

Abstract
Triple shape memory polymers (TSMPs) are a growing subset of a class of smart materials known as shape memory polymers, which are capable of changing shape and stiffness in response to a stimulus. A TSMP can change shapes twice and can fix two metastable shapes in addition to its permanent shape. In this work, a novel TSMP system comprised of both permanent covalent cross-links and supramolecular hydrogen bonding cross-links has been synthesized via a one-pot method. Triple shape properties arise from the combination of the glass transition of (meth)acrylate copolymers and the dissociation of self-complementary hydrogen bonding moieties, enabling broad and independent control of both glass transition temperature (Tg) and cross-link density. Specifically, ureidopyrimidone methacrylate and a novel monomer, ureidopyrimidone acrylate, were copolymerized with various alkyl acrylates and bisphenol A ethoxylate diacrylate. Control of Tg from 0 to 60 °C is demonstrated: concentration of hydrogen bonding moieties is varied from 0 to 40 wt %; concentration of the diacrylate is varied from 0 to 30 wt %. Toughness ranges from 0.06 to 0.14 MPa and is found to peak near 20 wt % of the supramolecular cross-linker. A widely tunable class of amorphous triple-shape memory polymers has been developed and characterized through dynamic and quasi-static thermomechanical testing to gain insights into the dynamics of supramolecular networks.
INTRODUCTION
Shape memory materials are a widely investigated class of smart materials capable of changing from one predetermined shape to another in response to a stimulus.1 This macro-scale phenomenon has been demonstrated in ceramics, metals and a range of polymeric systems, but results from very different mechanics in each material.2 Shape memory polymers (SMPs) can “remember” a metastable shape and upon application of a stimulus, such as heat or light, recover a globally stable shape.3,4 This memory phenomenon is referred to as dual-shape memory and is enabled by the presence of a switching phase and a cross-linking phase in a polymer network. The switching phase can be deformed under certain temporary conditions, such as high temperature, to program a metastable shape. Switching phases maintain this deformation through chain immobilization by reversible crystallization, vitrification or supramolecular interactions5,6 to hold this metastable shape until requisite activation energy facilitates a return to the original shape. The cross-linking phases serve as net points, which allow the deformation of chains, but maintain the relative positions of the chains. Cross-linking phases include covalent cross-links and physical cross-links, such as crystalline or glassy domains in phase separated polymers or strong supramolecular interactions.7 Cross-links can be incorporated during initial polymerization or through new alternative methods, including, for instance, the use of ionizing radiation to selectively cross-link SMPs after thermoplastic processing.8–10
The combined effect of a thermally activated switching phase and a cross-linking phase yields a polymer with the capability of deformation at high temperatures, fixation of the induced deformation when cooled below the transition temperature of the switching phase, and recovery of the deformation when heated above the transition temperature. This thermomechan-ical cycle describes the shape memory phenomenon in polymers. This effect is inherently different from the behavior exhibited by shape memory alloys, as it permits one-time large scale shape recovery. Deformation up to 800% has been shown to be fully recoverable in optimized acrylic SMPs as compared to roughly 8% for shape memory alloys.11 Shape memory has been demonstrated in a large number of polymer systems and has been investigated for use in biomedical and commodity applications.12–15 One inherent limitation of SMPs is that SMP-based devices will, when actuated, always recover toward a single predetermined shape that must be set at the time of molding or cross-linking.
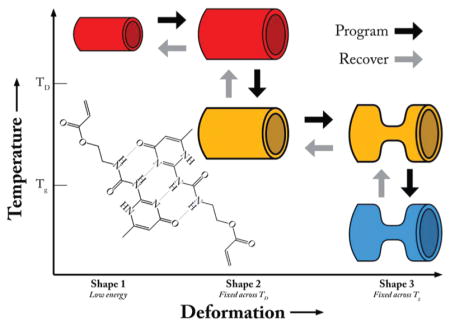
Triple-shape memory polymers (TSMPs) are an emerging class of polymers that have the capability of storing two metastable shapes in addition to a globally stable shape.16 These polymers contain a cross-linking phase, and two independent switching phases associated with two different transitions. Figure 1 is a schematic representing the programming and recovery cycles of a generic TSMP. Programming the TSMP consists of (1) heating the polymer above both transitions and inducing deformation, (2) maintaining the deformation while cooling the polymer to fix shape 2 at a temperature between the two transitions, (3) further deforming the polymer, and (4) maintaining the second deformation while cooling below both transitions, fixing shape 3. Upon heating without any applied load, the polymer will first recover shape 2 and, upon reaching the second transition, then recover shape 1. Bellin et al. have demonstrated the triple-shape memory effect in systems containing two crystalline switching phases, while others have combined an amorphous phase with a crystalline switching phase.17,18 Recently Xie has shown quadruple-shape memory utilizing the broad transition of perfluorosulfonic acid ionomers.19 Luo et al. have also demonstrated a system of composites capable of demonstrating the triple-shape memory effect.20 TSMPs stand to increase the complexity of devices utilizing the shape memory effect by enabling multistage complex recovery profiles. Recent work has elucidated the benefits of this approach and also certain limitations, especially a strong sensitivity to the specific conditions of the programming step.21
Figure 1.

Schematic of a triple shape memory polymer programming and recovery cycles. The triple shape cycle consists of heating (1 → 2) and programming a shape above both transitions (2 → 3); cooling to a temperature between the two transitions to fix this shape (3 → 4); further deforming the polymer (4 → 5); cooling to a temperature below both transitions to fix the third shape (5 → 6).
Supramolecular polymers are materials containing directional, intermolecular and highly reversible noncovalent interactions.22 There are many examples of the interactions that can be used for supramolecular engineering including hydrogen bonding, metal ion coordination and electrostatics. These interactions can be used to assemble monomers to form polymeric chains or assemble covalently linked chains into polymer networks through the formation of cross-links. Meijer et al. have demonstrated the unique ability of self-complementary arrays of hydrogen bonds in the formation of supramolecular polymers, specifically utilizing 2-ureido-4[1H]-pyrimidone (UPy) units.23 UPy has been proposed for a number of advanced applications due to its extremely high tendency to dimerize, with a dimerization constant greater than 106 M−1 in CHCl3.23 Specific areas of research include self-healing materials, biomimetic modular polymers, thermoplastic elastomers and SMPs.6,24–27 UPy functionalities have been included into thermoplastic segmented polyurethanes and thermoset acrylic elastomers (with Tg’s well below the intended use temperature) and used as the switching phase.6,28 Acrylic elastomers containing UPy show shape recovery approaching 100%, but show lower shape fixity, ~90%, as compared to switching segments based on vitrification or crystallization.6 To the knowledge of the authors, UPy has not previously been incorporated into acrylic systems with a glass transition near the intended use temperature or used in TSMPs.
This work seeks to combine the properties of supramolecular and covalent cross-links in an acrylic system to demonstrate amorphous TSMPs with tunable glass transitions and supra-molecular and covalent cross-link density. Specifically, UPy functionalized (meth)acrylate monomers were synthesized and polymerized with n-alkyl acrylates and a diacrylate cross-linker in a one-pot method. Thermomechanical properties of the synthesized system were evaluated by dynamic mechanical analysis, differential scanning calorimetry, and quasi-static tensile testing. The ability to serve as a TSMP was also evaluated for several samples.
MATERIALS AND METHODS
Methyl acrylate (MA), ethyl acrylate (EA), butyl acrylate (BA), bisphenol A ethoxylate diacrylate (BPAEDA) (Mw ~ 512 g/mol), anhydrous dimethyl sulfoxide (DMSO), 6-methylisocytosine, and 2,2-dimethoxy-2-phenyl acetophenone (DMPA) were purchased from Sigma-Aldrich. Propyl acrylate was purchased from Alfa Aesar. 2-isocyanatoethyl acrylate was purchased from Wako Chemicals. 2-Isocyanatoethyl methacrylate was purchased from TCI America. All chemicals were used as received without further purification.
One-Pot Synthesis of UPy Functionalized (Meth)acrylate Monomer and Polymer Films
Synthesis of (meth)acrylate functionalized monomer was adapted from Yamauchi et al. and is depicted in Figure 2.29 6-Methylisocytosine and 2-isocyantoethyl (meth)acrylate were added to DMSO in concentrations appropriate for the subsequent polymerization and heated to 150 °C until the isocytosine had completely dissolved. The solutions were held at 150 °C for 15 min and subsequently removed and quenched to room temperature, precipitating a white solid confirmed to be UPy(M)A by IR and 1D and 2D NMR spectroscopy. 1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) 11.95 (s, 1H), 10.50 (t, 1H, J = 4.7 Hz), 6.18–6.17 (m, 1H), 5.78 (t, 1H, J = 1.0 Hz), 5.54 (q, 1H, J = 1.6, J = 1.5 Hz), 4.27 (t, 2H, J = 5.6 Hz), 3.60–3.54 (q, 2H, J = 5.67 Hz), 2.23 (s, 3H), 1.93 (s, 3H). 13C NMR (300 MHz, CDCl3, 25 °C): δ (ppm) 172.8, 167.3, 156.8, 154.5, 148.2, 136.1, 125.8, 106.7, 63.1, 38.7, 18.9, 18.3. FTR-IR (ATR): ν = 2939 (broad), 1697, 1651, 1581, 1519, 1450, 1411, 1327, 1249, 1165, 1011, 941, 763, 740. ESI–TOF–MS (m/z) calcd, 280.12; observed, 281.1024 (M + H+). The appropriate n-alkyl acrylates, BPAEDA and DMPA were added and dissolved in the DMSO. 1.5 wt % (by monomer weight) DMPA was used as the initiator concentration for all samples. Total monomer concentration was fixed at 30 wt % for all samples. The monomer solution was then heated to 120 °C until UPy(M)A was completely solubilized, and the warm monomer mixture was injected between two glass slides (75 × 50 mm) separated by a glass spacer, 1.2 mm thick. Polymerization was performed using a cross-linking chamber with five overhead 365 nm UV bulbs (UVP via Cole-Parmer) for 120 min. The resulting gel was dried under vacuum at 110 °C until a constant weight was reached (approximately 12 h). Shrinkage of approximately 70% during drying was observed. Samples were dried on PTFE sheets to allow shrinkage without tearing during solvent removal. Upon removal from the vacuum oven, the samples were stored in a desiccated container and removed only for testing. It is expected all DMSO as well as any unreacted n-alkyl acrylates are removed by drying. Gel fractions were determined (n = 3) for samples containing 10, 20, and 30 wt % of both UPyA and UPyMA by repeated (3×) swelling and deswelling in chloroform for 1 week. After the third swelling, samples were dried for 12 h in ambient conditions and 24 h under vacuum at 70 °C to remove chloroform.
Figure 2.

Schematic of the synthesis procedure. Top: Reaction utilized to synthesize ureidopyrimidine functionalized acrylate monomer (UPyA). 2-Isocyanatoethyl acrylate is allowed to react with 6-methylisocytosine in DMSO. Bottom: The synthesized UPy(M)A, “A”, (UPyA shown) was mixed with an n-alkyl acrylate, “B” (methyl acrylate shown), and “C” bisphenol A ethoxylate diacrylate and photopolymerized using a photoinitiator in DMSO.
Differential Scanning Calorimetry
DSC was performed on a Mettler Toledo DSC 1 with an intracooler option. Samples were heated from room temperature to 100 °C, cooled to −50 °C, and subsequently heated to 200 °C. Data shown are of only the second heating ramp. All heating and cooling rates were fixed at 10 °C/min. Tests were conducted in a nitrogen atmosphere.
Dynamic Mechanical Analysis
DMA was performed on a Mettler Toledo DMA 861e/SDTA. Samples were cut into squares approximately ~0.4 mm thick and ~2 mm in dimension. The mode of deformation was shear, and strain was limited to a maximum of 0.29%. Samples were tested between −50 and +150 °C at a heating rate of 2 °C/min. A multiplexed frequency mode was run on all samples in which 1, 2, 5, and 10 Hz deformation occurs simultaneously; the frequency of deformation shown is 5 Hz, unless otherwise stated. For higher frequency measurements, a second multiplexed frequency test was run, where the frequencies probed were 10, 20, 50, and 100 Hz. Tests were conducted in a nitrogen atmosphere.
Uniaxial Tensile Testing
Tensile testing was conducted to failure on select samples (n ≥ 3) using a Lloyd-Ametek LR5KPlus with a 100 N load cell. The grips used were TG-22 self-tightening roller grips also supplied by Lloyd-Ametek. Tests were conducted at 10 mm/min inside a Eurotherm Thermal Chamber at the peak of loss modulus (G″) as determined by DMA at 1 Hz. Strain was measured using a long-travel laser extensometer. All stresses and strains reported are engineering stresses and strains, and toughness was measured as the integrated area under the stress–strain curve.
Shape Memory Testing
Shape memory testing consisted of free-strain recovery and constrained recovery. All samples were programmed using the same temperature-strain profile. All samples were programmed by (1) heating the polymer to the second peak of tan δ (determined by DMA at 1 Hz) and straining the sample to 20% in compression, (2) maintaining the deformation while cooling the polymer to the first peak of tan δ, fixing shape 2, (3) further deforming the polymer to 40% in compression, and (4) maintaining the second deformation while cooling below both transitions, fixing shape 3. For free-strain recovery, samples were unloaded at this temperature and then heated at 2 °C/min. For constrained recovery, 40% deformation was maintained and heated at 2 °C/min. All shape memory testing was performed on a TA Instruments Q800 DMA in DMA Strain Rate mode on 5 mm-diamater, 2 mm-thick cylindrical samples.
RESULTS
The gel fractions of samples containing 10, 20, and 30 wt % of both UPyA and UpyMA are reported in Table 1. All networks tested showed gel fractions greater than 88%. UPyA containing samples possessed higher gel fractions than samples containing UPyMA at equal UPy(M)A concentration.
Table 1.
Gel Fraction with Increased UPy(M)A Concentration
| sample | 10% UPyA | 20% UPyA | 30% UPyA | 10% UPyMA | 20% UPyMA | 30% UPyMA |
|---|---|---|---|---|---|---|
| gel fraction | 0.920 ± 0.004 | 0.934 ± 0.006 | 0.977 ± 0.005 | 0.882 ± 0.002 | 0.907 ± 0.011 | 0.935 ± 0.007 |
The thermomechanical properties of each synthesized network were characterized through DMA and DSC. Select samples were then further characterized through uniaxial tensile testing and shape memory testing. In Figures 3a,b, a 1:1 ratio of the linear monomers MA and BA was maintained in order to elucidate the effect of increased supramolecular cross-links at constant covalent cross-link density on the dynamic mechanical properties of the polymer system. Shear storage modulus from DMA is presented for samples containing UPyA or UPyMA. In all compositions tested, two transitions are present; the first transition is accompanied by a drastic decrease in modulus, from approximately 700 to 1 MPa. The second transition is accompanied by a much smaller decrease in modulus, approximately 0.9 MPa, near 80 °C. The first transition is strongly dependent in both breadth and temperature on the composition of UPy(M)A. The second transition, however, is nearly constant in peak temperature. From Figure 3, three modulus “regions” can be defined. The first region occurs below the low temperature transition, and represents the glassy modulus; the second occurs between the two transitions and will be denoted the “middle modulus” (GM); the third plateau occurs above both transitions and the rubbery modulus (GR).
Figure 3.

Dynamic mechanical analysis demonstrates the effects of (a) increasing supramolecular cross-links via UPyA concentration (upper), (b) increasing supramolecular cross-links via UPyMA concentration (middle), and (c) increasing covalent cross-links via BPAEDA concentration (lower). The glass transition increases strongly with added supramolecular cross-links. The plateau modulus increases with both supramolecular and covalent cross-link. Rubbery modulus increases with covalent cross-linking.
The effect of increasing covalent cross-linking at constant UPyMA concentration is evaluated in Figure 3c. In these samples, increasing BPAEDA is matched by an increasing BA:MA ratio in order to maintain the separation of the two transitions. It can be seen that increasing the covalent or supramolecular cross-linker concentration leads to an increase in GM and Gr. Gr increases from 0.07 to 0.47 MPa with an increase in BPAEDA concentration from 2 to 30 wt %.
Figure 4 serves as a comparison between samples made with UPyA and UPyMA. Figure 4a shows the difference in properties associated with the acrylate and methacrylate functionalized analogues of UPy. For low concentrations of UPy, it should be noted that two transitions are present for both UPyA and UPyMA. As the concentration of supra-molecular cross-linker increases, UPyA maintains to a large degree the separation of the two transitions, but UPyMA does not. Figure 4b shows DSC thermograms of representative samples containing UPy. It should be noted that a glass transition can be distinguished for the 20% UPyA sample but not for the UPyMA sample. There are no other apparent thermal events present in the thermograms over the temperature range studied.
Figure 4.

Comparison of the thermomechanical properties of samples containing the methacrylated and acrylated analogues of UPy by dynamic mechanical analysis of samples containing 5, 20, and 40% UPy (upper, a) and differential scanning calorimetry (Exo up) of samples containing 20% UPy (lower, b). The glass transition for high UPy concentrations is lower for UPyA, causing it to be more easily distinguished from the UPy dissociation.
The tunability of Tg of the studied system is presented in Figure 5. Figure 5a shows the storage modulus as a function of temperature for polymers containing different n-alkyl acrylates. It should be noted that GM and Gr are largely unaffected by the change of linear comonomers. Increasing the length of the alkyl side chain on the acrylate decreases the glass transition from 60 to 0 °C as can be seen in Figure 5b. The shape of the loss factor (tan δ) curve is common among samples with the methyl acrylate sample showing a larger area under the first peak and less area under the second peak than other samples tested.
Figure 5.

Dynamic mechanical analysis showing the effect of different n-alkyl acrylate comonomers on the (a) storage modulus and (b) loss factor (tan δ) as a function of temperature. The glass transition is lowered with increasing alkyl side group length.
Figure 6 contains loss factor curves for a single representative sample, tested over two decades of frequency between −50 and +200 °C. The peaks associated with both transitions are shown to be frequency dependent. It should be noted that while the first peak varies with frequency by approximately 7 °C/decade of frequency, the second varies by 27 °C/decade of frequency. The quasi-static tensile response of the samples as a function of increased supramolecular cross-linker (UPyMA) concentration is shown in Figure 7a. Samples were tested at the lower temperature peak of loss modulus as determined by DMA at 1 Hz; the testing temperature for each sample is shown in Figure 7c. Strain-to-failure decreases with increased UPyMA while stress-at-failure increases. The toughness of the samples, measured as the integrated area under the engineering stress–strain curve is shown for each sample in Figure 7b. A peak in toughness is seen around 15% UPyMA.
Figure 6.

Dynamic mechanical analysis showing the effect of deformation frequency on the loss factor peaks associated with the two transitions. While both transitions are frequency dependent, the dissociation of UPy shows a much larger frequency response than does the glass transition.
Figure 7.

Quasi-static tensile testing-to-failure results for samples with increased supramolecular cross-linking. Failure strain decreases while failure stress increases with increased UPy content (upper, a). Toughness peaks around 15 wt % UPy. All tests were conducted at the peak of loss modulus from DMA (1 Hz) (middle, b). Peak of loss modulus as determined by DMA at 1 Hz for samples that were subsequently quasi-statically strained to failure (lower, c).
Figure 8 presents shape recovery behavior of samples containing UPyMA in both free-strain and constrained recovery conditions. All samples were programmed by (1) heating the polymer to the second peak of tan δ (determined by DMA at 1 Hz) and straining the sample to 20% in compression, (2) maintaining the deformation while cooling the polymer to the first peak of tan δ, fixing shape 2, (3) further deforming the polymer to 40% in compression and (4) maintaining the second deformation while cooling below both transitions, fixing shape 3. In Figure 8a, each sample was released and allowed to recover without an applied load while heating. With increased UPyMA concentration, the onset of recovery occurs at higher temperatures and the separation of the two recovery events decreases. Final recovery exceeded 95% of the induced deformation for all samples tested. Figure 8b shows the stress generated through sample recovery for a sample containing 10% UPyMA. The peak stress generated is 0.81 MPa and occurs near physiological temperature for the selected system. An effort was made to demonstrate the versatility of the fabricated TSMP system for fixing arbitrary deformations, as compared to the uniaxial triple-shape memory effect that was quantitatively described in Figure 8, Figure 9 shows the recovery of a sample that was twisted and textured on half the sample with a clamp at 90 °C, cooled to 37 °C untwisted and bent and subsequently cooled to 20 °C,
Figure 8.

Recovery behavior of samples with between 5 and 20 wt % UPyMA: (a) free-strain recovery and (b) constrained recovery of a sample with 10% UPyMA. Samples were heated to 130 °C, strained to 20% in compression, cooled to Tg, further strained to 40% of the original length, and cooled to −20 °C while maintaining the applied deformation.
Figure 9.

Sequential photographs taken of the recovery of a sample containing 10% UPyMA demonstrating the triple-shape memory effect. Recovery proceeded from the first temporary shape (bent and textured) (a) to the second temporary shape (twisted and textured) (b) and to the permanent shape (flat and smooth) (c).
DISCUSSION
Demonstration of a tunable SMP (or TSMP) system requires wide and, where possible, independent control of several key properties, namely the activation temperature(s), determined by the temperature of the transition(s), and recovery characteristics, determined by the cross-link density(ies). This work attempts to demonstrate wide and independent control of the glass transition and covalent cross-link density in a system where a supramolecular network is formed by self-complementary hydrogen bonding. The specific effects of supra-molecular cross-links on the thermomechanical properties of a covalently cross-linked system are investigated in quasi-static and dynamic regimes to understand the interplay between covalent and supramolecular network phenomena.
Figure 3 demonstrates control over the supramolecular and covalent cross-link densities in a system where the glass transition and UPy dissociation occur between 0 and 100 °C, which is across the useful range for many applications. The acrylated and methacrylated analogues of UPy introduce thermoreversible cross-links into the covalent network. As shown in Table 1, Gel fraction was above 88% for all measured samples, indicating a high level of network formation. Reactivity of the n-alkyl acrylates is likely to be closer to UPyA than to UPyMA leading to slightly better network incorporation for UPyA. Increasing UPy content also led to more complete network formation. The effect of the presence of UPy on polymerization and termination events is beyond the scope of this work, but should be considered in future work. This is demonstrated by the presence of a transition above the glass transition. Figure 4b and Figure 6 demonstrate that this UPy-mediated transition is both frequency dependent and not associated with a distinct enthalpic event, indicating the second transition is not crystalline in nature. Increasing UPy(M)A or BPAEDA concentration increases GM, due to the increase of total cross-link density. Quantifying the increase in GM proves difficult beyond 10 wt % UPy(M)A, as the glass transition increases in temperature and broadens significantly, causing the two transitions to overlap.
In order to study independent glass and UPy dissociation transitions, UPyA was synthesized and characterized. The temperature at which the UPy dissociation transition occurs is unaffected by the change from methacrylate to acrylate, but the Tg does not increase as rapidly with UPyA, thereby allowing for the two transitions to remain separated for compositions with less than 20 wt % UPyA. Figure 4 directly compares samples made with UPyA and UPyMA. The glass transition associated with UPyA-containing samples is consistently lower than that for equivalent concentrations of UPyMA, which allows the two transitions to be distinguished by DMA. The cost and lack of commercial availability of 2-isocyanatoethyl acrylate as compared to 2-isocyanatoethyl methacrylate, however, led to the selection of UPyMA for further synthesis and thermo-mechanical testing. The 10 wt % UPyMA sample was selected as a test composition to study the effect of covalent cross-link density. Increasing the covalent cross-link density at constant supramolecular cross-link density and approximately constant Tg was achieved by varying the MA:BA ratio, to decrease proportionally with the increase in BPAEDA concentration (Figure 3c).
Figure 5 demonstrates the utility of the selected n-alkyl acrylate system for changing the first activation temperature of the TSMP. By varying the length of the alkyl side group from 1 to 4 carbons, the glass transition is tuned over a range of 60 °C at an approximately constant GM and GR. While this study was limited to 4 commercial alkyl acrylate comonomers, the authors recognize the potential of this system for further tuning, by utilizing the plethora of available (meth)acrylate monomers. The same MA-BA system characterized in Figure 3b was selected for quasi-static tensile testing at the first peak of loss modulus from DMA at 1 Hz (Figure 7). As the UPyMA concentration increases, stress-at-failure increases while strain-to-failure decreases. This behavior is consistent with the view of the supramolecular hydrogen bonding serving as cross-links. A peak in toughness is seen around 15 wt % UPyMA. It is proposed that this sharp increase in toughness is due to the increasing likelihood of the hydrogen bonds to break and reform, thus dissipating energy as the temperature increases and the concentration of the hydrogen bonding moiety increases. This effect is difficult to isolate, as testing was carried out at a relative temperature to the first transition, but the testing temperature increased relative to the second transition, with increasing UPyMA. The peak of loss modulus increases dramatically between 10 and 20 wt %, as shown in Figure 7c, thus allowing for significant toughening from the hydrogen bonding. Beyond 20 wt % the toughness decreases as strain-to-failure is severely limited by the increased supra-molecular interactions without a concomitant increase in stress-at-failure.
The dynamic nature of the supramolecular networks is probed further in Figure 6 by DMA at frequencies between 1 and 100 Hz. The UPy dissociation transition shifts ~4× further along the temperature scale than does the glass transition, as a function of frequency. This can be attributed to the constant breaking and reforming of the hydrogen bonds along the UPy moieties. This strong frequency dependence elucidates the truly dynamic nature of the supramolecular network, which must persist in order to serve as a cross-linking phase. It is important to consider that shape recovery exists in the quasi-static domain and that the persistence of a cross-link must be evaluated under similar deformation conditions. The free-strain recovery of selected systems shown in Figure 8a demonstrates the importance of the differences between quasi-static shape recovery and dynamic mechanical deformation. Samples containing more than 10 wt % UPyMA do not show two distinct transitions but instead undergo a single broad recovery. Interestingly, this transition in properties occurs in the same composition range as the abrupt change in toughness shown in Figure 7b. Since both changes in properties can be attributed to the dynamic nature of the supramolecular cross-links, we propose a common mechanism for both properties. Segmental motion begins at a much higher temperature with increasing UPy, and the likelihood of a UPy molecule being able to disassociate and find a different UPy moiety to bond with increases with increasing UPy. The supramolecular network does not persist under quasi-static conditions for compositions of UPy beyond 10 wt % for the studied system. Despite this lack of an effective supramolecular network for high compositions of UPy, triple shape properties can be clearly distinguished for the 5% and 10% samples. Stress recovery for the 10% UPyMA sample under constraint shows a single recovery peak, as is commonly seen for SMPs strained near the Tg.
The described material system has several characteristics that are expected to influence future work in this area. The highly dynamic nature of the supramolecular network is the most defining of these characteristics. This leads to poor shape fidelity above the glass transition, which at high UPy concentrations can lead to a loss in high quality triple shape properties. Despite these limitations, enabling large-scale shape recovery while maintaining a deformation that was induced postpolymerization, such as a surface texturing, is demonstrated Figure 9. This supramolecular network may also show interesting cyclic properties, due to its ability to sustain nonpermanent damage, and should be investigated in future work. Another major hurdle this system must face is a current lack of processability. Although a one-pot synthetic procedure was developed for this work, any device would be subjected to the constraints of the polymers’ thermoset nature and the fact that the materials must be made as a gel. This significantly hinders processing for geometries that are not sheets or films, as the materials cannot be melt-processed and large cross sections complicate the removal of solvent and lead to warping. Although challenges and further investigation remain, a systematic approach of optimizing thermomechanical programming and creative synthetic approaches would enable a highly tunable TSMP system based on widely available (meth)acrylate monomers and synthetically available supramolecular cross-links.
CONCLUSIONS
A system of acrylic triple shape memory polymers that utilizes the glass transition and the dissociation of self-complementary hydrogen bonding moieties has been synthesized and characterized. A one-pot synthetic procedure was developed in which a (meth)acrylated 2-ureido-4[1H]pyrimidone monomer was first synthesized and then subsequently copolymerized with desired ratios of n-alkyl acrylates and a bisphenol A-based diacrylate cross-linker. The resulting samples had both supramolecular and covalent network properties. The glass transition was varied between 0 and 60 °C, and covalent cross-link density was varied quasi-independently from the glass transition. The effect of increased supramolecular cross-linking on the thermomechanical properties of the polymer networks was also demonstrated, with results providing insights into the dynamics of UPy supramolecular cross-linking. Finally, triple-shape capability was shown in an amorphous monophasic polymer system. Accurate control of the transitions and thermomechanics of TSMPs make them candidate materials for specialty applications that demand multiple high-strain shape changes after deployment without intermediate mechanical retraining.
Acknowledgments
The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry (K.L.W.), Grant No. A-0001. This work was partially supported by the National Institutes of Health/National Institute of Biomedical Imaging and Bioengineering, Grant R01EB000462. This material is based also partially based upon work supported by a National Science Foundation Graduate Research Fellowship under Grant 2011113646.
References
- 1.Lendlein A, Langer R. Science. 2002;296(5573):1673–1676. doi: 10.1126/science.1066102. [DOI] [PubMed] [Google Scholar]
- 2.Feninat FE, Laroche G, Fiset M, Mantovani D. Adv Eng Mater. 2002;4(3):91–104. [Google Scholar]
- 3.Mather P, Luo X, Rousseau I. Annu Rev Mater Res. 2009;39:445–471. [Google Scholar]
- 4.Lendlein A, Jiang H, Junger O, Langer R. Nature. 2005;434(7035):879–882. doi: 10.1038/nature03496. [DOI] [PubMed] [Google Scholar]
- 5.Liu C, Qin H, Mather PT. J Mater Chem. 2007;17:1543–1558. [Google Scholar]
- 6.Li J, Viveros J, Wrue M, Anthamatten M. Adv Mater. 2007;19(19):2851. [Google Scholar]
- 7.Tobushi H, Hara H, Yamada E, Hayashi S. Smart Mater Struct. 1996;5:483–491. [Google Scholar]
- 8.Voit W, Ware T, Gall K. Polymer. 2010;51(15):3551–3559. [Google Scholar]
- 9.Ware T, Voit W, Gall K. Radiat Phys Chem. 2010;79(4):446–453. [Google Scholar]
- 10.Hearon K, Gall K, Ware T, Maitland DJ, Bearinger JP, Wilson TS. J Appl Polym Sci. 2011;121(1):144–153. doi: 10.1002/app.33428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voit W, Ware T, Dasari RR, Smith P, Danz L, Simon D, Barlow S, Marder SR, Gall K. Adv Funct Mater. 2010;20(1):162–171. [Google Scholar]
- 12.Gall K, Yakacki CM, Liu Y, Shandas R, Willett N, Anseth KS. J Biomed Mater Res, Part A. 2005;73A(3):339–348. doi: 10.1002/jbm.a.30296. [DOI] [PubMed] [Google Scholar]
- 13.Baer G, Wilson TS, Matthews DL, Maitland DJ. J Appl Polym Sci. 2007;103(6):3882–3892. [Google Scholar]
- 14.Yakacki CM, Shandas R, Lanning C, Rech B, Eckstein A, Gall K. Biomaterials. 2007;28(14):2255–2263. doi: 10.1016/j.biomaterials.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small IVW, Singhal P, Wilson TS, Maitland DJ. J Mater Chem. 2010;20(17):3356–3366. doi: 10.1039/B923717H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behl M, Lendlein A. J Mater Chem. 2010;20(17):3335–3345. [Google Scholar]
- 17.Bellin I, Kelch S, Langer R, Lendlein A. Proc Natl Acad Sci USA. 2006;103(48):18043–18047. doi: 10.1073/pnas.0608586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu G, Ding X, Cao Y, Zheng Z, Peng Y. Macromol Rapid Commun. 2005;26(8):649–652. [Google Scholar]
- 19.Xie T. Nature. 2010;464(7286):267–270. doi: 10.1038/nature08863. [DOI] [PubMed] [Google Scholar]
- 20.Luo X, Mather PT. Adv Funct Mater. 2010;20(16):2649–2656. [Google Scholar]
- 21.Li J, Xie T. Macromolecules. 2010;44(1):175–180. [Google Scholar]
- 22.Lehn JM. Polym Int. 2002;51(10):825–839. [Google Scholar]
- 23.Sijbesma RP, Beijer FH, Brunsveld L, Folmer BJB, Hirschberg JHKK, Lange RFM, Lowe JKL, Meijer EW. Science. 1997;278(5343):1601–1604. doi: 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- 24.Bergman S, Wudl F. J Mater Chem. 2008;18(1):41–62. [Google Scholar]
- 25.Wietor JL, Dimopoulos A, Govaert LE, van Benthem RATM, de With G, Sijbesma RP. Macromolecules. 2009;42(17):6640–6646. [Google Scholar]
- 26.Berl V, Schmutz M, Krische MJ, Khoury RG, Lehn JM. Chem— Eur J. 2002;8(5):1227–1244. doi: 10.1002/1521-3765(20020301)8:5<1227::aid-chem1227>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Kushner A, Gabuchian V, Johnson E, Guan Z. J Am Chem Soc. 2007;129(46):14110–14111. doi: 10.1021/ja0742176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Hu J, Liu Y. Eur Phys J E: Soft Matter Biol Phys. 2009;28(1):3–10. doi: 10.1140/epje/i2008-10395-2. [DOI] [PubMed] [Google Scholar]
- 29.Yamauchi K, Lizotte J, Long T. Macromolecules. 2003;36(4):1083–1088. [Google Scholar]