75. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- 30.Constantinou-Kokotou V, Peristeraki A, Kokotos CG, Six DA, Dennis EA. Synthesis and activity of 2-oxoamides containing long chain beta-amino acids. J Pept Sci. 2005;11:431–5. doi: 10.1002/psc.628. [DOI] [PubMed] [Google Scholar]
- 31.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. Inhibition of group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells, and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 32.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human group IVA phospholipase A2. J Med Chem. 2002;45:2891–2893. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 33.Mouchlis VD, Barbayianni E, Mavromoustakos TM, Kokotos G. The application of rational design on phospholipase A2 inhibitors. Curr Med Chem. 2011;18:2566–82. doi: 10.2174/092986711795933678. [DOI] [PubMed] [Google Scholar]
- 34.Mouchlis VD, Magrioti V, Barbayianni E, Cermak N, Oslund RC, Mavromoustakos TM, Gelb MH, Kokotos G. Inhibition of secreted phospholipases A2 by 2-oxoamides based on α-amino acids: Synthesis, in vitro evaluation and molecular docking calculations. Bioorg Med Chem. 2011;19:735–743. doi: 10.1016/j.bmc.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mouchlis VD, Mavromoustakos TM, Kokotos G. Molecular Docking and 3D-QSAR CoMFA Studies on Indole Inhibitors of GIIA Secreted Phospholipase A2. J Chem Inf Model. 2010;50:1589–1601. doi: 10.1021/ci100217k. [DOI] [PubMed] [Google Scholar]
- 36.Mouchlis VD, Mavromoustakos TM, Kokotos G. Design of new secreted phospholipase A2 inhibitors based on docking calculations by modifying the pharmacophore segments of the FPL67047XX inhibitor. J Comput-Aided Mol Des. 2010;24:107–115. doi: 10.1007/s10822-010-9319-7. [DOI] [PubMed] [Google Scholar]
- 37.Pintore M, Mombelli E, Wechman C, Chretien JR. 3D QSAR Study of Human PLA2 Inhibitors. A Modeling Approach to Select New and Specific Anti-Inflammatory Drugs. Anti-Inflammatory Anti-Allergy Agents Med Chem. 2006;5:175–187. [Google Scholar]
- 38.Gopalsamy A, Yang H, Ellingboe JW, McKew JC, Tam S, Joseph-McCarthy D, Zhang W, Shen M, Clark JD. 1,2,4-Oxadiazolidin-3,5-diones and 1,3,5-triazin-2,4,6-triones as cytosolic phospholipase A2α inhibitors. Bioorg Med Chem Lett. 2006;16:2978–2981. doi: 10.1016/j.bmcl.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 39.Burke JE, Babakhani A, Gorfe AA, Kokotos G, Li S, Woods VL, Jr, McCammon JA, Dennis EA. Location of inhibitors bound to group IVA phospholipase A2 determined by molecular dynamics and deuterium exchange mass spectrometry. J Am Chem Soc. 2009;131:8083–8091. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacroModel, version 9.7. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 41.Kokotos G, Verger R, Chiou A. Synthesis of 2-Oxo amide triacylglycerol analogues and study of their inhibition effect on pancreatic and gastric lipases. Chemistry. 2000;6:4211–7. doi: 10.1002/1521-3765(20001117)6:22<4211::aid-chem4211>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 42.Schrödinger Suite 2009 Protein Preparation Wizard; Epik version 2.0. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]; Impact version 5.5. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]; Prime version 2.1. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 43.SYBYL, molecular modeling software packages, version 8.0, 2007. Tripos Inc; 1699 South Hanley Rd., St. Louis, MO 63144–2917: [Google Scholar]
- 44.Kirkpatrick S, Gelatt CD, Jr, Vecchi MP. Optimization by Simulated Annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 45.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 46.Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput-Aided Mol Des. 1996;10:427–40. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 47.Epik, version 2.0. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 48.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J Comput-Aided Mol Des. 2007;21:681–91. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 49.Clark M, Crammer DR, III, Van Opdenbosch N. Validation of the general purpose tripos 5.2 force field. J Comput Chem. 1989;10:982–1012. [Google Scholar]
- 50.Powell DJM. Restart procedures for the conjugate gradient method. Math Program. 1977;12:241–254. [Google Scholar]
- 51.SYBYL/Surflex-Dock, molecular modeling software packages, version 8.0. Tripos Inc; 1699 South Hanley Rd., St. Louis, MO 63144–2917: 2007. [Google Scholar]
- 52.Welch W, Ruppert J, Jain AN. Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol. 1996;3:449–62. doi: 10.1016/s1074-5521(96)90093-9. [DOI] [PubMed] [Google Scholar]
- 53.Polak E, Ribiere G. Note sur la Convergence de Méthodes de Directions Conjuguées. Revenue Francaise Informat Recherche Operationelle, Serie Rouge. 1969;16:35–43. [Google Scholar]
- 54.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 55.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 56.Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 57.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.