

Abstract
A current hypothesis for the pathology of Alzheimer’s disease (AD) proposes that amyloid-β (Aβ) peptides induce uncontrolled, neurotoxic ion flux across cellular membranes. The mechanism of ion flux is not fully understood because no experiment-based Aβ channel structures at atomic resolution are currently available (only a few polymorphic states have been predicted by computational models). Structural models and experimental evidence lend support to the view that the Aβ channel is an assembly of loosely associated mobile β-sheet subunits. Here, using planar lipid bilayers and molecular dynamics (MD) simulations, we show that amino acid substitutions can be used to infer which residues are essential for channel structure. We created two Aβ1–42 peptides with point mutations: F19P and F20C. The substitution of Phe19 with Pro inhibited channel conductance. MD simulation suggests a collapsed pore of F19P channels at the lower bilayer leaflet. The kinks at the Pro residues in the pore-lining β-strands induce blockage of the solvated pore by the N-termini of the chains. The cysteine mutant is capable of forming channels, and the conductance behavior of F20C channels is similar to that of the wild type. Overall, the mutational analysis of the channel activity performed in this work tests the proposition that the channels consist of a β-sheet rich organization, with the charged/polar central strand containing the mutation sites lining the pore, and the C-terminal strands facing the hydrophobic lipid tails. A detailed understanding of channel formation and its structure should aid studies of drug design aiming to control unregulated Aβ-dependent ion fluxes.
The increase in life expectancy in the modern era has increased the population subject to neurodegenerative diseases such as Alzheimer’s disease (AD), occurring late in life.1−3 AD is characterized clinically by memory loss and pathologically by the presence of intracellular neurofibrillary tangles and extracellular senile plaques. These plaques are insoluble amyloid deposits composed primarily of aggregates of amyloid-β (Aβ) in their fibril form. Although Aβ is found in large fibrils in the brain, the mechanism by which Aβ causes neurotoxicity is not fully understood. Fibrils are no longer considered the main toxic agent in AD; rather, Aβ oligomers have been shown to be most damaging to cells.4−11 The unstable nature of oligomeric structures complicates efforts aimed at characterizing the Aβ toxic species.12−15
To be cytotoxic, Aβ aggregates must interact with the cell surface by either a receptor or the membrane itself.1,3,12,15−19 The channel hypothesis suggests that Aβ is cytotoxic because of its ability to form ion channel-like pores, inducing an unregulated ionic flux across cellular membranes.10,12,20−31 The ionic fluxes produced by Aβ create a state of calcium dysregulation, leading to cell death. Understanding the molecular mechanics by which Aβ induces unregulated ionic fluxes has become crucial to AD pathology. Cellular membranes are very complex, involving many variables that are difficult to isolate and control. Consequently, to isolate and study the Aβ-dependent ionic fluxes across a controlled membrane, we use model bilayers or the planar lipid bilayer (PLB) technique. Both Aβ1–40 and Aβ1–42 peptides have been shown to exhibit channel-like activity in PLBs. This activity shows a wide range of heterogeneous conductances. Ionic fluxes resulting from Aβ channels have been shown to be inhibited by metal ions such as Zn2+.27,29,32
Polymorphism in the Aβ channel structure makes it difficult to examine the relative role of specific amino acids in channel structure and activity. The Aβ peptide has several point mutations occurring naturally, and some are related to familial forms of AD. Their mutation sites are particularly clustered at Aβ peptide positions 22 and 23: Arctic (E22G),33,34 Dutch (E22Q),35 Italian (E22K),36 and Iowa (D23N).37 These mutants have a strong tendency to form fibrillar aggregates on a lipid membrane38 and are associated with cerebral amyloid angiopathy and AD.39,40 Unlike the disease-related Aβ mutants, an artificially designed substitution of Phe19 with Pro (F19P) in the Aβ17–42 (p3) channel prevented bilayer channel activity and cellular toxicity.28 Proline has been defined as a β-sheet breaker on the basis of the U-shaped p3 structural information with the β-strand–turn−β-strand motif.41 In the pore-lining region of p3, the Phe19 side chains are π-stacked between two β-sheets, holding tightly the U shape morphology. However, the kink produced by Pro19 introduces into each peptide an unbalanced force on the pore-lining β-strands that move toward the interior pore and consequently inhibit ions crossing through the pore. The nonconductive bilayer behavior seen in electrophysiological recordings and low fluorescence intensities in cell calcium imaging suggest that the p3-F19P mutant is a nontoxic species.
This work focuses on testing the activity of Aβ1–42 models, as a growing body of evidence points toward oligomers rich in β-sheet structure as the species toxic to neurons. Structural models of Aβ channels allow the design of experiments in which Aβ1–42 amino acid substitutions can be used to interrogate these models,28,42−46 which in turn provides further insight for optimizing the models. Here we examined non-natural Aβ1–42 amino acid substitutions for their Aβ channel membrane activity to gain information about the structural requirements for Aβ channel formation and structure. The results gathered are used to refine the structure(s) of the Aβ pore. The ultimate goal is to understand which residues are necessary structurally and which line the pore and whether those residues are water accessible. This work presents the conductance behavior of two mutants, F19P and F20C, and its implications in the first step of such a study. Structural information relating to the membrane is needed to map the channel structure(s) and aid drug design, with the aim of controlling unregulated Aβ ion fluxes.
Materials and Methods
Materials
We obtained wild-type Aβ1–42 and F19P and F20C mutants from Bachem Inc. The phospholipids 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids. All other chemicals were purchased from Sigma-Aldrich.
Formation of Planar Lipid Bilayers
We prepared planar lipid bilayers by using the so-called “folded technique”, which employs apposition of lipid monolayers.31,47−49 We used a Teflon film with a hole diameter of 120 μm. We pretreated the Teflon film with hexadecane in pentane. Then using high-vacuum grease (Corning), we secured the film to a Teflon chamber separating two 4 mL bath solutions. As the electrolyte, we used 150 mM KCl, 10 mM Hepes (pH 7.4), and 1 mM MgCl2. All bilayers were prepared with a 1:1 (w/w) mixture of DOPS and DOPE lipids at a concentration of 20–25 mg/mL in pentane. This phospholipid solution was added to each chamber. We formed the bilayer by raising the buffer in each chamber until the hole on the Teflon film was completely submerged in the electrolyte buffer. Lipid monolayers in each chamber came into contact over the hole to form the lipid bilayer.31,47
Planar Lipid Bilayer Recordings
We measured the current using Ag/AgCl electrode pellets placed in each compartment of the PLB chamber. Before performing electrical recordings, we verified that the bilayer was stable for several minutes and that the system capacitance was >70 pF. When both criteria were met, we added the specific Aβ1–42 peptide to the cis (hot wire) chamber and stirred the contents for a few minutes and if no activity was present every 15–30 min. Frozen aliquots of 20 μg of each Aβ1–42 peptide (wild type, F19P, or F20C) at 0.5 or 1 mg/mL in water were used. Aliquots were thawed only once. Aβ peptide concentrations in the bilayer chamber ranged from 0.5 to 18 μM. We recorded all current traces using the voltage clamp mode. We used the amplifiers built-in filter cutoff set at 2 or 3 kHz for BC535 or EPC-7, respectively. All current recordings were acquired at a sampling frequency of 15 kHz. We used a custom-made LabVIEW program to record the current traces and Clampfit 10.2 to analyze current versus time traces.31,42,47,49 For representation in figures, we filtered the recorded current versus time traces with a digital Gaussian low-pass filter with a cutoff frequency of 50 Hz unless noted otherwise.
Membrane Stability
Before performing the experiments with Aβ peptides in folded bilayers, we determined that the lipids used [1:1 (w/w) DOPS/DOPE] were stable over long periods of time. We chose DOPS/DOPE bilayers because Aβ requires a negatively charged membrane. Both PE and PS are enriched in brain lipids, and in previous studies, this membrane composition was shown to be stable. We performed seven PLB experiments using the folded technique in which no peptide was added. These experiments were conducted for up to 4 h. Periodic capacitance measurements monitored membrane quality and stability. We found that in these seven experiments, the average bilayer conductance (in the ±100 mV range) was 0.86 ± 0.40 pS (n = 7). The lowest conductance we obtained was 0.32 pS and the highest 1.36 pS. These conductances corresponded to membrane resistances of 3125 and 735 GΩ, respectively. By verifying the stability of our membranes prior to addition of the Aβ peptide, we ensured that the values measured for the membranes fell within the range measured in these control experiments. The stability of all seven peptide-free DOPS/DOPE bilayers exceeded 4 h. After data had been recorded for 4 h, the bilayers were further examined for integrity by adding, to both sides of the bilayer, gramicidin A (gA), a peptide known to form well-defined ion channels. As expected, we obtained gA channel activity, further demonstrating that DOPS/DOPE bilayers were stable after 4 h.
Molecular Dynamics (MD) Simulations
Aβ1–42 barrels were simulated using two U-shaped monomer conformations: Aβ1–42 as defined in the pentamer on the basis of hydrogen–deuterium exchange NMR data, side chain packing constraints from pairwise mutagenesis, solid-state NMR, and EM [Protein Data Bank (PDB) entry 2BEG]41 and Aβ1–40 based on the solid-state NMR model of small protofibrils.50 However, both conformers miss the N-terminal coordinates due to conformational disorder. We used the N-terminal coordinates obtained from the solution NMR structure of Aβ1–16; however, removing the Zn2+ (PDB entry 1ZE7).51 This structure was used to fill in the missing N-terminal portion of the peptides. For each combination of the N-terminal structure with the U-shaped motifs, two Aβ1–42 conformers were generated. Conformer 1 has a turn at Ser26–Ile31, and conformer 2 has a turn at Asp23–Gly29. In the latter conformer, two C-terminal residues, Ile41 and Ala42, were added to create Aβ1–42.
Two mutant coordinates were obtained by replacing Phe19 with Pro19 (F19P) and Phe20 with Cys20 (F20C). Thus, six Aβ1–42 monomers, including wild type, F19P, and F20C for each conformer, were used to construct the β-barrel structure. These Aβ conformers were inclined ∼37° relative to the pore axis44 and then rotated 18 times with respect to the pore axis creating Aβ barrels. A unit cell containing two layers of lipids was constructed. The Aβ barrels were embedded in an anionic lipid bilayer containing DOPS and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) (1:2 DOPS:POPE molar ratio). For the bilayer construction, we closely follow previous β-sheet channel simulations.13,14,28,42−44,52,53 The anionic lipid bilayer containing a total of 420 lipids constitutes the unit cell with TIP3P waters, added at both sides. The system contains Mg2+, K+, Ca2+, and Zn2+ at the same concentration (25 mM) to satisfy a total cation concentration near 100 mM. CHARMM54 was used to construct the set of starting points and to relax the systems to a production-ready stage. For production runs, the NAMD code55 on a Biowulf cluster (http://biowulf.nih.gov) at the National Institutes of Health was used for the starting point with the same CHARMM27 force field. Averages were taken after 20 ns discarding initial transients. Analysis was performed with the CHARMM programming package.54
Results and Discussion
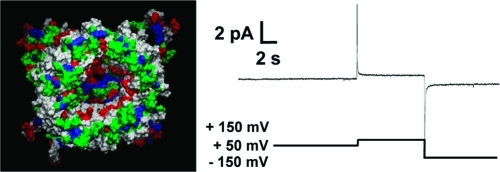
The “Aβ ion channel hypothesis” suggests that Aβ forms ion channel-like pores in lipid membranes causing transmembrane currents via Aβ ion channels.10,15,22,24−27,31,56−58 Upon addition of the Aβ peptide to one side of the PLB chamber, Aβ must first bind to the bilayer and then likely undergo a conformational change that helps the Aβ peptide overcome the barrier to insert or slip into the bilayer. Once in the bilayer, monomers or oligomers need to interact with one another to form a porelike structure. There are three different types of ion channel activity that have been described for Aβ channels.22 The first type of channel activity is the “bursting” fast cation channel that, as its name implies, is a short burst of activity that gives a nonlinear current–voltage relationship. The second type of channel activity is the so-called “spiky” fast cation channel that is similar to a burst of activity; however, the spikes are short-lived compared to bursting activity. Lastly, the third type of channel activity is the “step” or “steplike” activity. With the steplike behavior, a clear, defined jump in current is seen as a channel opens and closes. We observed these types of current activity for wild-type Aβ1–42 in the DOPS/DOPE lipid bilayers. Figure 1A illustrates bursting and spiky behavior of the wild-type Aβ1–42 channels. At −80 mV, these channels began to exhibit the so-called “bursting fast cation channel”. The only truly apparent difference between bursting and spiky behavior is that the bursting channel activity is characterized by the absence of the long closures of channels.22 As we increased the applied potential from −80 to −50 mV in 10 mV steps, the magnitude of the current decreased, as well. The inset of Figure 1A shows a higher time resolution of the steplike activity seen with Aβ1–42 channels. These steps are much less frequent in folded bilayers than in painted bilayers.
Figure 1.

Current vs time trace of wild-type Aβ1–42 showing channel-like activity. (A) The channel activity shown here depicts the more frequently seen spikes and bursts for Aβ1–42 in the folded bilayers used. The inset, indicated by the red bar, shows steplike activity at a higher time resolution. (B) The Aβ1–42 channel is inhibited by Zn2+. Two continuous current vs time traces totaling 30 min of recording with 4.5 μM wild-type Aβ1–42. We added Zn2+ to final concentration of 2 mM and stirred the mixture. The activity was not immediately inhibited and decreased gradually. The voltage vs time plot shown below follows the changes in applied potential to the current vs time trace above. The vertical line marked with the letter C indicates a capacitance measurement during the recording. The electrolyte contained 150 mM KCl, 10 mM Hepes (pH 7.4), and 1 mM MgCl2. The bilayer was made by the folded technique, using a 1:1 (w/w) DOPS/DOPE lipid solution in pentane. Peptide was added to the cis side (hot wire), while the trans side was the virtual ground.
Figure 1B depicts 30 min of recording and the ability of zinc to block a portion of the Aβ1–42 activity as previously shown.27,31,32,60 Zn2+ appears to inhibit Aβ1–42 in two sites; it binds to the N-terminal histidines (His6, His13, and His14) on the peptide,51 inducing a conformational change, and it has been shown also to inhibit the current activity induced by the Aβ17–42 fragment (p3), a fragment without histidines.28 The arrow in Figure 1B points to the time at which zinc was added to the same chamber. The channels are not immediately blocked by zinc (Figure 1B), yet as the experiment progressed, we observed decreased conductances.
The electrical recordings show the ionic current with multiple conductances; this heterogeneous nature is typical of Aβ ion channels and other channel-forming amyloids. One possible explanation for this behavior is that the channels consist of distinct oligomeric species, forming channel-like structures.10,28,45,61 Given the different conductances of the Aβ channels, this could explain the variance in conductance measured in the electrical recordings. In 13 experiments with wild-type Aβ1–42, we measured channel activity in six cases, making the frequency of channel activity 46%, which is comparable to reports from previous work with Aβ1–42.27,42,62,63 Interestingly, we also found that different lots of Aβ1–42 differentially affected the percentage of channel activity observed. This suggests that the aggregation state of Aβ1–42 affects channel activity. Our results show that Aβ1–42 activity occurred at concentrations as low as 0.5–1 μM. We did not exceed concentrations of 4.5 μM with Aβ1–42. Channel-like behavior was never observed in bilayers prior to the addition of the peptide, or without its addition.
Although work showing channel activity of Aβ1–42 has previously been done,23,27,31,42,62,63 one prerequisite for this work was to characterize Aβ1–42 activity in our membrane/buffer system. We also needed a reference to which we could compare our results with those of the Aβ1–42 mutants. To the best of our knowledge, this is the second report showing Aβ channel-like activity with folded bilayers,31,42 and only one report exists using the tip-dip method.20 Most groups studying Aβ ion channel formation in model membranes used painted bilayers.10,22,23,26,27,63 Generally, solvent free bilayers (folded and tip-dip) are considered better models of bilayers, because the bilayers are thinner and contain smaller amounts of nonbiological solvents in the hydrophobic core. We found that Aβ1–42 in folded bilayers generally exhibited more spiky and bursting types of activity (short-lived) than steplike activity. The combined results of the membrane-only experiments and Aβ1–42 activity in DOPS/DOPE folded bilayers validate a system for testing the activity of Aβ1–42 mutants. To gather structural information about Aβ ion channels, we studied amino acid substitutions of Aβ1–42. By studying these mutants, we aim to infer structural features of Aβ and its function in membranes. This functional approach provides structural information that cannot be elucidated by other techniques.
The F19P Mutant Does Not Exhibit Channel-like Activity
The amino acid proline (Pro or P) is under-represented in β-sheets of proteins with known structure.64 Thermodynamic studies of amino acid replacements identify proline as the amino acid least compatible with β-sheet structure.64−66 Consequently, proline mutagenesis of Aβ has been intensively studied, specifically the ability of these types of mutants to form fibrils.64−66 Proline is energetically unfavorable in the extended cross-β-sheet structure and, as a result, inhibits amyloid aggregation.64−66 The substitution with proline introduces a “kink” into the strands of the U-shaped peptide. In previous work, we proposed that the Aβ17–42 fragment (p3) with the F19P substitution (p3-F19P) exhibited no channel activity.28 We show here that Aβ1–42 F19P exhibits no channel activity, suggesting that the Pro substitution at this position hinders the formation of Aβ-conducting structures. Figure 2 shows a current versus time trace of the nonconductive F19P mutant. At voltages as high as ±150 mV, there was still no visible conductance from channel formation (see the inset of Figure 2).
Figure 2.

Representative current vs time trace of Aβ1–42 F19P. (A) The trace shows F19P at 4.5 μM with no activity. The inset shows the lack of activity for applied voltages as high as ±150 mV. These 4 h bilayer experiments were repeated 10 times with no channel-like activity observed. (B and C) The Aβ1–42 F19P mutant may form collapsed channels. Current vs time trace showing the low-conductance, steplike activity for F19P at 4.5 μM. The calculated conductance for all steps shown was (B) ∼4.6 pS at an applied voltage of 50 mV and (C) ∼2.2 pS at an applied voltage of −50 mV. Note that this is the only activity observed after we had recorded data for >40 h. Current traces shown in panels B and C were filtered at 10 Hz and could barely be seen otherwise. The electrolyte contained 150 mM KCl, 10 mM Hepes (pH 7.4), and 1 mM MgCl2. The bilayer consisted of a 1:1 (w/w) DOPS/DOPE mixture and was made by the folding technique.
We conducted these experiments for up to 4 h and then verified again the integrity of bilayers by adding gramicidin A, a well-established pore former.49 F19P was tested in folded bilayers (n = 10) at concentrations ranging from 4.5 to 13.5 μM. After data had been recorded for more than 40 h (n = 10, 4 h each) with F19P, we observed only 100 s of channel-like activity with very low conductance. The activities are presented in Figure 2B,C. The pores shown in Figure 2B,C have conductances of 4.6 and 2.2 pS (below the level for gA in this membrane/electrolyte system), compared to the wide range of higher conductances for wild-type Aβ1–42, generally between 50 pS and 1 nS (Figure 1). F19P was also tested in DOPS/POPE painted bilayers [n = 7 (data not shown)] and showed no sign of instability for extended periods of time, with an average of 104 ± 40 min. In these painted bilayers, the lowest recorded point of bilayer instability was 75 min and the highest 190 min. This is comparable to the normal life span of these bilayers without any addition of peptide. However, the unusually low and unique conductance measurements (Figure 2) might suggest that either F19P forms collapsed pores or this mutant is impaired in its binding and/or insertion into bilayers. The most rigorous interpretation of the F19P results is that F19P does not form conducting structures in the bilayers, which suggests that the β-sheet structure of the U-turn is necessary for Aβ1–42 pore formation (see below).
The F20C Mutant Behaves Like the Wild Type in Lipid Bilayers
For the Aβ1–42 F20C mutant, we substituted Phe20 with Cys. To the best of our knowledge, the membrane behavior of the F20C mutant has not been described previously. We found that the F20C mutant conducts in a manner somewhat comparable to that of the wild type, in good agreement with the model presented here (see below). We performed seven experiments with F20C and observed activity in four (57%). In all experiments, we added F20C directly to one side of the PLB chamber. We initially added the mutant to a final concentration of 4.5 μM and if needed added additional 4.5 μM mutant every 45 min until a final concentration of 13.5 μM was reached. Under these conditions, F20C activity appears mostly as spikes and bursts and occasionally as short-lived steplike activity. Spiky and bursting activities are shown in the Supporting Information.
Figure 3 presents a 1 h current versus time trace recording of a continuous activity by F20C. During this time, we added Zn2+ ions and observed inhibition of this activity. For the sake of simplicity, the figure is presented in 15 min panels. Figure 3A presents a current trace that can be described in four smaller parts that closely follow the voltage plot under the trace. Initially, (i) the applied voltage is −50 mV showing channel-like activity, followed by (ii) a gradual decrease in the applied potential. Once there is no current (at −1.5 mV), (iii) the voltage is set to zero and sustained at this level. Finally, (iv) the applied voltage is increased to 30 mV. In Figure 3B, we added 0.5 mM Zn2+ to the same side of F20C. Following the addition of Zn2+, the activity begins to subside, although not immediately. In the next 30 min (Figure 3C,D), the amplitude and frequency of membrane conductance decrease and ultimately appear to be inhibited by Zn2+. This result shows that F20C activity is sensitive to Zn2+ ions. The replacement of Phe20 with Cys in Aβ1–42 does not preclude channel formation. This region of the Aβ peptide is central to its ability to form fibrils. We observed precipitates in Aβ1–42 F20C residual aliquots. In fact, a scanning cysteine mutagenesis study of Aβ1–40 found that the F20C mutant was accessible to alkylation in the fibril state, indicating that F20 is exposed to the solvent in fibers.67
Figure 3.

Current vs time trace of channel-like activity of Aβ1–42 F20C. Channels formed by F20C predominantly exhibited spiky and bursting activity. The Aβ1–42 F20C mutant channel-like activity is inhibited by Zn2+ ions. (A) Channel-like activity of Aβ1–42 F20C shows large sustained bursts and spikes with fast openings and closings. This panel shows changes in voltage as indicated by the voltage plot below the current trace. Starting at −50 mV, we gradually reduced the applied potential first to −1.5 mV and then to 0 mV followed by an increase to 30 mV. (B) We added Zn2+ to a final concentration of 0.5 mM and stirred the mixture. The activity decreased gradually as shown in panels C and D. (C) We changed the voltage bias to −50 mV, and after an additional 15 min (D), we changed the applied potential to 50 mV. At this point, the channel activity is mostly inhibited with occasional events. The figure shows four continuous 15 min traces totaling 1 h of continuous recording. The vertical lines marked with C in panels A, B, and D indicate a capacitance measurement during the recording. The electrolyte contained 150 mM KCl, 10 mM Hepes (pH 7.4), and 1 mM MgCl2. The bilayer consisted of a 1:1 (w/w) DOPS/DOPE mixture. The Aβ1–42 F20C concentration was 9 μM.
Both F19P and F20C Mutants Form Channel-like Structures but Have Different Pore Morphologies
For two Aβ1–42 conformers, we performed 100 ns explicit MD simulations on the wild-type, F19P, and F20C barrels embedded in an anionic lipid bilayer composed of DOPS and POPE (1:2 molar ratio). Conformer 1 barrels have a turn at Ser26–Ile31, and conformer 2 barrels have a turn at Asp23–Gly29. Both Aβ conformers can be divided into four domains: the N-terminal chain (residues 1–16 and 1–8 for conformers 1 and 2, respectively), pore-lining β-stand (residues 17–25 and 9–22 for conformers 1 and 2, respectively), turn (residues 26–31 and 23–29 for conformers 1 and 2, respectively), and C-terminal β-strand (residues 32–42 and 30–42 for conformers 1 and 2, respectively). Both point mutations were in the pore-lining β-stand. All Aβ barrels were initially designed as a perfect annular shape. However, the initial annular conformation is gradually lost via relaxation of the lipid bilayer, and environmentally relaxed peptides can be observed after 30 ns. The membrane-embedded U-shaped portions of the Aβ barrels reach equilibration after the initial transient state, while the extramembranous N-termini of the peptides are disordered. Heterogeneous channel conformations as observed in the average Aβ barrels structure (Figure 4) are similar to the structure of Aβ channels with various sequences in our previous simulations.13,14,28,42−44,52,53 However, although the outer shapes of Aβ barrels are similar to each other, the morphology of the solvated pore is quite different. For the wild-type Aβ barrels, both conformers preserve the solvated pore, wide enough for ions to enter and exit at the same time. The averaged pore diameters are ∼1.83 and ∼1.86 nm for the conformer 1 and 2 Aβ barrels, respectively. However, both conformers of each mutant significantly decrease the size of the solvated pore. For F19P, the averaged pore diameters are ∼1.48 and ∼1.69 nm for the conformer 1 and 2 Aβ barrels, respectively, and for F20C, they are ∼1.67 and ∼1.69 nm for the conformer 1 and 2 Aβ barrels, respectively. In particular, the conformer 1 F19P mutant completely blocks the channel mouth in the lower bilayer leaflet (Figure 4C).
Figure 4.

Cartoons representing snapshots of the averaged Aβ barrel structures over the simulations for the (A) conformer 1 and (B) conformer 2 wild-type Aβ1–42 barrels, (C) conformer 1 and (D) conformer 2 F19P barrels, and (E) conformer 1 and (F) conformer 2 F20C barrels. In the channel structures, hydrophobic residues are colored white, polar and Gly residues are green, positively charged residues blue, and negatively charged residues red.
The pore diameter was calculated using HOLE.68 Figure 5 shows the pore diameter measured along the pore axis for the averaged barrel conformations. The averaged pore heights calculated from the program are ∼4.1, ∼5.6, and ∼5.2 nm for conformer 1 and ∼4.5, ∼5.9, and ∼5.3 nm for conformer 2 wild-type, F19P, and F20C barrels, respectively. The pore heights for the wild-type Aβ barrels match the bilayer thickness, while both mutant barrels have longer and narrower pores than the wild type. Especially, both F19P barrels have a collapsed pore due to interacting N-terminal chains at the lower channel mouth blocking the entry into the pore (Figure 5A,B). The N-terminal chains contain several charged residues; they normally stretch toward the lipid headgroups, and only a few chains can fluctuate toward the channel mouth in wild-type barrels. However, in the F19P barrels, the N-terminal chains easily stick together because of kinks produced by the Pro19 residues in the pore-lining β-stands. No kinks can be observed in the F20C barrels, but still the F20C mutants provide a less stable and smaller pore than wild-type Aβ1–42.
Figure 5.

Pore diameters measured for the averaged Aβ barrel conformations as a function of the distance along the pore center axis for the (A) conformer 1 and (B) conformer 2 Aβ1–42 barrels. In each conformer, black, blue, and red lines represent the pore diameters for the wild-type, F19P, and F20C barrels, respectively. (C and D) Change in total charge in the pore as a function of simulation time for (C) conformer 1 and (D) conformer 2 Aβ barrels. The following pore heights with cutoffs along the pore axis were used: −10 nm < z < 10 nm for conformer 1 Aβ barrels, and −15 nm < z < 15 nm for conformer 2 Aβ barrels.
In the wild-type Aβ barrels, we observe that few ions cross through the water pore, but most ions spend time at the binding sites and are frequently near the channel mouth during the simulation. However, in F19P barrels, we found that no ions cross through the water pore during the simulations, although ions can spend time at the binding sites located in the upper channel mouth. The behavior of ions in the F20C pores is similar to those in the wild-type pores. To observe the fluctuation of the ion across the pore, we calculated the change in the total charge in the pore as a function of simulation time (Figure 5C,D). For the conformer 1 Aβ barrels, a pore height cutoff along the pore axis is |z| < 10 nm, while for the conformer 2 Aβ barrels, the cutoff is |z| < 15 nm. With these cutoffs, charge fluctuations involve only contributions of ions fluctuating in the middle of the pore. Apparently, the wild-type pores have larger charge fluctuations than any mutant pore, because the wild-type Aβ barrels have the wider pore. The electrical charge fluctuation due to movement of a number of ions across the pore appears to resemble the single-channel conductance, although the measured time frame is significantly limited.
We provide experimental evidence that amino acid substitutions can have a direct and profound impact on Aβ1–42 membrane activity measurements. This functional tool can be coupled with molecular dynamics (MD) models to gain valuable insights into structural aspects of the Aβ1–42 channel structure. The initial results presented in this work suggest that (i) the nature of the bilayer affects the type of Aβ1–42 activity. In folded DOPS/DOPE bilayers, we rarely observed steplike activity with wild-type Aβ1–42. To the best of our knowledge, this is the second report showing Aβ1–42 channel-like activity with folded bilayers.31,42 (ii) Results with the Aβ1–42 F19P mutant may imply that the β-sheet U turn is needed for activity in bilayers. The fact that F19P has been previously shown not to form fibers64,66 provides further information about the structural significance of this residue. The lack of membrane activity for F19P suggests that a β-sheet in the U turn is necessary for Aβ1–42 pore formation. Generally, Pro substitutions form a kink hindering either β-sheets or α-helices. Additional amino acid substitutions at position Phe19 would be needed to exclude other Aβ1–42 secondary structures,69 which could be embedded in the bilayer. The F19P peptide could be used as a structure-impaired negative control in experiments aiming to address toxicity or other structural features of Aβ1–42. In model bilayers, the heterogeneous activity of Aβ1–42 has been at times interpreted as nonspecific membrane perturbations by the peptide. The lack of F19P activity (n = 17; 10 folded and 7 painted) lends support to the electrical activity most likely occurring through a defined structure(s), i.e., pore formation. While this conclusion is strictly applicable for only the folded and painted bilayers with the electrolyte used in this study, we may expect similar behavior in other lipid compositions where Aβ1–42 exhibits bilayer activity.
These results led us to suggest that, like p3-F19P, the Aβ1–42 F19P mutant might form a collapsed pore that is nonconductive. Further support for this suggestion was obtained by the MD simulations in which we observed that the pore is blocked by the N-terminal chains due to kinks at Pro residues in the pore-lining β-strands. We observed F19P minor channel conductances in one experiment. This F19P activity is inconclusive, because this activity was seen in 100 s out of 40 h of recording, yet we did not see such activity in 28 h in identical bilayers without peptide. The MD model proposed in this study, together with the PLB results, suggests a collapsed pore. Results with F19P are negative (no bilayer activity); therefore, its implications are inferential, and other experimental approaches are needed to exclude the possibility that the F19P peptide might simply not insert or bind the membrane or that the peptide binds and inserts itself into the membrane and forms a collapsed pore.
Here, we present evidence showing that Aβ1–42 F20C forms ion channels with membrane activity comparable to that of the wild type, and this activity can be inhibited by low millimolar concentrations of Zn2+ like the wild type. Cysteine residues have a reactive sulfhydryl group that if in the proximity of other monomers could form −S–S– bridges with neighboring, in-register monomers, somewhat stabilizing the pore structure. This possibility remains unaddressed in this study. In future work, we would like to react the F20C residue with MTS reagents.70 These experiments are often difficult without prior knowledge of the pore structure.71,72 If we succeed, we will be able to experimentally determine if the Phe20 residue lines the pore of the ion channel as suggested by the MD models presented here.
Conclusion
To summarize, we demonstrate the applicability of a combined approach using PLB and MD modeling toward testing Aβ1–42 pore structures. We do this by testing the membrane activity of designed amino acid substitutions in the Aβ peptide. These substitutions aim to experimentally probe the structural requirements for Aβ1–42 pore formation and modulation. By comprehensively studying Aβ mutants, we believe that it is possible to develop a clearer picture of conducting Aβ structure in bilayers; this should aid drug design aiming to ameliorate or prevent channel-dependent Aβ toxicity.
Acknowledgments
All simulations were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health.
Glossary
Abbreviations:
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- PLB
planar lipid bilayers
- MD
molecular dynamics
- Pro or P
proline
- Phe or F
phenylalanine
- Cys or C
cysteine
- DOPS
1,2-dioleoyl-sn-glycero-3-phosphoserine
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine.
Supporting Information Available
Two monomer conformers and the position of each mutation as well as the starting point and conformation for each Aβ1–42 β-barrel and an additional Aβ1–42 F20C current trace showing activity in DOPS/DOPE bilayers. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by the National Institutes of Health (National Institute on Aging Grant AG028709 to R.L.). This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health (NIH), under Contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Author Contributions
R.C., H.J., and S.A.K. contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Selkoe D. J. (1991) The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (1989) Amyloid β protein precursor and the pathogenesis of Alzheimer’s disease. Cell 58, 611–612. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (1989) Aging, amyloid, and Alzheimer’s disease. N. Engl. J. Med. 320, 1484–1487. [DOI] [PubMed] [Google Scholar]
- Ferretti M. T.; Bruno M. A.; Ducatenzeiler A.; Klein W. L.; Cuello A. C. (2011) Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging DOI: 10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Klein W. L. (2002) Aβ toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 41, 345–352. [DOI] [PubMed] [Google Scholar]
- Umeda T.; Tomiyama T.; Sakama N.; Tanaka S.; Lambert M. P.; Klein W. L.; Mori H. (2011) Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042. [DOI] [PubMed] [Google Scholar]
- Simakova O.; Arispe N. J. (2006) Early and late cytotoxic effects of external application of the Alzheimer’s Aβ result from the initial formation and function of Aβ ion channels. Biochemistry 45, 5907–5915. [DOI] [PubMed] [Google Scholar]
- Bhatia R.; Lin H.; Lal R. (2000) Fresh and globular amyloid β protein (1–42) induces rapid cellular degeneration: Evidence for AβP channel-mediated cellular toxicity. FASEB J. 14, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Zhu Y. J.; Lin H.; Lal R. (2000) Fresh and nonfibrillar amyloid β protein(1–40) induces rapid cellular degeneration in aged human fibroblasts: Evidence for AβP-channel-mediated cellular toxicity. FASEB J. 14, 1244–1254. [DOI] [PubMed] [Google Scholar]
- Quist A. (2005) Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ(1–42) are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan B. L.; Azimov R.; Azimova R. (2004) Amyloid peptide channels. J. Membr. Biol. 202, 1–10. [DOI] [PubMed] [Google Scholar]
- Jang H.; Zheng J.; Nussinov R. (2007) Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 93, 1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H.; Arce F. T.; Capone R.; Ramachandran S.; Lal R.; Nussinov R. (2009) Misfolded Amyloid Ion Channels Present Mobile β-Sheet Subunits in Contrast to Conventional Ion Channels. Biophys. J. 97, 3029–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan B. L.; Hirakura Y.; Azimov R.; Azimova R.; Lin M. C. (2002) The channel hypothesis of Alzheimer’s disease: Current status. Peptides 23, 1311–1315. [DOI] [PubMed] [Google Scholar]
- Simakova O.; Arispe N. J. (2007) The Cell-Selective Neurotoxicity of the Alzheimer’s Aβ Peptide Is Determined by Surface Phosphatidylserine and Cytosolic ATP Levels. Membrane Binding Is Required for Aβ Toxicity. J. Neurosci. 27, 13719–13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (1991) Alzheimer’s disease. In the beginning. Nature 354, 432–433. [DOI] [PubMed] [Google Scholar]
- Hardy J. (2002) The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. (1992) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Arispe N.; Pollard H. B.; Rojas E. (1993) Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. U.S.A. 90, 10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A.; Smith M.; Parker I. (2011) Single-channel Ca2+ imaging implicates Aβ1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 195, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourie J. I.; Henry C. L.; Farrelly P. (2001) Diversity of amyloid β protein fragment [1–40]-formed channels. Cell. Mol. Neurobiol. 21, 255–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadi R.; Farrelly P. V.; Kenna B. L.; Curtain C. C.; Masters C. L.; Cappai R.; Barnham K. J.; Kourie J. I. (2003) Cu2+-induced modification of the kinetics of Aβ(1–42) channels. Am. J. Physiol. 285, C873–C880. [DOI] [PubMed] [Google Scholar]
- Arispe N.; Pollard H. B.; Rojas E. (1994) The ability of amyloid β-protein [AβP(1–40)] to form Ca2+ channels provides a mechanism for neuronal death in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 747, 256–266. [DOI] [PubMed] [Google Scholar]
- Arispe N.; Pollard H. B.; Rojas E. (1994) β-Amyloid Ca2+-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 140, 119–125. [DOI] [PubMed] [Google Scholar]
- Arispe N.; Rojas E.; Pollard H. B. (1993) Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U.S.A. 90, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakura Y.; Lin M. C.; Kagan B. L. (1999) Alzheimer amyloid Aβ1–42 channels: Effects of solvent, pH, and Congo Red. J. Neurosci. Res. 57, 458–466. [DOI] [PubMed] [Google Scholar]
- Jang H.; Arce F. T.; Ramachandran S.; Capone R.; Azimova R.; Kagan B. L.; Nussinov R.; Lal R. (2010) Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 6538–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S. K.; Quist A. P.; Lal R. (1998) Amyloid β protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 273, 13379–13382. [DOI] [PubMed] [Google Scholar]
- Lin M. C.; Kagan B. L. (2002) Electrophysiologic properties of channels induced by Aβ25–35 in planar lipid bilayers. Peptides 23, 1215–1228. [DOI] [PubMed] [Google Scholar]
- Capone R.; Quiroz F. G.; Prangkio P.; Saluja I.; Sauer A. M.; Bautista M. R.; Turner R. S.; Yang J.; Mayer M. (2009) Amyloid-β-Induced Ion Flux in Artificial Lipid Bilayers and Neuronal Cells: Resolving a Controversy. Neurotox. Res. 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N.; Pollard H. B.; Rojas E. (1996) Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc. Natl. Acad. Sci. U.S.A. 93, 1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamino K.; Orr H. T.; Payami H.; Wijsman E. M.; Alonso M. E.; Pulst S. M.; Anderson L.; Odahl S.; Nemens E.; White J. A.; Sadovnick A. D.; Ball M. J.; Kaye J.; Warren A.; McInnis M.; Antonarakis S. E.; Korenberg J. R.; Sharma V.; Kukull W.; Larson E.; Heston L. L.; Martin G. M.; Bird T. D.; Schellenberg G. D. (1992) Linkage and Mutational Analysis of Alzheimer-disease kindreds for APP Gene Region. Am. J. Hum. Genet. 51, 998–1014. [PMC free article] [PubMed] [Google Scholar]
- Nilsberth C.; Westlind-Danielsson A.; Eckman C. B.; Condron M. M.; Axelman K.; Forsell C.; Stenh C.; Luthman J.; Teplow D. B.; Younkin S. G.; Naslund J.; Lannfelt L. (2001) The 'Arctic' APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 4, 887–893. [DOI] [PubMed] [Google Scholar]
- Levy E.; Carman M. D.; Fernandezmadrid I. J.; Power M. D.; Lieberburg I.; Vanduinen S. G.; Bots G.; Luyendijk W.; Frangione B. (1990) Mutation of the Alzheimer’s Disease Amyloid Gene in Hereditary Cerebral-Hemorrhage, Dutch type. Science 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- Miravalle L.; Tokuda T.; Chiarle R.; Giaccone G.; Bugiani O.; Tagliavini F.; Frangione B.; Ghiso J. (2000) Substitutions at codon 22 of Alzheimer’s Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 275, 27110–27116. [DOI] [PubMed] [Google Scholar]
- Grabowski T. J.; Cho H. S.; Vonsattel J. P. G.; Rebeck G. W.; Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705. [DOI] [PubMed] [Google Scholar]
- Pifer P. M.; Yates E. A.; Legleiter J. (2011) Point Mutations in Aβ Result in the Formation of Distinct Polymorphic Aggregates in the Presence of Lipid Bilayers. PLoS One 6, e16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K.; Irie K.; Morimoto A.; Ohigashi H.; Shindo M.; Nagao M.; Shimizu T.; Shirasawa T. (2002) Synthesis, aggregation, neurotoxicity, and secondary structure of various Aβ1–42 mutants of familial Alzheimer’s disease at positions 21–23. Biochem. Biophys. Res. Commun. 294, 5–10. [DOI] [PubMed] [Google Scholar]
- Murakami K.; Irie K.; Morimoto A.; Ohigashi H.; Shindo M.; Nagao M.; Shimizu T.; Shirasawa T. (2003) Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: Implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J. Biol. Chem. 278, 46179–46187. [DOI] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Doeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone R., Jang H., Kotler S. A., Connelly L., Teran A. F., Ramachandran S., Kagan B. L., Nussinov R., and Lal R. (2011) All-d-Enantiomer of β-Amyloid Peptide Forms Ion Channels in Lipid Bilayers (submitted for publication). [DOI] [PMC free article] [PubMed]
- Connelly L., Teran A. F., Jang H., Capone R., Kotler S. A., Ramachandran S., Kagan B. L., Nussinov R., and Lal R. (2011) Atomic Force Microscopy and MD Simulations Reveal Pore-Like Structures of All-d-Enantiomer of Alzheimer’s β-Amyloid Peptide: Relevance to the Ion Channel Mechanism of AD Pathology. J. Phys. Chem. B, in press, DOI: 10.1021/jp2108126. [DOI] [PMC free article] [PubMed]
- Jang H.; Arce F. T.; Ramachandran S.; Capone R.; Lal R.; Nussinov R. (2010) β-Barrel Topology of Alzheimer’s β-Amyloid Ion Channels. J. Mol. Biol. 404, 917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafrir Y.; Durell S.; Arispe N.; Guy H. R. (2010) Models of membrane-bound Alzheimer’s Aβ peptide assemblies. Proteins 78, 3473–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strodel B.; Lee J. W. L.; Whittleston C. S.; Wales D. J. (2010) Transmembrane Structures for Alzheimer’s Aβ(1–42) Oligomers. J. Am. Chem. Soc. 132, 13300–13312. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Kriebel J. K.; Tosteson M. T.; Whitesides G. M. (2003) Microfabricated Teflon membranes for low-noise recordings of ion channels in planar lipid bilayers. Biophys. J. 85, 2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montal M.; Mueller P. (1972) Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Natl. Acad. Sci. U.S.A. 69, 3561–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone R.; Blake S.; Restrepo M. R.; Yang J.; Mayer M. (2007) Designing nanosensors based on charged derivatives of gramicidin A. J. Am. Chem. Soc. 129, 9737–9745. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirah S.; Kozin S. A.; Mazur A. K.; Blond A.; Cheminant M.; Segalas-Milazzo I.; Debey P.; Rebuffat S. (2006) Structural changes of region 1–16 of the Alzheimer disease amyloid β-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 281, 2151–2161. [DOI] [PubMed] [Google Scholar]
- Jang H.; Zheng J.; Lal R.; Nussinov R. (2008) New structures help the modeling of toxic amyloid β ion channels. Trends Biochem. Sci. 33, 91–100. [DOI] [PubMed] [Google Scholar]
- Jang H.; Arce F. T.; Ramachandran S.; Capone R.; Lal R.; Nussinov R. (2010) Structural Convergence Among Diverse, Toxic β-Sheet Ion Channels. J. Phys. Chem. B 114, 9445–9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. (2009) CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N.; Diaz J. C.; Simakova O. (2007) Aβ ion channels. Prospects for treating Alzheimer’s disease with Aβ channel blockers. Biochim. Biophys. Acta 1768, 1952–1965. [DOI] [PubMed] [Google Scholar]
- Pollard H. B.; Rojas E.; Arispe N. (1993) A new hypothesis for the mechanism of amyloid toxicity, based on the calcium channel activity of amyloid β protein (AβP) in phospholipid bilayer membranes. Ann. N.Y. Acad. Sci. 695, 165–168. [DOI] [PubMed] [Google Scholar]
- Kagan L. B.; Dobson C. M. (2005) Amyloidosis and Protein Folding. Science 307, 42–43. [DOI] [PubMed] [Google Scholar]
- Kawahara M.; Arispe N.; Kuroda Y.; Rojas E. (1997) Alzheimer’s disease amyloid β-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys. J. 73, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Bhatia R.; Lal R. (2001) Amyloid β protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J. 15, 2433–2444. [DOI] [PubMed] [Google Scholar]
- Diaz J. C.; Simakova O.; Jacobson K. A.; Arispe N.; Pollard H. B. (2009) Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 3348–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakura Y.; Lin M. C.; Kagan B. (1999) Erratum: Hirakura, Y., Lin, M.-C., Kagan, B. L. 1999. Alzheimer amyloid Aβ1–42 channels: Effects of solvent, pH, and Congo red. J. Neurosci. Res. 57:458–466. J. Neurosci. Res 58, 726. [DOI] [PubMed] [Google Scholar]
- Williams A. D.; Portelius E.; Kheterpal I.; Guo J. T.; Cook K. D.; Xu Y.; Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842. [DOI] [PubMed] [Google Scholar]
- Cannon M. J.; Williams A. D.; Wetzel R.; Myszka D. G. (2004) Kinetic analysis of β-amyloid fibril elongation. Anal. Biochem. 328, 67–75. [DOI] [PubMed] [Google Scholar]
- Wood S. J.; Wetzel R.; Martin J. D.; Hurle M. R. (1995) Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide β/A4. Biochemistry 34, 724–730. [DOI] [PubMed] [Google Scholar]
- Shivaprasad S.; Wetzel R. (2006) Scanning cysteine mutagenesis analysis of Aβ(1–40) amyloid fibrils. J. Biol. Chem. 281, 993–1000. [DOI] [PubMed] [Google Scholar]
- Smart O. S.; Goodfellow J. M.; Wallace B. A. (1993) The Pore Dimensions of Gramicidin A. Biophys. J. 65, 2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan S.; Brender J. R.; Lee S. Y.; Ramamoorthy A. (2011) A partially folded structure of amyloid-β(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov O. V.; Merzlyak P. G.; Yuldasheva L. N.; Rodrigues C. G.; Bhakdi S.; Valeva A. (2000) Electrophysiological evidence for heptameric stoichiometry of ion channels formed by Staphylococcus aureus α-toxin in planar lipid bilayers. Mol. Microbiol. 37, 1372–1378. [DOI] [PubMed] [Google Scholar]
- Mindell J. A.; Zhan H. J.; Huynh P. D.; Collier R. J.; Finkelstein A. (1994) Reaction of Diphtheria-Toxin Channels with Sulfhydryl-Specific Reagents: Observation of Chemical Reactions at the Single Molecule Level. Proc. Natl. Acad. Sci. U.S.A. 91, 5272–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh P. D.; Cui C.; Zhan H. J.; Oh K. J.; Collier R. J.; Finkelstein A. (1997) Probing the structure of the diphtheria toxin channel: Reactivity in planar lipid bilayer membranes of cysteine-substituted mutant channels with methanethiosulfonate derivatives. J. Gen. Physiol. 110, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.