

Abstract
Protein databases have become a crucial part of modern biology. Huge amounts of data for protein structures, functions, and particularly sequences are being generated. Searching databases is often the first step in the study of a new protein. Comparison between proteins or between protein families provides information about the relationship between proteins within a genome or across different species, and hence offers much more information than can be obtained by studying only an isolated protein. In addition, secondary databases derived from experimental databases are also widely available. These databases reorganize and annotate the data or provide predictions. The use of multiple databases often helps researchers understand the structure and function of a protein. Although some protein databases are widely known, they are far from being fully utilized in the protein science community. This unit provides a starting point for readers to explore the potential of protein databases on the Internet.
Keywords: Bioinformatics, Biological Databases, Protein Analysis, Protein Modeling
INTRODUCTION
Protein databases have become a crucial part of modern biology. Huge amounts of data for protein structures, functions, and particularly sequences are being generated. These data cannot be handled without using computer databases. Searching databases is often the first step in the study of a new protein. Without the prior knowledge obtained from such searches, known information about the protein could be missed, or an experiment could be repeated unnecessarily. Comparison between proteins and protein classification provide information about the relationship between proteins within a genome or across different species, and hence offer much more information than can be obtained by studying only an isolated protein. In this sense, protein comparison through databases allows one to view life as a forest instead of individual trees. In addition, secondary databases derived from experimental databases are also widely available. These databases reorganize and annotate the data or provide predictions. The use of multiple databases often helps researchers understand evolution, structure, and function of a protein.
Protein databases are especially powered by the Internet. Unlike traditional media, such as the CD-ROM, the Internet allows databases to be easily maintained and frequently updated with minimum cost. Researchers with limited resources can afford to set up their own databases and disseminate their data quickly. Notably, many small databases on specific types of proteins, such as the EF-Hand Calcium-Binding Proteins Data Library (http://structbio.vanderbilt.edu/cabp_database/), are widely available. Users worldwide can easily access the most up-to-date version through a user-friendly interface. Most protein databases have interactive search engines so that users can specify their needs and obtain the related information interactively. Many protein databases also allow submitters to deposit data, and database servers can check the format of the data and provide immediate feedback.
Although some protein databases are widely known, they are far from being fully utilized in the protein science community. This unit provides a starting point for readers to explore the potential of protein databases on the Internet. Databases for different aspects of proteins are discussed with the focus on sequence, structure, and family. The strengths and weaknesses of the databases are addressed. For Web addresses of the databases discussed in this unit, see Internet Resources and Table 19.4.1. From hundreds of on-line protein databases, several major databases are discussed as examples to illustrate their features and how they can be used effectively. Most other protein databases can be explored in a similar way.
Table 19.4.1.
Web Addresses and Sizes of Selected Protein Databases
NA, size not available at time of printing. The data are as of September 2011.
PROTEIN SEQUENCE DATABASES
Thanks to the Human Genome Project and other sequencing efforts, new sequences have been generated at a prodigious rate. These sequences provide a rich information source and are the core of the revolutionary movement toward “large-scale biology.” The protein sequences can be computationally annotated from these genomic sequences. Various databases contain protein sequences with different focuses. Among all protein sequence databases, UniProt (UniProt Consortium, 2011) is the most widely used one. It provides more annotations than any other sequence database with a minimal level of redundancy through human input or integration with other databases. UniProtKB has three components: (1) Protein knowledgebase, including Swiss-Prot (manually annotated and reviewed) and TrEMBL (automatically annotated) (Bairoch and Apweiler, 1999); (2) UniRef (sequence clusters for fast sequence similarity searches); and (3) UniParc (sequence archive for keeping track of sequences and their identifiers). In addition to Swiss-Prot and TrEMBL, UniProtKB includes information from Protein Sequence Database (PSD) in the Protein Identification Resource (PIR; Barker et al., 1999), which builds a complete and non-redundant database from a number of protein and nucleic acid sequence databases together with bibliographic and annotated information. The National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov) also provides rich information and a number of useful tools for protein sequences. For example, the nr protein database is used for BLAST search (Altschul et al., 1997), which is described in UNIT 2.5 of this book. It includes entries from the non-redundant GenBank (Benson et al., 1999) translations, UniProt, PIR, Protein Research Foundation (PRF) in Japan, and the Protein Data Bank (PDB). Only entries with absolutely identical sequences are merged.
Most of the sequence databases have a sequence search tool and cross-references to entries of other protein and gene databases. Many sequence databases, such as UniProt, also provide text searching using, for instance, protein names or key words. To study a new protein, the author recommends first performing a sequence search using BLAST in nr if the protein sequence is available. The search often gives entry names in the protein databases included in nr. Even when the protein is not found in nr, it is likely that a homologous protein will be hit, which can often lead to some useful information, such as the function of the query protein. If the sequence of the query protein is unavailable, doing a text search in UniProt usually identifies the protein. UniProt is probably the place to obtain the most information about a protein if it can be found in UniProt. However, some additional information may be found by checking other sequence databases. For example, the Kyoto Encyclopedia of Genes and Genomes (KEGG; Ogata et al., 1999) annotates some gene entries with information about metabolic and regulatory pathways. One can also study proteins based on gene models (predicted protein sequences) from many species-specific genome resources, such as Mouse Genome Database (MGD, http://www.informatics.jax.org), FlyBase (a resource for Drosophila genes, http://flybase.org), WormBase (a resource for C. elegans, http://www.wormbase.org), Saccharomyces Genome Database (SGD, http://www.yeastgenome.org), Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org), and Soybean Knowledge Base (SoyKB, http://soykb.org). Although predicted sequences generated by computational gene-finding tools in these resources may contain errors, a large number of proteins are covered and are often reliable enough to provide useful information. When the protein of interest is from a species that is not covered by any of these databases, it is likely that some information can be retrieved from its homolog of a model organism in one of the databases.
UniProt, as a curated protein sequence database, offers a portal to a wide range of annotations, covering areas such as function, family, domain parsing, post-translational modifications, and variants. UniProt can be accessed at http://www.uniprot.org.
Human vitronectin is used here as an example for searching protein sequence databases. To locate the UniProt entry for this protein, one can search either the entry name (VTNC_HUMAN) or the accession number (P04004) obtained from a BLAST search. Alternatively, one can use the full-text search at the UniProt Web page to search by protein name (human vitronectin) or key words (e.g., serum spreading, as vitronectin is also called serum spreading factor s-protein). A combination of several entries can be used in a search.
The entry name in UniProt has the general format X_Y, where X is a mnemonic code of up to four characters indicating the protein name (in this case, VTNC), and Y is a mnemonic species identification code of up to five characters for the biological source of the protein. Some codes used for Y are full English names, e.g., HORSE, HUMAN, MAIZE, MOUSE, PIG, RAT, SHEEP, YEAST (baker’s yeast, Saccharomyces cerevisiae), and WHEAT. Some are abbreviations, including BOVIN (bovine), CHICK (chicken), ECOLI (Escherichia coli), PEA (garden pea, Pisum sativum), RABIT (rabbit), SOYBN (soybean, Glycine max), and TOBAC (common tobacco, Nicotina tabacum).
An entry name may have several accession numbers if they have been merged. An accession number is always conserved from release to release and, therefore, allows unambiguous citation.
Each entry contains the following items shown in table format in the NiceProt View layout: (1) name and origin, (2) protein attributes, (3) general annotation (comments), (4) ontologies (gene functions), (5) binary protein-protein interactions, (6) sequence annotation (features), (7) sequence, (8) references (literature citation), (9) web resources, (10) cross-references (links to other databases), (11) entry information, and (12) relevant documents. The text in the general annotation entry provides a function annotation for the protein (e.g., “Vitronectin is a cell adhesion and spreading factor found in serum and tissues. Vitronectins interact with glycosaminoglycans and proteoglycans…”). The “Cross-references” entry lists the annotations of the protein by other databases, such as GeneCards (Rebhan et al., 1998) and InterPro (Apweiler et al., 2001). GeneCards, a database of human genes, shows chromosomal location and the involvement of the protein in certain diseases (if applicable). InterPro contains predictive protein “signatures”, such as functional domains, repeats and important sites. Clicking the link to InterPro from UniProt leads to a nice graphic view for domain parsing, as shown in Figure 19.4.1 for vitronectin.
Figure 19.4.1.

Annotation of human vitronectin by InterPro.
Various research results are given under sequence annotation (features). Some of the sample features items for VTNC_HUMAN are as follows:
| Feature key | Position (s) | Length | Description |
|---|---|---|---|
| Signal peptide | 1–19 | 19 | Ref.8 Ref.9 |
| Chain | 20–398 | 379 | Vitronentin V65 subunit |
| Peptide | 20–63 | 44 | Somatomedin-B (Ref. 8) |
| Domain | 161–204 | 44 | Hemopexin-like 1 |
| Motif | 64–66 | 3 | Cell attachment site |
| Site | 398–399 | 2 | Cleavage. |
| Modified residue | 75 | 1 | Sulfotyrosine (Ref. 22) |
| Glycosylation | 86 | 1 | N-linked (GlcNAc…) |
| Disulfide bond | 24 ← → 40 | Alternative (by similarity) | |
| Natural variant | 122 | 1 | A→S.[dbSNP:rs2227741] |
| Sequence conflict | 50 | 50 | C → N AA sequence |
Here, “peptide” represents an active peptide in the mature protein, “modified residue” indicates a post-translationally modified residue, and “sequence conflict” shows that different papers report differing sequences.
PROTEIN STRUCTURAL DATABASES
Searching structure databases is becoming more and more popular in molecular biology. The three-dimensional structures of proteins not only define their biological functions, but also hold a key in rational drug design. Traditionally, protein structures were solved at a low-throughput mode. However, advances in new technologies, such as synchrotron radiation sources and high-resolution nuclear magnetic resonance (NMR), accelerate the rate of protein structure determination substantially. The only international repository for the processing and distribution of protein structures is the PDB (Bernstein et al., 1977). The structures in the PDB were determined experimentally by X-ray crystallography, NMR, electron microscopy, etc. Theoretical models have been removed from PDB, effective July 2, 2002, based on the new PDB policy. The PDB also contains some structures of chemical ligands and nucleotides. Each PDB entry is represented by a four-character identifier (PDB ID), where the first character is always a number from 0 to 9 (e.g., 1cau, 256b). The PDB can be accessed at http://www.rcsb.org/pdb/or http://www.pdb.org.
The PDB offers a broad range of search methods, from PDB ID and keywords to structural features and binding ligands. The PDB stores structural information in two formats: the PDB file format (Bernstein et al., 1977) and the macromolecular crystallographic information file (mmCIF) format (Bourne et al., 1997). The PDB file format is still the dominant format used in the protein community. It contains three parts: annotations, coordinates, and connectivities. The connectivity part, which shows chemical connectivities between atoms, is optional. It is listed at the end of the PDB file, beginning the line with the key word CONECT. The coordinate part uses each line for a three-dimensional coordinate of an atom, starting from ATOM (for standard amino acids) or HETATM (for nonstandard groups). The following shows an example of the PDB file format:
| HEADER | OXIDOREDUCTASE | (OXYGEN(A)) | 14-JUN-89 | 1GOX | 1GOX | 3 | |||||
| COMPND | GLYCOLATE | OXIDASE | (E.C.1.1.3.1) | 1GOX | 4 | ||||||
| … | |||||||||||
| ATOM | 232 | N | ALA | 29 | 54.035 | 4.332 | 19.352 | 1.00 | 23.93 | 1GOX | 374 |
| ATOM | 233 | CA | ALA | 29 | 52.992 | 65.356 | 19.569 | 1.00 | 24.74 | 1GOX | 375 |
| ATOM | 234 | C | ALA | 29 | 53.519 | 66.762 | 19.309 | 1.00 | 25.43 | 1GOX | 376 |
| ATOM | 235 | O | ALA | 29 | 54.648 | 67.179 | 19.655 | 1.00 | 25.66 | 1GOX | 377 |
| ATOM | 236 | C | BALA | 29 | 52.433 | 65.340 | 20.993 | 1.00 | 24.54 | 1GOX | 378 |
| … | |||||||||||
| HETATM | 3165 | O | HOH | 658 | 62.480 | 62.480 | 0.000 | 0.50 | 65.79N | 1GOX | 3170 |
| CONECT | 2837 | 2838 | 2854 | 1GOX | 3171 | ||||||
Each line shows the atom serial number, atom type, residue type, chain identifier (in case of multi-chain structure), residue serial number, orthogonal coordinates (three values), occupancy, temperature factor, and segment identifier.
The annotation part of the PDB file format contains dozens of possible record types, including: HEADER (name of protein and release date), COMPND (molecular contents of the entry), SOURCE (biological source), AUTHOR (list of contributors), SSBOND (disulfide bonds), SLTBRG (salt bridges), SITE (groups comprising important sites), HET (nonstandard groups or residues [heterogens]), MODRES (modifications to standard residues), SEQRES (primary sequence of backbone residues), HELIX (helical substructures), SHEET (sheet substructures), and REMARK (other information and comments).
The PDB allows a user to view a molecule structure interactively through Jmol (Hanson, 2010), PDB SimpleViewer, PDB ProteinWorkshop, and RCSB-Kiosk, when the browser is configured to support these free rendering tools. The PDB provides related information about the protein, such as secondary structure assignment and geometry. Each PDB entry also links to a wide range of annotations from secondary databases, including (1) summary and display databases such as Structural Biology Knowledgebase (SBKB, http://sbkb.org), PISA (Protein Interfaces, Surfaces and Assemblies; Krissinel and Henrick, 2007), Molecular Modelling Database (MMDB; Marchler-Bauer et al., 1999) in Entrez, PDBsum (Laskowski et al., 1997), Jena Library of Biological Macromolecules (JenaLib, http://www.fli-leibniz.de/IMAGE.html), PDBWiki (a community annotated knowledge base of biological molecular structures, http://pdbwiki.org), and Proteopedia (a collaborative 3D-encyclopedia of proteins and other molecules; Prilusky et al., 2011); (2) domain annotation from SCOP (Murzin et al., 1995), CATH (Orengo et al., 1997), and Pfam (Finn et al., 2010); (3) structure comparison to other proteins using various methods; (4) the MEDLINE bibliography; (5) protein movements recorded in Database of Macromolecular Movement (MolMovDB; Gerstein and Krebs, 1998); and (6) geometry analyses of the protein, such as CSU Contacts of Structural Units (Sobolev et al., 1999) and castP Identification of Protein Pockets & Cavities (Liang et al., 1998).
In addition to PDB and its linking databases, other structure-related databases can also provide useful information. For example, pdbLight (http://mufold.org/pdblight.php) integrates protein sequence and structure data from multiple sources for protein structure prediction and analysis, together with predicted SCOP classification for the weekly updated PDB structures. BioMagResBank (BMRB; University of Wisconsin, 1999) is a repository for NMR spectroscopy data on proteins, peptides, and nucleic acids. Particularly, it provides partial NMR data (e.g., chemical shifts) before the full structure is solved. Protein Model Portal (PMP; Arnold et al., 2009) provides predicted structural models and their quality assessments for a large number of proteins.
PROTEIN FAMILY DATABASES
Introduction
Proteins can be classified according to their sequence, evolutionary, structural, or functional relationships. A protein in the context of its family is much more informative than the single protein itself. For example, residues conserved across the family often indicate special functional roles. Two proteins classified in the same functional family may suggest that they share similar structures, even when their sequences do not have significant similarity.
There is no unique way to classify proteins into families. Boundaries between different families may be subjective. The choice of classification system depends in part on the problem; in general, the author suggests looking into classification systems from different databases and comparing them. Three types of classification methods are widely adopted based upon the similarity of sequence, structure, or function. Sequence-based methods are applicable to any proteins whose sequences are known, while structure-based methods are limited to the proteins of known structures, and function-based methods depend on the functions of proteins being annotated. Sequence- and structure-based classifications can be automated and are scalable to high-throughput data, whereas function-based classification is typically carried out manually. Structure- and function-based methods are more reliable, while sequence-based methods may result in a false positive result when sequence similarity is weak (i.e., two proteins are classified into one family by chance rather than by any biological significance). In addition, since protein structure and function are better conserved than sequence, two proteins having similar structures or similar functions may not be identified through sequence-based methods.
Databases for Sequence-Based Protein Families
Sequence-based protein families are classified according to a profile derived from a multiple-sequence alignment. The profile can be shown across a long domain (tens of residues or more) or can be revealed in short sequence motifs. Classification methods based on profiles across long domains tend to be more reliable but less sensitive than those based on short sequence motifs.
Several sequence-based methods focus more on profiles across long domains, including Pfam (Finn et al., 2010), ProDom (Corpet et al., 1999), and Clusters of Orthologous Group (COG; Tatusov et al., 1997). These methods differ in the techniques used to construct families. Pfam builds multiple-sequence alignments of many common protein domains using hidden Markov models. The ProDom protein domain database consists of homologous domains based on recursive PSI-BLAST searches (UNIT 2.5). COG aims toward finding ancient conserved domains by delineating families of orthologs across a wide phylogenetic range. SMART (Simple Modular Architecture Research Tool; Letunic et al., 2009) collects domain families, which are annotated with respect to phyletic distributions, functional class, three-dimensional structures and functionally important residues. It can be used for identification and annotation of genetically mobile domains and analysis of domain architectures. The iProClass database (Wu et al., 2004) combines multiple sources of information for protein classification. One can use all these databases for a comprehensive analysis or choose one of them based on the purpose of the study. Various sequence-based protein families have different focuses. For example, Pfam focuses on function, ProDom on sequence domain, and COG on evolution.
The following shows an example of Pfam for the GRIP domain (accession number PF01465). Pfam lists some useful functional information for the entry as follows:
“The GRIP (golgin-97, RanBp2alpha, Imh1p and p230/golgin-245) domain is found in many large coiled-coil proteins. It has been shown to be sufficient for targeting to the Golgi. The GRIP domain contains a completely conserved tyrosine residue. At least some of these domains have been shown to bind to GTPase Arl1, see structures in [4,5].”
In addition, Pfam gives the alignment among the family members.
One can identify some features of the family through this pattern (i.e., from particularly conserved residues at specific alignment positions).
Some methods are based on “fingerprints” of small conserved motifs in sequences, as with PROSITE (Hofmann et al., 1999), PRINTS (Attwood et al., 1999), and BLOCKS (Heniko et al., 1999). In protein sequence families, some regions have been better conserved than others during evolution. These regions are generally important for the function of a protein or for the maintenance of its three-dimensional structure or function. The fingerprints may be used to assign a newly sequenced protein to a specific family. Fingerprints are derived from gapped alignments in PROSITE and PRINTS, but are derived from ungapped alignments (corresponding to the highly conserved regions in proteins) in BLOCKS. A fingerprint in PRINTS may contain several motifs from PROSITE, and thus may be more flexible and powerful than a single PROSITE motif. Therefore, PRINTS can provide a useful adjunct to PROSITE. It should be noted that some functionally unrelated proteins may be classified together due to chance matches in short motifs.
Databases for Structure-Based Protein Families
The hierarchical relationship among proteins can be clearly revealed in structures through structure-structure comparison. Structure families often provide more information on the relationship between proteins than what sequence families can offer, particularly when two proteins share a similar structure but no significant sequence identity. Figure 19.4.2 shows an example of a structure-structure alignment between two proteins. Sometimes, sequence similarity between two proteins exists but is not strong enough to produce an unambiguous alignment. In this case, the alignment between two structures can generate better alignment in terms of biological significance, and thus may pinpoint the evolutionary relationship and active sites more accurately.
Figure 19.4.2.
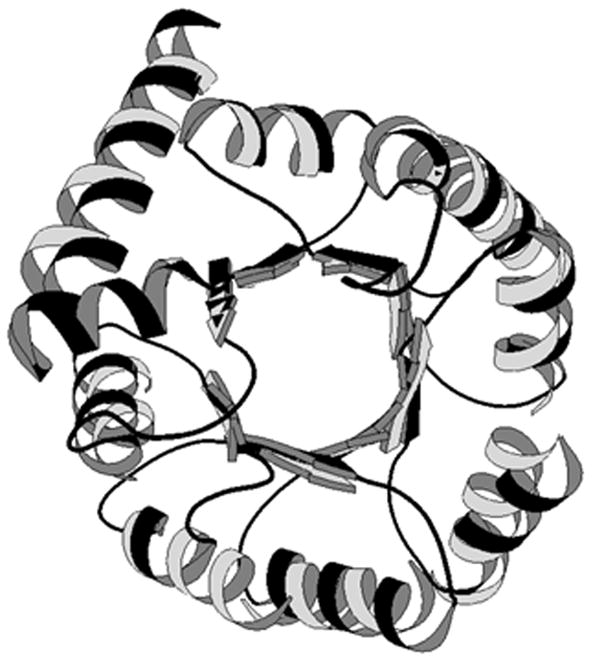
Structure superposition between glycolate oxidase(1gox, in black) and inosine monophosphate dehydrogenase (1ak5, in gray). This figure was made using MOLSCRIPT (Kraulis, 1991).
Different structure-structure comparison methods yield different structure families. CATH (Class, Architecture, Topology and Homologous superfamily; Orengo et al., 1997) is a hierarchical classification of protein domain structures. CE (Combinatorial Extension of the optimal path; Shindyalov and Bourne, 1998) provides structural neighbors of the PDB entries with structure-structure alignments and three-dimensional superposition. FSSP (Fold classification based on Structure-Structure alignment of Proteins; Holm and Sander, 1996) features a protein family tree and a domain dictionary, in addition to whole-chain-based classification, sequence neighbors, and multiple structure alignments. SCOP (Structural Classification of Proteins; Murzin et al., 1995) uses augmented manual classification, class, fold, superfamily, and family classification. VAST (Vector Alignment Search Tool; Gibrat et al., 1996) contains representative structure alignments and three-dimensional superposition. Among these five databases, SCOP provides more function-related information. However, due to the manual work involved, SCOP is not updated as frequently as the others (as of September 2011, it was last updated for the PDB release on June, 2009), whereas FSSP and CATH follow the PDB updates closely.
SCOP is used here as an example to show the features of structure-based families. SCOP can be accessed through its home server in the UK (http://scop.mrc-lmb.cam.ac.uk/scop/). SCOP describes the hierarchical relationship among proteins through the major levels of (homologous) family, superfamily, and fold. Proteins are clustered together into a (homologous) family if they have significant sequence similarity. Different families that have low sequence similarity but whose structural and functional features suggest a common evolutionary origin are placed together in a superfamily. Different superfamilies are categorized into a fold if they have the same major secondary structures in the same arrangement and with the same topological connections (the peripheral elements of secondary structure and turn regions may differ in size and conformation). Two superfamilies in the same fold may not have a common evolutionary origin. Their structural similarities may arise from the physics and chemistry of proteins favoring certain packing arrangements and chain topologies (Murzin et al., 1995). Figure 19.4.3 shows the SCOP interface using an example of protein 1gox in the PDB.
Figure 19.4.3.

An example of the SCOP interface when searching the structure of 1gox in the PDB.
Databases for Function-Based Protein Families
There are various protein functional families classified from different perspectives. The ENZYME data bank (Bairoch, 1993) contains the following data for each enzyme: EC number, recommended name, alternative names, catalytic activity, cofactors, pointers to the UniProt entry, and pointers to any disease associated with a deficiency of the enzyme. BRENDA (Scheer et al., 2011) collects extensive enzyme functional data. Catalytic Site Atlas (Porter et al., 2004) is a database of three-dimensional enzyme active sites derived from PDB structures. Various gene ontologies, such as Gene Ontology (GO; The Gene Ontology Consortium, 2000) and KEGG, also organize proteins into functional categories. Annotation and analysis by these ontologies for a given list of genes can be carried out using tools and databases such as DAVID (Database for Annotation, Visualization and Integrated Discovery; Huang et al., 2009). In addition, there are a growing number of databases dedicated to special types of proteins, such as G-protein-coupled receptors, transporters, and protein kinases, as shown in Table 19.4.1.
OTHER DATABASES
Protein Modification Databases
There are a number of databases for protein post-translational modifications. O-GlycBase (Gupta et al., 1999) collected, experimentally verified O- or C-glycosylation sites. Plant Protein Phosphorylation Database (P3DB; Gao et al., 2009) condenses phosphoproteomics information (including experimental phosphorylation sites) from various plants. Compendium of protein lysine acetylation (CPLA; Liu et al., 2010) includes manually curated lysine acetylated substrates with their sites.
Protein Localization Databases
A number of databases are available to describe protein subcellular localization and targeting. These databases are for various species, such as eSLDB (eukaryotic Subcellular Localization database) for general eukaryotes (Pierleoni et al., 2007), LOCATE for human and minor (Sprenger et al., 2008), SUBA for Arabidopsis (Heazlewood et al., 2007), and PSORTdb for bacteria and archaea (Yu et al., 2011). Some databases focus on special organelles, such as Organelle DB (Wiwatwattana and Kumar, 2005) and Centrosome:db (Nogales-Cadenas et al., 2009).
Protein Binding Databases
Protein binding includes protein-substrate docking and protein-protein association. ReLiBase (Hendlich, 1998) is a database system for analyzing receptor-ligand complexes in the PDB. BindingDB (Liu et al., 2007) describe many interactions between drug-target proteins and small, drug-like molecules. As protein-protein interactions are measured in large scales, there are many protein interaction databases. An early one is Database of Interacting Proteins (DIP; Xenarios et al., 2000). Some later databases are more comprehensive. For example, Biological General Repository for Interaction Datasets (BioGRID; Stark et al., 2011) includes protein–protein and genetic interactions for all major model organism species; STRING (Search Tool for the Retrieval of Interacting Genes/Proteins; Jensen et al., 2009) covers known and predicted protein interactions for many species, as well as direct (physical) and indirect (functional) associations. Furthermore, some protein interaction databases are based on protein structures, such as 3D Complex (Levy et al., 2006), DOMMINO (http://dommino.org), etc.
Protein Energetics Databases
There are few databases for protein energetics, due to the low-throughput nature of the data source. One useful energetics database can be found in ProTherm (Thermodynamic Database for Proteins and Mutants; Gromiha et al., 1999). It contains thermodynamic data on mutations, including Gibbs free energy, enthalpy, heat capacity, and transition temperature. Another database is 3D-footprint (Contreras-Moreira, 2010), which provides estimates of binding specificity for protein-DNA complexes in PDB.
Bibliographic Databases
Searching for protein information through traditional bibliographic databases, such as MEDLINE or Grateful Med, can be rewarding. In addition, some bibliographic reference databases dedicated to proteins may provide certain information more directly. For example, iProLINK (integrated Protein Literature, INformation and Knowledge; Hu et al., 2004) provides literature information on proteins and their features or properties.
Combined Databases
By integrating different types of protein databases together, a database of databases (or a data warehouse) can be built. Such combined databases not only serve as a “one-stop shop,” but also provide cross-references between entries in different databases. One example of such databases is SRS (Sequence Retrieval System; Etzold et al., 1996), which is a comprehensive database for molecular biology. The home server at http://srs.ebi.ac.uk supports many biological databases, including almost all the major protein/genetic databases. As an indexing system, it provides fast access to different databases through searches by sequence or by key words from various data fields. SRS also builds indices using cross-references between databases. An entry from one database can be linked to other databases that contain the entry. However, it should be noted that the contents of SRS might lag behind the other databases in updating (i.e., some new entries in the original databases may not be included in SRS).
SUMMARY
This unit reviews some of major protein databases on the Internet and shows what kind of information users can expect from protein databases. Although all technical procedures cannot be described here, most of the protein databases are easy to use and provide detailed on-line manuals so that even users with little computer skill can learn them quickly. Readers are encouraged to study additional protein databases that are not covered in this unit. For example, the portals listed in “INTERNET RESOURCES” give links to many other protein databases. Furthermore, the journal “Nucleic Acids Research” has a Database issue every year, which describes many high-quality, well-maintained protein databases.
Protein databases may not always be easily accessible or usable through the Internet. Sometimes a database server may be down or the Internet connection may be interrupted. For a frequent user, it may be worthwhile to install the database on a local machine. On the other hand, it must be kept in mind that a mirror site or a local copy may contain an older version of the database than the one on the home server.
It is important to assess the quality of the data. There are three types of data in protein databases. (1) Experimental data are generally very reliable. However, some entries may contain errors (e.g., some protein sequences) or may be based on low-resolution data (e.g., some protein structures determined by NMR). (2) Annotation data uses computational techniques on experimental data, for example, secondary structure assignment and domain partition in structure. These data depend on the quality of the experimental data and the computational methods used. Different methods may yield different results. (3) Prediction data includes, for example, sequence domain parsing and three-dimensional structure prediction. No matter how good the method, the results are still predictions and should be subjected to experimental verification. In addition, different methods typically give different predictions.
While protein databases on the Internet become indispensable resources for studying proteins, caution is needed when using the data from databases to draw a conclusion. The qualities of databases vary significantly. Some databases are not well maintained and contain obsolete information. It is not rare to see some protein databases disappear after a few years. In addition, the data in some databases are not carefully validated and may not be reliable. It is worthwhile to check the same type of data from different databases and compare them. It is sometimes necessary to use additional computational tools (e.g., tools to assess the quality of a structure) for further analysis.
INTERNET RESOURCES
The Web addresses of the databases mentioned in this unit are listed in Table 19.4.1. Readers can find more protein databases and related bioinformatics tools in the following Web pages, which collect a large number of useful links:
http://bioinformatics.ca/links_directory/ (Bioinformatics Links Directory)
http://www.biophys.uni-duesseldorf.de/BioNet/Pedro/research_tools.html (Pedro’s biomolecular research tools)
http://www.expasy.org (SIB Bioinformatics Resource Portal)
http://www.123genomics.com (Genomics, Proteomics and Bioinformatics Knowledge Base)
http://bioinformatics.ws/index.php/Bioinformatics_tools_and_algorithms (Bioinformatics tools and algorithms)
Acknowledgments
This work is partially supported by National Institutes of Health Grant R21/R33-GM078601.
Literature Cited
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Kiefer F, Kopp J, Battey JN, Podvinec M, Westbrook JD, Berman HM, Bordoli L, Schwede T. The protein model portal. J Struct Funct Genomics. 2009;10:1–8. doi: 10.1007/s10969-008-9048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, Durbin R, Falquet L, Fleischmann W, Gouzy J, Hermjakob H, Hulo N, Jonassen I, Kahn D, Kanapin A, Karavidopoulou Y, Lopez R, Marx B, Mulder NJ, Oinn TM, Pagni M, Servant F, Sigrist CJ, Zdobnov EM. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucl Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood TK, Flower DR, Lewis AP, Mabey JE, Morgan SR, Scordis P, Selley J, Wright W. PRINTS prepares for the new millennium. Nucl Acids Res. 1999;27:220–225. doi: 10.1093/nar/27.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. The ENZYME data bank. Nucl Acids Res. 1993;21:3155–3156. doi: 10.1093/nar/21.13.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The UniProt protein sequence data bank and its supplement TrEMBL in 1999. Nucl Acids Res. 1999;27:49–54. doi: 10.1093/nar/27.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker WC, Garavelli JS, McGarvey PB, Marzec CR, Orcutt BC, Srinivasarao GY, Yeh LL, Ledley RS, Mewes H, Pfeiffer F, Tsugita A, Wu C. The PIR-international protein sequence database. Nucl Acids Res. 1999;27:39–42. doi: 10.1093/nar/27.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF, Rapp BA, Wheeler DL. Genbank. Nucl Acids Res. 1999;27:12–17. doi: 10.1093/nar/27.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank: A computer based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Bourne P, Berman H, Watenpaugh K, Westbrook J, Fitzgerald P. The macromolecular crystallographic information file (mmCIF) Methods Enzymol. 1997;277:571–590. doi: 10.1016/s0076-6879(97)77032-0. [DOI] [PubMed] [Google Scholar]
- Contreras-Moreira B. 3D-footprint: a database for the structural analysis of protein-DNA complexes. Nucl Acids Res. 2010;38(Database issue):D91–97. doi: 10.1093/nar/gkp781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F, Gouzy J, Kahn D. Recent improvements of the ProDom database of protein domain families. Nucl Acids Res. 1999;27:263–267. doi: 10.1093/nar/27.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzold T, Ulyanov A, Argos P. SRS: Information retrieval system for molecular biology data banks. Methods Enzymol. 1996;266:114–128. doi: 10.1016/s0076-6879(96)66010-8. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucl Acids Res. 2010;38(Database issue):D211–22. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Agrawal GK, Thelen JJ, Xu D. P3DB: a plant protein phosphorylation database. Nucl Acids Res. 2009;37(Database issue):D960–D962. doi: 10.1093/nar/gkn733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M, Krebs W. A database of macromolecular motions. Nucl Acids Res. 1998;26:4280–4290. doi: 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opinion Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Gromiha MM, An J, Kono H, Oobatake M, Uedaira H, Sarai A. Protherm: Thermodynamic database for proteins and mutants. Nucl Acids Res. 1999;27:286–288. doi: 10.1093/nar/27.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Birch H, Rapacki K, Brunak S, Hansen JE. O-GLYCBASE version 4.0: a revised database of O-glycosylated proteins. Nucl Acids Res. 1999;27:370–372. doi: 10.1093/nar/27.1.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RM. Jmol – a paradigm shift in crystallographic visualization. Journal of Applied Crystallography. 2010;43:1250–1260. [Google Scholar]
- Hendlich M. Databases for protein-ligand complexes. Acta Crystallogr, Sect D. 1998;1:1178–1182. doi: 10.1107/s0907444998007124. [DOI] [PubMed] [Google Scholar]
- Heniko JG, Heniko S, Pietrokovski S. New features of the blocks database servers. Nucl Acids Res. 1999;27:226–228. doi: 10.1093/nar/27.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heazlewood JL, Verboom RE, Tonti-Filippini J, Small I, Millar AH. SUBA: the Arabidopsis Subcellular Database. Nucl Acids Res. 2007;35(Database issue):D213–218. doi: 10.1093/nar/gkl863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Falquet L, Bairoch A. The PROSITE database, its status in 1999. Nucl Acids Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Hu ZZ, Mani I, Hermoso V, Liu H, Wu CH. iProLINK: an integrated protein resource for literature mining. Comput Biol Chem. 2004;28:409–416. doi: 10.1016/j.compbiolchem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucl Acids Res. 2009;37(Database issue):D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P. MOLSCRIPT—a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Krissinel K, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: A web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci. 1997;22:488–490. doi: 10.1016/s0968-0004(97)01140-7. [DOI] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucl Acids Res. 2009;37(Database issue):D229–232. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Science. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucl Acids Res. 2007;35(Database issue):D198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Cao J, Gao X, Zhou Y, Wen L, Yang X, Yao X, Ren J, Xue Y. CPLA 1.0: an integrated database of protein lysine acetylation. Nucl Acids Res. 2011;39(Database issue):D1029–1034. doi: 10.1093/nar/gkq939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Addess KJ, Chappey C, Geer L, Madej T, Matsuo Y, Wang Y, Bryant SH. MMDB: Entrez’s 3D structure database. Nucl Acids Res. 1999;27:240–243. doi: 10.1093/nar/27.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nogales-Cadenas R, Abascal F, Díez-Pérez J, Carazo JM, Pascual-Montano A. CentrosomeDB: a human centrosomal proteins database. Nucl Acids Res. 2009;37(Database issue):D175–80. doi: 10.1093/nar/gkn815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucl Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo CA, Michie AD, Jones DT, Swindells MB, Thornton JM. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL, Fariselli P, Casadio R. eSLDB: eukaryotic subcellular localization database. Nucl Acids Res. 2007;35(Database issue):D208–212. doi: 10.1093/nar/gkl775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucl Acids Res. 2004;32(Database issue):D129–33. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilusky J, Hodis E, Canner D, Decatur WA, Oberholser K, Martz E, Berchanski A, Harel M, Sussman JL. Proteopedia: A status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. Journal of Structural Biology. 2011;175:244–252. doi: 10.1016/j.jsb.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: A novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics. 1998;14:656–664. doi: 10.1093/bioinformatics/14.8.656. [DOI] [PubMed] [Google Scholar]
- Scheer M, Grote A, Chang A, Schomburg I, Munaretto C, Rother M, Söhngen C, Stelzer M, Thiele J, Schomburg D. BRENDA, the enzyme information system in 2011. Nucl Acids Res. 2011;39(Database issue):D670–676. doi: 10.1093/nar/gkq1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- Sprenger J, Lynn Fink J, Karunaratne S, Hanson K, Hamilton NA, Teasdale RD. LOCATE: a mammalian protein subcellular localization database. Nucl Acids Res. 2008;36(Database issue):D230–3. doi: 10.1093/nar/gkm950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, Reguly T, Rust JM, Winter A, Dolinski K, Tyers M. The BioGRID Interaction Database: 2011 update. Nucl Acids Res. 2011;39(Database issue):D698–704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucl Acids Res. 2011;39(Database issue):D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Wisconsin. BioMagResBank. University of Wisconsin; Madison, Wis: 1999. [Google Scholar]
- Vriend G. WHAT IF: A molecular modelling and drug design program. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Wiwatwattana N, Kumar A. Organelle DB: a cross-species database of protein localization and function. Nucl Acids Res. 2005;33(Database issue):D598–604. doi: 10.1093/nar/gki071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Huang H, Nikolskaya A, Hu Z, Barker WC. The iProClass integrated database for protein functional analysis. Comput Biol Chem. 2004;28:87–96. doi: 10.1016/j.compbiolchem.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D. DIP: the database of interacting proteins. Nucl Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Laird MR, Spencer C, Brinkman FS. PSORTdb--an expanded, auto-updated, user-friendly protein subcellular localization database for Bacteria and Archaea. Nucl Acids Res. 2011;39(Database issue):D241–244. doi: 10.1093/nar/gkq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]