

Abstract
Nanoparticles interfere with protein amyloid formation. Catalysis of the process may occur due to increased local protein concentration and nucleation on the nanoparticle surface, whereas tight binding or a large particle/protein surface area may lead to inhibition of protein aggregation. Here we show a clear correlation between the intrinsic protein stability and the nanoparticle effect on the aggregation rate. The results were reached for a series of five mutants of single-chain monellin differing in intrinsic stability toward denaturation, for which a correlation between protein stability and aggregation propensity has been previously documented by Szczepankiewicz et al. [Mol. Biosyst.20107 (2), 521–532]. The aggregation process was monitored by thioflavin T fluorescence in the absence and presence of copolymeric nanoparticles with different hydrophobic characters. For mutants with a high intrinsic stability and low intrinsic aggregation rate, we find that amyloid fibril formation is accelerated by nanoparticles. For mutants with a low intrinsic stability and high intrinsic aggregation rate, we find the opposite—a retardation of amyloid fibril formation by nanoparticles. Moreover, both catalytic and inhibitory effects are most pronounced with the least hydrophobic nanoparticles, which have a larger surface accessibility of hydrogen-bonding groups in the polymer backbone.
Introduction
The formation of well-defined cross-β-sheet structures known as amyloid fibrils is now considered a generic feature of polypeptides.1,2 Some of the proteins with a high propensity to form such structures are associated with severe diseases such as Alzheimer's, Parkinson's, diabetes type 2, etc.3 The propensity of a given peptide to form amyloid aggregates is strongly influenced by the nature of the amino acid sequence as well as the properties of the environment.4,5 The amino acid sequence may favor or disfavor aggregation due to charge, hydrophobicity, spacing, and patterns of polar and nonpolar residues as well as secondary structure propensity.5−10 The aggregation propensity of globular proteins is often inversely related to the stability of the native state.6 Destabilization of the native state is recognized as a primary mechanism for familial or sporadic mutations to exert their pathogenic potential in amyloid diseases.10−13 Amino acid substitutions that destabilize the native state increase the aggregation propensity by increasing the population of unfolded species.11,14
Protein adsorption is the first event that occurs when a foreign material is introduced into a physiological fluid.15 Proteins are highly surface active and have a high affinity for surfaces.15,16 Adsorption is usually fast and depends on the balance of a number of enthalpic and entropic changes, such as partial dehydration of the protein or sorbent surface, redistribution of charged groups on the surface, and conformational changes in the protein. The contribution of each process will depend on the nature of the protein and surface.17 The adsorption of globular proteins on a solid surface is related to the stability of its native structure. Proteins with high structural stability act like hard particles, and the adsorption is mainly governed by hydrophobic and electrostatic effects. In the case of proteins with low structural stability, the ability of structural arrangement on the particle surface is an extra factor for protein adsorption, allowing attractive interactions with hydrophilic and oppositely charged surfaces.18 Protein adsorption can have diverse effects on the conformation of the protein. Once more, the extent of conformational modification would depend on the stability of the native structure. Adsorption may be with little structural change or cause denaturation depending also on the character of the surface.16,19−21
The stability of the native state of a protein will hence govern not only the propensity to form amyloid fibrils but also the interaction between the protein and surfaces. Globular proteins tend to minimize the exposure of their hydrophobic groups, but the protein exterior is often partially hydrophobic. Dehydration of hydrophobic parts of the protein and surface is driven by a gain in entropy and therefore favors adsorption.17,22,23 Modifications that lead to increased exposure of the protein hydrophobic core will thus favor adsorption on a hydrophobic surface. Moreover, proteins with low solubility adsorb strongly to suitable surfaces. Therefore, a mutation which reduces the solubility of a protein modifies the strength of the protein–particle interaction.24
The fibrillization process of amyloid proteins can be affected by several factors, including solution properties such as the ionic strength, pH, temperature, presence of chaperones, peptide inhibitors, etc.25−31 The presence of foreign surfaces in the system, in the form of engineered nanoparticles, shows a profound effect on the formation of amyloids.30,32−36 For example, copolymeric nanoparticles accelerate the fibrillization of β2-microglobulin by promoting nucleation on the particle surface.36 On the other hand, the same nanoparticle type produces the opposite effect on β-amyloid peptide (Aβ)32 and islet amyloid polypeptide (IAPP).37 In those cases the retardation of the process was explained by the depletion of monomers from solution due to the adsorption on the particle surface. Dual behavior was observed for amino-modified polystyrene particles, which accelerate or retard the amyloid formation of Aβ peptide depending on the relative concentration of peptide and nanoparticle surface.38 Diverse studies have shown the effect of other nanoparticles, such as Teflon or fluorinated polymeric nanoparticles, on the conformation of β-amyloid. Shifting of the secondary structure toward a more β-sheet rich structure upon binding on the particle surface enhances the risk of forming amyloid fibrils.33,34 However, no systematic study on the effect of mutations on the interaction between nanoparticles and amyloid proteins is found in the literature.
In this work we have explored the fibrillization kinetics of single-chain monellin mutants in the presence of model copolymeric nanoparticles of N-isopropylacrylamine (NiPAM) and N-tert-butylacrylamide (BAM) with different ratios between the monomers. Monellin (MN) is a sweet-tasting plant protein composed of two subunit chains. This protein shows amyloidogenic properties and forms amyloid fibrils under certain conditions.39,40 Recombinant single-chain MN (scMN), with MNA (subdomain A) covalently linked to MNB (subdomain B), has retained sweetness and increased stability against thermal denaturation.41 The structures of monellin and scMN superimpose, with MNA forming three β-strands and MNB forming two β-strands separated by an α-helix. The five scMN mutants studied here were retrieved from a phage display library on the basis of their surface activity. All modifications are confined to subdomain A and alter the electrostatic charge of subdomain A or the hydrophobic interactions between the subdomains.
The results show that the fibrillization of the mutants is affected by the presence of particles, but the type and extent of the effect vary depending on the mutation in a systematic manner. Thus, the amino acid sequence and, therefore, physicochemical characteristics of mutants have an effect on both the fibrillization pattern and its modulation by nanoparticles. This indicates the existence of different modes of interaction between the particle and protein.
Materials and Methods
Production and Purification of Single-Chain Monellin Mutants
Production and purification were performed as described elsewhere.42
Particle Preparation
Nanoparticles were kindly supplied by Dr. Iseult Lynch, University College Dublin. NiPAM/BAM copolymer particles were synthesized in sodium dodecyl sulfate (SDS) micelles as previously described.36 Particles were produced at different comonomer ratios (85:15 and 50:50 NiPAM/BAM) and with a diameter of 40 nm (at 37 °C). After polymerization, the particles were dialyzed against Milli-Q H2O for at least 14 days with daily changes of H2O to eliminate any trace of SDS as corroborated by proton NMR. Particles were lyophilized and stored until use. Stock solutions were prepared by dissolving particles in ice-cold Milli-Q water.
Thioflavin T Fluorescence Assay
Aggregation kinetics were monitored using thioflavin T (ThT; Calbiochem) as a probe. The fluorescence signal was measured at regular intervals using a Molecular Devices SpectraMax M2 microplate reader (Sunnyvale, CA) with excitation and emission at 440 and 480 nm, respectively. Each experimental point is an average of the fluorescence signal of six wells containing aliquots of the same solution. Samples for aggregation experiments were prepared as follows. Lyophilized protein was dissolved in 10 mM sodium phosphate buffer, 0.02% NaN3, pH 3.5, to a concentration of 1 mg/mL. The solution was vortexed and centrifuged to eliminate any solids. After dilution in the same buffer, the solution was aliquoted in a 96-well black fluorescence plate, NUNC 96 MicroWell black polypropylene plate, and ThT stock solution and particle stock solutions were added. Typical concentrations in these aggregation experiments are 0.20 mg/mL protein and 18 μM ThT. The nanoparticle concentration ranges from 0.9 to 30 μg/mL (from 8.1 to 270 pM as previously calculated32). The plates were incubated at 37 °C and shaken at 700 rpm using a VorTemp 56 incubator/shaker with an orbit of 3 mm (Labnet International, Berkshire, U.K.). Aggregation curves were normalized to the maximum intensity for each mutant to facilitate comparison between mutants. A sigmoidal fit was used to obtain the kinetic parameters of the process. An empirical sigmoidal equation was used:43
| 1 |
where y is the fluorescence intensity at time t, y0 and ymax are the initial and maximum fluorescence intensities, respectively, t1/2 is the time required to reach half the maximum intensity, and k is the apparent first-order aggregation constant. The lag time is defined as lag time = t1/2 – 2/k.
Transmission Electron Microscopy
Negative-staining transmission electron microscopy (TEM) samples were prepared as follows. A 10 μL volume of sample was applied to a formwar/carbon-coated grid for 2 min and blotted with filter paper. The sample was stained with 2% uranyl acetate aqueous solution for 2 min and blotted. Finally, the grids were washed twice with filtered (0.2 μm) water and air-dried. Images were obtained using a JEOL 2000 transmission electron microscope operating at 80 V. Intensity measurements between replicates typically deviate less than 10%.
Results
Fibrillization experiments of the parent protein scMN and the mutants were performed in the absence and presence of 85:15 and 50:50 NiPAM/BAM particles at different concentrations. The monomer ratio of the particles establishes their physicochemical properties, such as the phase transition temperature and hydrophobic character. The fibrillization reaction was monitored by means of a ThT fluorescence assay. ThT is a dye that specifically binds amyloid aggregates, and upon binding, the quantum yield of fluorescence increases, thus allowing quantitative assessment of the presence of fibrillar species. As reported previously,42 each mutant shows a different aggregation tendency (Figure 1). The parent protein presents no aggregation during the length of the experiment (up to 4 days) in the presence or absence of nanoparticles, while the formation rate differs for all the mutants. All of the aggregation experiments exhibit the characteristics of a typical amyloid fibrillization profile: a sigmoidal curve with an initial lag phase, where no increase of fluorescence is observed because no amyloid aggregates are formed, a rapid elongation phase, where the fluorescence intensity increases as the aggregates form, and an equilibrium plateau phase, where the maximum fluorescence remains stable. Self-assembly of amyloid has the appearance of a nucleation-dependent mechanism with a characteristic sigmoidal reaction profile as shown by scMN.31,43,44 The lag time is defined as the time required to reach a certain fraction of monomer in fibrillar form. After the lag time, the aggregation proceeds rapidly, showing a sharp increase of fluorescence intensity in the ThT assay.
Figure 1.

Fibrillization kinetics of single-chain monellin (scMN parent and mutants) at 37 °C monitored by temporal development of ThT fluorescence at different 85:15 NiPAM/BAM nanoparticle concentrations: (black squares) 0, (blue circles) 0.9, (blue-green triangles) 1.8, (green inverted triangles) 3.5, (yellow left-pointing triangles) 7, (red right-pointing triangles) 15, and (purple hexagons) 30 μg/mL (8.1–270 pM). The concentration of scMN is 0.2 mg/mL (17.5–18.7 μM depending on the mutant). The lines represent the best fittings to eq 1.
A different response to the addition of the particles is observed for each mutant. The aggregation rates are affected in different directions depending on the mutant character. There are also differences (significant and persistent between experiments) between the values of the equilibrium plateau in the presence or absence of particles. To facilitate the comparison between fibrillization rates, the curves are normalized (see the Materials and Methods). In general, the effect of 85:15 NiPAM/BAM nanoparticles is more pronounced than for the 50:50 NiPAM/BAM nanoparticles. For that reason, we will mainly focus here on the effect of 85:15 nanoparticles, while the aggregation kinetics in the presence of 50:50 nanoparticles for all the mutants can be found in the Supporting Information (Figure S1).
Figure 1 shows the fibrillization traces for the scMN parent and mutants in the presence of different concentrations of 85:15 NiPAM/BAM particles. Figure 2A shows the lag times (plotted as the ratio of the lag time at each particle concentration and the lag time in the absence of particles) as a function of the nanoparticle concentration. The parent scMN shows no fibrillization for 6000 min, and no nanoparticle effect is observed in that time range. In the case of scMN13 and scMN14, the presence of the particles accelerates the fibrillization process. In both cases, the reaction kinetics in the absence of particles is quite slow and there is not a consistent pattern between the particle concentration and shortening of the lag time. For the rest of the mutants, scMN10, scMN11, and scMM12, the presence of the particles increases the lag time. In these cases there is a clear correlation between the concentration and the rate of fibrillization; the higher the concentration of particles, the slower the fibrillization process. The fibrillization of scMN10 and scMN14 is the most sensitive to particle addition. Just a small amount of particles, 1.8 μg/mL, produces a significant variation in the lag phase. Acceleration of the fibrillization after the addition of particles is observed for the most stable mutants (scMN13, scMN14) for which fibrillization in the absence of the particles is quite slow. On the other hand, for the mutants with faster kinetics (scMN10, scMN11, and scMN12), the process is slowed in the presence of the particles.
Figure 2.

(A) Ratio of lag times in the presence and absence of nanoparticles versus particle concentration. (B) Lag time versus thermal denaturation midpoint, Tm, for the different mutants.42 The straight line indicates the observed linear correlation. Key: (■) scMN10, (●) scMN11, (▲) scMN12, (◆) scMN13, (▼) scMN14, and (★) parent scMN.
The formation of the fibrils in the presence or absence of particles was confirmed by transmission electronic microscopy (Figure 3). Samples were prepared after 4000 min of incubation to ensure formation of fibrils in the presence and absence of nanoparticles. Amyloid-like aggregates were found in all cases except for the parent protein. For the case of scMN10, it also appears that the presence of particles changes the final morphology of the fibrils and leads to the formation of shorter and stickier fibrils.
Figure 3.

Negative-staining TEM images for samples prepared in the absence or presence of 30 mg/mL (270 pM) 85:15 NiPAM/BAM nanoparticles for parent and mutant single-chain monellin. The scale bar indicates 200 nm.
Discussion
The fibrillization process of the single-chain mutants of the sweet-tasting protein monellin is profoundly affected by the presence of polymeric nanoparticles. This set of mutants constitutes an interesting model system to understand protein characteristics and its effect on fibrillization. The observed variation of behavior in the nanoparticle–scMN system arises from the physicochemical characteristics of the mutants and how these properties govern the adsorption of the monomeric/oligomeric species on the particle surface. The conformational stability of each mutant affects both the rate of the fibrillization process relative to the parent protein and the effect of nanoparticles reporting on specific interactions between the protein and the nanoparticle surface.
As reported before,42 all the mutants are folded proteins, but their far-UV circular dichroism spectra differ from that of the parent protein. The parent protein has five β-strands and one α-helix and a predominantly β-sheet spectrum, while for the mutants the helical segment is more apparent in the spectra. The near-UV CD spectra indicate a higher conformational flexibility for the mutants than for the parent protein. Moreover, thermal denaturation studies show that the mutants are less stable than the parent protein (Figure 2B).42 The increased exposure of aggregation-prone segments, which are protected in the parent protein, favors the aggregation into amyloid fibrils. Residues that are changed or missing in all mutants are in the A chain in close contact with the B chain. Those residues seem to establish important interactions between the two chains. Weakening these interactions may lead to the destabilization of the protein and the increase of aggregation-prone segments.42
Figure 2B shows the correlation found between the lag times of fibrillization and the apparent melting temperature. Clearly, as the stability against thermal denaturation increases, the lag time for fibrillization increases as well. This indicates that the most stable proteins have a lower tendency to unfold and aggregate into amyloid fibrils. This was corroborated with the calculation of the aggregation propensity of each mutant based on the amino acid sequences.42 A similar correlation between stability and fibrillization time was reported previously for mutants of the immunoglobulin light chain14 and acylphosphatase.11
The effect on fibrillization is also dependent on the type of nanoparticle used. The 50:50 NiPAM/BAM nanoparticles have a lower effect on scMN fibrillization than the most hydrophilic 85:15 NiPAM/BAM nanoparticles. Similar results were obtained previously for the aggregation of β2-microglobulin36 and Aβ peptide.32 These studies indicate a stronger interaction between protein and particles that have more accessible hydrogen bond groups. The higher propensity to form hydrogen bonds for the 85:15 particles may explain why these particles are more potent in disturbing the fibrillization process of single-chain monellin.
The nanoparticle effect is observed not only in the fibrillization kinetics, but also on the final fibrillar material. For example, in the case of scMN10, the morphology of the fibrils differs from the morphology of the fibrils formed in the absence of particles. This observation may indicate an influence of nanoparticles on the packing and formation of the mature fibrils.43,45 These differences in morphology in the presence or absence of particles can be the reason for the different equilibrium values of the ThT fluorescence intensity. Changes in structure and packing can enhance or decrease the binding, and therefore fluorescence signal, of ThT, resulting in different plateau values.45
The different kinetic effects observed cannot be explained by a kinetic competition between the two processes of amyloid fibril formation and nanoparticle–protein complex formation. In a kinetic competition situation, the most affected mutant should be the one with the longest lag phase, which would allow a longer interaction between the oligomeric, prefibrillar species and the nanoparticles.26 However, in the case of scMN, two mutants exhibit the biggest effect by the addition of the particles, scMN10, which has the fastest intrinsic aggregation kinetics, and scMn14, which has the slowest. Besides, this hypothesis would not explain acceleration of the fibrillization process. The observed dual behavior suggests that the interaction with the nanoparticle occurs at the monomer and oligomer levels.
During the lag phase for fibrillization, soluble oligomers of different sizes are formed and are in equilibrium with monomeric species. Amino acid replacements that destabilize the native fold are known to favor the process of aggregation.2 As we reported before, a correlation between the aggregation propensity and the stability of the mutants is found.42 Protein destabilization causes an increase in the population of partially unfolded conformations that expose the aggregation-prone regions and increase the probability of intermolecular interactions.9 In that context, the population of oligomers during the lag phase for the more stable mutants scMn14 and scMn15 should be lower than for the most unstable mutants scMn10, scMN11, and scMN12.
The observed nanoparticle effect on single-chain monellin fibrillization can be divided into two categories, acceleration or retardation of the fibrillization process caused mainly by a modification of the lag time (Figure 2).
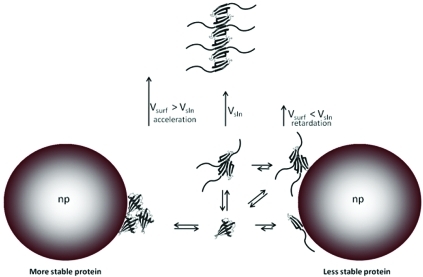
Acceleration of the fibrillization in the presence of nanoparticles for the most stable mutants (Figure 2B) can be explained by taking into account the physicochemical characteristics of the protein that govern the interactions with the nanoparticle surface. It is well-known from the studies of protein adsorption on flat surfaces that the physicochemical characteristics of the protein strongly affect the adsorption process. For example, it is widely considered that “soft proteins” would interact strongly with surfaces due to the gain of entropy coming from the changes in conformation upon adsorption. More flexible proteins can form many noncovalent interactions with the surface by modification of the secondary structure, increasing the strength of the adsorption and the residence time on the surface. In general, changes in the protein conformation are the main driving forces in the adsorption of protein on prohibited surfaces, i.e., hydrophilic or bearing the same charge as the protein. “Hard proteins” retain the secondary structure, and therefore, the adsorption depends on the surface–surface interactions, either hydrophobic or electrostatic interactions. In the case of noncharged surfaces, the main drive of the interaction is the gain of entropy by the release of hydration water on the protein and particle surface.15,23 For scMN14 and scMN15, the addition of particles causes the adsorption on the particle surface of monomers and oligomers in a weak fashion without loss of their structural conformation. Therefore, the proteins would be concentrated in a reduced volume but would keep their amyloidogenic propensity. This crowding effect would favor the nucleation and therefore speed up the fibrillization process (Scheme 1). Similar effects were observed in the case of β2-microglobulin, where we hypothesized that the nanoparticle promotes nucleation and consequently fibril formation.36 Similar crowding effects have been reported previously7 where the fibrillization kinetics is accelerated by raising the local concentration of an amyloid protein in the presence of dextran.
Scheme 1. Different Modes of Nanoparticle–Protein Interactions and Their Effects on the Fibrillization Rate.

Retardation of the fibrillization due to the presence of nanoparticles for the less stable mutants is observed (Figure 2B). A similar effect was reported for the fibrillization of diverse amyloid proteins (Aβ protein32,38 and IAPP37) in the presence of nanoparticles, and depletion of the monomer from the bulk was proposed as a possible cause. However, in this case, the analysis of the ratio between the nanoparticle surface and the maximum protein surface coverage discards a relevant effect of nanoparticles at the scMN monomer level. scMN has an approximate cross-section area of (5–12) × 10–18 m2. Therefore, a single particle 40 nm in diameter (surface area 5 × 10–15 m2) can accommodate a maximum of 1000 nondenaturated proteins arranged in a single layer. Taking into account the total surface area in solution exposed by the particle at the highest concentration used (0.84 m2), only 2% of the protein molecules are needed to cover the particle as a single layer. Multiple layers can be formed, but this implies a high local concentration of proteins that will enhance fibrillization instead of hinder it. Increases of 5–10-fold in lag times are observed for the highest particle concentration. Correlations between lag time and protein concentration have been reported for several systems.46,47 Even assuming a strong scaling factor, a decrease of 2% in concentration may lead to only a marginal increase in lag time. Therefore, we postulate that nanoparticles interact with early aggregates of scMN, disturbing their growth into fibrils. This is not unexpected if we consider that the population of oligomers for those unstable mutants must be significantly higher than for more stable ones due to stronger intermolecular interactions (see above). Nanoparticles can deplete and/or dissolve prefibrillar oligomers or small fibrillar aggregates, which are metastable and present in relatively small populations (Scheme 1). To obtain an inhibitory effect by depletion, nanoparticles must bind preferentially lowly abundant oligomeric species in a way that blocks further growth. Adsorption of abundant intermediate oligomers must imply destabilization and dissolution of oligomers into monomers or oligomers of smaller size. As explained before, “soft” flexible proteins interact strongly with surfaces since they can establish many noncovalent interactions, resulting in long resident times. If the nanoparticle–monomer interactions are stronger than the monomer–monomer interactions, the oligomers adsorbed on the particle surface will dissolve into monomers and/or oligomers of smaller size. Overall, the equilibrium between monomers and oligomers will be shifted toward the monomer side due to the presence of the particles. This implies an elongation of the lag phase and therefore a retardation of the whole aggregation process.
Conclusions
Two factors control the effect of nanoparticles on amyloid formation: first the surface chemistry of the nanoparticles and second the protein intrinsic stability. Those factors determine the specific interactions between nanoparticles and protein which lead to dual effects on the fibrillization process of amyloid proteins. Polymeric nanoparticles interact at both the monomer and oligomer levels, acting either as microreactors or as destabilizing agents, which causes acceleration or retardation of amyloid formation.
Acknowledgments
This study was supported by the Swedish Research Council (VR; to S.L. and C.C.-L.) with its Linneaus Centre Organizing Molecular Matter (S.L.), the Crafoord Foundation (S.L.), and the Irish Research Council for Science, Engineering and Technology (IRCSET; C.C.-L.).
Supporting Information Available
Figure S1 showing the fibrillization kinetics of different scMN mutants in the presence and absence of different concentrations of 85:15 and 50:50 NiPAM/BAM nanoparticles. This material is available free of charge via the Internet at http://pubs.acs.org/.
Supplementary Material
References
- Chiti F.; Webster P.; Taddei N.; Clark A.; Stefani M.; Ramponi G.; Dobson C. M. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (7), 3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M. Nature 2003, 426 (6968), 884–890. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Trends Biochem. Sci. 1999, 24 (9), 329–332. [DOI] [PubMed] [Google Scholar]
- Christopeit T.; Hortschansky P.; Schroeckh V.; Guhrs K.; Zandomeneghi G.; Fandrich M. Protein Sci. 2005, 14 (8), 2125–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Stefani M.; Taddei N.; Ramponi G.; Dobson C. M. Nature 2003, 424 (6950), 805–808. [DOI] [PubMed] [Google Scholar]
- Dubay K. F.; Pawar A. P.; Chiti F.; Zurdo J.; Dobson C. M.; Vendruscolo M. J. Mol. Biol. 2004, 341 (5), 1317–1326. [DOI] [PubMed] [Google Scholar]
- van den Berg B.; Ellis R. J.; Dobson C. M. EMBO J. 1999, 18 (24), 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M. J.; Sievers S. A.; Karanicolas J.; Ivanova M. I.; Baker D.; Eisenberg D. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (11), 4074–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Calamai M.; Taddei N.; Stefani M.; Ramponi G.; Dobson C. M. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 16419–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Taddei N.; Baroni F.; Capanni C.; Stefani M.; Ramponi G.; Dobson C. M. Nat. Struct. Biol. 2002, 9 (2), 137–143. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Taddei N.; Bucciantini M.; White P.; Ramponi G.; Dobson C. M. EMBO J. 2000, 19 (7), 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurle M. R.; Helms L. R.; Li L.; Chan W. N.; Wetzel R. Proc. Natl. Acad. Sci. U.S.A. 1994, 91 (12), 5446–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Alvarado M.; Merkel J. S.; Regan L. Proc. Natl. Acad. Sci. U.S.A. 2000, 97 (16), 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden E. M.; Randles E. G.; Aboagye A. K.; Thompson J. R.; Ramirez-Alvarado M. J. Biol. Chem. 2008, 283 (45), 30950–30956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. J.; Clegg R. E.; Leavesley D. I.; Pearcy M. J. Tissue Eng. 2005, 11 (1–2), 1–18. [DOI] [PubMed] [Google Scholar]
- Norde W.; Giacomelli C. E. Macromol. Symp. 1999, 145, 125–136. [Google Scholar]
- Haynes C. A.; Sliwinsky E.; Norde W. J. Colloid Interface Sci. 1994, 164 (2), 394–409. [Google Scholar]
- Arai T.; Norde W. Colloids Surf. 1990, 51, 1–15. [Google Scholar]
- Wertz C. F.; Santore M. M. Langmuir 1999, 15 (26), 8884–8894. [Google Scholar]
- Giacomelli C. E.; Norde W. J. Colloid Interface Sci. 2001, 233 (2), 234–240. [DOI] [PubMed] [Google Scholar]
- Cukalevski R.; Lundqvist M.; Oslakovic C.; Dahlbäck B.; Linse S.; Cedervall T.. Langmuir 2011, 27(23), 14360–14369. [DOI] [PubMed] [Google Scholar]
- Norde W. Adv. Colloid Interface Sci. 1986, 25 (4), 267–340. [DOI] [PubMed] [Google Scholar]
- Norde W.; Fraaye J.; Lyklema J. ACS Symp. Ser. 1987, 343, 36–47. [Google Scholar]
- Phillips D. C.; York R. L.; Mermut O.; McCrea K. R.; Ward R. S.; Somorjai G. A. J. Phys. Chem. C 2007, 111 (1), 255–261. [Google Scholar]
- Ghosh J. G.; Houck S. A.; Clark J. I. Int. J. Biochem. Cell Biol. 2008, 40 (5), 954–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekas A.; Adda C. G.; Aquilina J. A.; Barnham K. J.; Sunde M.; Galatis D.; Williamson N. A.; Masters C. L.; Anders R. F.; Robinson C. V.; Cappai R.; Carver J. A. J. Mol. Biol. 2004, 340 (5), 1167–1183. [DOI] [PubMed] [Google Scholar]
- Gordon D. J.; Sciarretta K. L.; Meredith S. C. Biochemistry 2001, 40 (28), 8237–8245. [DOI] [PubMed] [Google Scholar]
- Soto C.; Sigurdsson E. M.; Morelli L.; Kumar R. A.; Castano E. M.; Frangione B. Nat. Med. 1998, 4 (7), 822–826. [DOI] [PubMed] [Google Scholar]
- Gazit E. FEBS J. 2005, 272 (23), 5971–5978. [DOI] [PubMed] [Google Scholar]
- Kim J. E.; Lee M. Biochem. Biophys. Res. Commun. 2003, 303 (2), 576–579. [DOI] [PubMed] [Google Scholar]
- Necula M.; Kayed R.; Milton S.; Glabe C. G. J. Biol. Chem. 2007, 282 (14), 10311–10324. [DOI] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Quinlan-Pluck F.; Lynch I.; Lindman S.; Minogue A. M.; Thulin E.; Walsh D. M.; Dawson K. A.; Linse S. J. Am. Chem. Soc. 2008, 130 (46), 15437–15443. [DOI] [PubMed] [Google Scholar]
- Giacomelli C. E.; Norde W. Biomacromolecules 2003, 4 (6), 1719–1726. [DOI] [PubMed] [Google Scholar]
- Rocha S.; Thuneman A. F.; Pereira M. D.; Coelho M.; Mohwald H.; Brezesinski G. Biophys. Chem. 2008, 137 (1), 35–42. [DOI] [PubMed] [Google Scholar]
- Giacomelli C. E.; Norde W. Macromol. Biosci. 2005, 5 (5), 401–407. [DOI] [PubMed] [Google Scholar]
- Linse S.; Cabaleiro-Lago C.; Xue W. F.; Lynch I.; Lindman S.; Thulin E.; Radford S. E.; Dawson K. A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (21), 8691–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Lynch I.; Dawson K. A.; Linse S. Langmuir 2010, 26 (5), 3453–3461. [DOI] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Quinlan-Pluck F.; Lynch I.; Dawson K. A.; Linse S. ACS Chem. Neurosci. 2010, 1 (4), 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T. Protein Sci. 2001, 10 (10), 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T.; Murata K.; Nagayama K. FEBS Lett. 1999, 454 (1–2), 122–126. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Kang C. H.; Kim R.; Cho J. M.; Lee Y. B.; Lee T. K. Protein Eng. 1989, 2 (8), 571–575. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz O.; Cabaleiro-Lago C.; Tartaglia G. G.; Vendruscolo M.; Hunter T.; Hunter G. J.; Nilsson H.; Thulin E.; Linse S. Mol. Biosyst. 2011, 7 (2), 521–532. [DOI] [PubMed] [Google Scholar]
- Nielsen L.; Khurana R.; Coats A.; Frokjaer S.; Brange J.; Vyas S.; Uversky V. N.; Fink A. L. Biochemistry 2001, 40 (20), 6036–6046. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Nayak A.; Sethuraman A.; Belfort G.; McRae G. J. Biophys. J. 2007, 92 (10), 3448–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen J. S.; Dikov D.; Flink J. L.; Hjuler H. A.; Christiansen G.; Otzen D. E. J. Mol. Biol. 2006, 355 (3), 501–523. [DOI] [PubMed] [Google Scholar]
- Hellstrand E.; Boland B.; Walsh D. M.; Linse S. ACS Chem. Neurosci. 2010, 1 (1), 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J; Waudby C. A.; Devlin G. L.; Cohen S. I. A.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. Science 2009, 326 (5959), 1533–1537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.