

Abstract
The potential for interactions with current medications should always be considered when administering or prescribing any drug. Considering the staggering number of drugs patients may be taking, this task can be daunting. Fortunately, drug classes employed in dental practice are relatively few in number and therapy is generally brief in duration. While this reduces the volume of potential interactions, there are still a significant number to be considered. This article will review basic principles of drug interactions and highlight those of greatest concern in dental practice.
Key Words: Drug interactions, CYP450, Drug potentiation, Drug synergism
INTRODUCTION AND OVERVIEW OF INTERACTIONS
It is certain that the dentist or physician cannot be expected to memorize the staggering number of pharmaceuticals available and the equally daunting potential for drug interactions. However, prescribers should recognize that patients often come to them medicated with scores of drugs acquired from multiple sources. A meticulous drug history should include examination of the patient's prescribed medications as well as over‐the‐counter (OTC) drugs and health food supplements. When prescribing a medication, one is always encouraged to check with any number of references or tables regarding risk for interactions. Nevertheless, general principles of drug interactions should be understood and the major risks for interactions should be appreciated for the principal drug classes prescribed.
When two or more drugs are taken concurrently, they may influence one another in a manner that results in either an enhanced or diminished intensity of effect produced by any of the drugs taken alone. When the intensity is reduced, the interaction is generally described as antagonistic. If the intensity is increased, the interaction is described variably as summation, potentiation, or synergism. These latter terms are frequently used interchangeably without regard to precise definition, but they are intended to differ in the actual magnitude of the resultant effect. These differences are distinguished in Table 1 and will be used in this article.
Table 1.
Terminology of Drug Interactions

While the context of this article is to address drug interactions that are undesirable, it should be emphasized that not all drug interactions are detrimental. The reversal of opioid‐induced respiratory depression and the enhanced effects of regimens for sedation or management of hypertension are examples. An understanding of the basic mechanisms by which drugs may interact provides a conceptual framework for prevention. By convention, drug interactions are categorized as pharmaceutical, pharmacodynamic, and pharmacokinetic.
Pharmaceutical Drug Interactions
Pharmaceutical interactions occur before drugs are actually administered to the patient and generally represent incompatibilities of drugs administered by intravenous infusion. These incompatibilities manifest as an increase in measured haze or turbidity, particulates, and color changes. Ultimate consequences are not established but at the very least are presumed to increase the potential for vein irritation. There are very few incompatibilities among the conventional sedatives and opioids used for sedation and general anesthesia. These include the benzodiazepines, methohexital, propofol, fentanyl, meperidine, and nalbuphine. Diazepam is a notable exception and should not be introduced into the intravenous (IV) infusion with any medication other than physiologic solutions. This is an important consideration during continuous propofol infusions because diazepam causes emulsion damage with free oil formation.1
There are several concerns when adding additional drug classes to the IV infusion. These include certain antibiotics, glucocorticosteroids, and antihistamine‐antiemetics.1 The principal incompatibilities among these agents are summarized in Table 2. When administering any of these drugs in combination, the IV line should be cleared of the previous drug before introducing another having an incompatibility. In addition to those presented in the table, neither propofol nor nalbuphine should be mixed with methylprednisolone, nor should the antiemetic ondansetron be mixed with methohexital. Otherwise, these adjuncts pose no incompatibilities with conventional opioids or sedatives.
Table 2.
Incompatibilities of Adjunctive Drugs*

Pharmacodynamic Drug Interactions
Pharmacodynamic interactions relate to actual drug actions or effects on a targeted organ or system. These interactions may occur at identical receptor sites or by diverse mechanisms on identical or related organs. It should be reemphasized that interactions may be desirable or undesirable. A diuretic promotes sodium and water excretion by the kidney, while a beta blocker reduces cardiac output. Their interaction provides a greater reduction in blood pressure than either can impart alone. A further example is the enhanced effect produced by combining drugs that depress the central nervous system. This may be a therapeutic goal of sedation regimens or a deleterious consequence of combining ethanol with antihistamines.
Pharmacokinetic Drug Interactions
Pharmacokinetic interactions occur when one drug alters the delivery of another to its target. Drugs may alter the absorption, distribution, or elimination of one another. A particular drug may have no action on a targeted tissue whatsoever but may modify another drug's influence by varying its concentration available to produce an effect. This interaction may result either in diminished effects or drug potentiation. It is rare for any enhancement to reach the level of synergism. In terms of absorption, the most familiar is the delayed absorption of tetracycline antibiotics by divalent cations found in antacids and dairy products. The principal mechanism regarding distribution relates to protein‐binding of drugs while circulating in serum. While protein‐bound, a drug is not available to distribute into targeted tissues. A drug that is capable of displacing a bound drug from protein increases the amount available for distribution and may enhance its effect. One example is the potential for furosemide (Lasix) to displace warfarin (Coumadin) and increase the risk for bleeding. This mechanism is unlikely to have any significance with any of the drugs prescribed in dental practice.
By far the most frequent and significant pharmacokinetic drug interactions relate to drug metabolism or biotransformation. These interactions occur when one drug induces or inhibits the enzyme responsible for activation or inactivation of another drug. This issue is the current focus of most literature on the topic of drug interactions and should be understood in greater detail.
The term parent drug reflects the molecular structure of a drug when administered. Parent drugs may be active pharmacologically or inactive (prodrugs) and require biotransformation to an active metabolite. Therefore, biotransformation may be required to activate or inactivate a parent drug. These reactions occur primarily in the liver and may proceed in either or both of two phases. Phase I reactions consist of oxidation, reduction, and hydrolysis, while Phase II reactions involve conjugations that generally confer water solubility to the metabolite to expedite renal excretion. Enzymes associated with Phase I reactions are the target of most drug interactions. These enzymes are collectively regarded as the hepatic microsomal system based on their location within endoplasmic reticulum of cells comprising intestinal mucosa and liver.
Nomenclature for the microsomal enzymes has evolved into a sophisticated system labeled as the cytochrome P450 enzymes. The term cytochrome is derived from the color of liver cells (dark red) attributed to the iron content of the enzymes, and P450 refers to ultraviolet light wavelength absorbed by the enzymes. The numbers and letters following CYP refer to the families, subfamilies, and specific genes responsible for synthesis of the particular enzymes (eg, CYP3A4 and CYP2D6). Although three large families of enzymes have been fully classified, those belonging to the 3A and 2D subfamilies account for most of the well‐identified drug interactions. Of these, CYP 2D6 and 3A4 carry the most relevance for interactions in dental practice (Figure 1).
Figure 1.
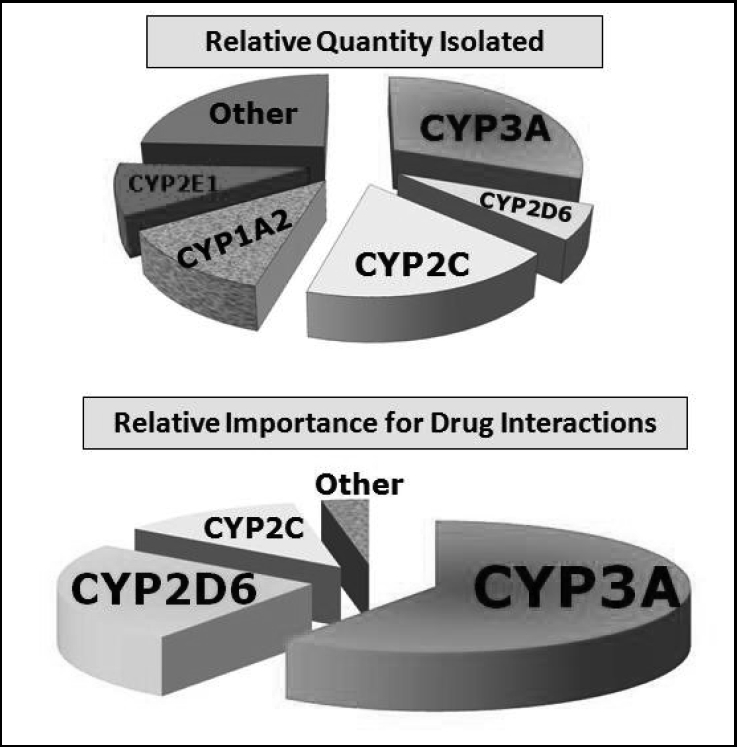
Cytochrome P450 families of enzymes. These pie graphs provide a conceptualization of the relative importance of the microsomal enzymes. The graph on the top reflects the relative quantities of enzymes that have been isolated, while the graph on the bottom reflects their relative importance for drug interactions. For example, while small in overall quantity, CYP2D6 enzymes have major significance for drug interactions.
Drugs that undergo hepatic biotransformation are frequently substrates for the same enzymes. While requiring these enzymes for their own metabolism, they also may induce or inhibit enzyme activity on other drugs taken concurrently. These influences are established in vitro and their clinical significance frequently lacks confirmation. Nevertheless, stunning lists of enzymes and substrates have been assembled, but any attempt to glean their relevance is daunting.2,3 An abridged summary of such information is presented in Table 3. When an interaction occurs, enzyme induction generally proceeds more slowly than enzyme inhibition. Whereas interactions based on induction may require 7–10 days to manifest clinically, those based on inhibition can manifest following a single dose or two, especially when the targeted drug has a short half life.4 For this reason, considerations regarding enzyme inhibition are more germane for the short‐term drug therapy typical in dental practice.
Table 3.
Selected CYP450 Enzymes and Their Substrates. Most Relevant for Drugs Prescribed in Dental Practice*

DRUG INTERACTIONS IN DENTAL PRACTICE
In dental practice, drug interactions are not as voluminous as they are in medical practice. This is based on the fact that most drug therapy is short‐term and the number of drug classes prescribed is small in comparison. For this reason, we will address potential interactions according to the class of drug commonly prescribed in dental practice.
Nonopioid Analgesics
Nonsteroidal anti‐inflammatory drugs (NSAIDs) may reduce the effectiveness of most classes of antihypertensive medications; calcium channel blockers are a notable exception. The interaction is pharmacodynamic and is believed to be related to a reduction of prostaglandins within the kidney that support its role in blood pressure regulation.5 There is no evidence that short‐term use of NSAIDs (3–5 days) carries significant risk. In the rare event that postoperative analgesics must be continued for more than 5 days, hypertensive patients should return to the office for blood pressure assessment. If pressure has elevated more than 10% above baseline, it would be wise to replace the NSAID with acetaminophen.
Ibuprofen has been found to competitively inhibit the antiplatelet influence of aspirin.6,7 It is the only NSAID implicated in this interaction, but diclofenac and the selective COX‐2 inhibitors are the only agents that have been confirmed not to interact.7 An empiric solution to this problem is predicated on the fact that the antiplatelet influence of low‐dose aspirin occurs when it contacts platelets within the hepatic portal system following absorption.8 Simply advise the patient to take their daily aspirin upon rising and delay the first dose of ibuprofen for 1–2 hours. By this time the antiplatelet influence of aspirin will have been established.9 This entire issue may eventually prove moot because its actual clinical relevance has been challenged impressively. Cryer et al10 found thromboxane inhibition by aspirin was reduced only 1% after 10 days of concurrent ibuprofen use, and Patel and Goldberg11 found no increase in incidence of myocardial infarction over a 10‐year period in patients with coronary disease taking ibuprofen with low‐dose aspirin.
The most common side effect, excluding dyspepsia, of NSAIDs is their erosive effect on gastrointestinal (GI) mucosa. This is particularly so in elderly patients. Although a negligible amount of bleeding occurs in many patients, it can become significant in those receiving antithrombotic therapy. For this reason, NSAIDs should be avoided in patients taking anticoagulants such as warfarin (Coumadin) or enoxaparin (Lovenox), or powerful antiplatelet drugs such as clopidogrel (Plavix). Although NSAIDs have some degree of antiplatelet activity, this action is not the explanation for concern. Rather, it is the potential for more significant bleeding from NSAID‐induced GI damage.12 Short‐term use of NSAIDs (3–5 days) is probably not a concern with monotherapy using low‐dose aspirin unless the patient is elderly.
With regard to enhanced risk for GI mucosal injury, there has been recent concern for patients taking selective serotonin reuptake inhibitor (SSRI) antidepressants and NSAIDs. These antidepressants are also known to produce GI injury, and this risk may be accentuated following prolonged use of NSAIDs. Short‐term use is not likely a concern, but caution may be advised for patients with a prior history of mucosal injury.13,14
Finally, serum levels of lithium and methotrexate are elevated during concurrent consumption of NSAIDs. To prevent toxicity, NSAIDs should be avoided in patients medicated with these agents, particularly those taking high‐dose regimens.
Acetaminophen is the preferred alternative when NSAIDs must be avoided. It shares none of the side effects or potential drug interactions. However, it is potentially hepatotoxic and this risk is increased with liver dysfunction or other drugs that may alter its biotransformation. Acetaminophen‐induced hepatotoxicity is attributed to a metabolite that cannot be adequately conjugated when dosages exceed 200–250 mg/kg in a 24‐hour period.15,16 This metabolite also inhibits activity of vitamin K which may account for a spattering of reports where prolonged use of acetaminophen has potentiated the anticoagulant effects of warfarin.17 Provided daily doses of acetaminophen do not exceed 4 g per day, the normal liver has adequate amounts of glutathione conjugant to clear this toxic metabolite. However, the dose should be reduced for patients who are poorly nourished, suffer liver dysfunction, or are treated with other hepatotoxic medications. Ethanol, carbamazepine (Tegretol), and phenytoin (Dilantin) are known to induce hepatic enzymes that create the toxic metabolite of acetaminophen. Whereas short‐term use is not a concern, it is probably wise to reduce daily amounts of acetaminophen during extended use.
Opioid Analgesics
Codeine and its derivatives are the most commonly prescribed agents for postoperative pain. Codeine is a prodrug. Roughly 10% of the administered parent drug is converted by CYP2D6 to the active metabolite morphine.18 Hydrocodone is active but a portion of its analgesic effect is also attributed to the active metabolite, hydromorphone. Drugs that inhibit CYP2D6 will diminish the analgesic effect of these products. Implicated inhibitors include the widely‐prescribed SSRI antidepressants such as fluoxetine (Prozac) and paroxetine (Paxil).19 In contrast, dexamethasone is a CYP2D6 inducer and will enhance conversion to active metabolites. The analgesic effect of oxycodone is almost entirely attributed to the parent drug because only scant amounts are demethylated to oxymorphone.20 This makes it the better choice for patients taking medications known to inhibit CYP2D6.
Meperidine and propoxyphene must never be used for patients taking drugs that inhibit monoamine oxidase (MAO). This includes phenelzine (Nardil) used for depression and selegiline (Eldepryl) used for Parkinson's disease. Their combination can lead to a striking excitatory reaction referred to as “acute serotonin syndrome” that manifests as delirium, hyperthermia, and convulsions.18 This reaction occurs when combining drugs that elevate serotonin levels. Metabolites of both meperidine and propoxyphene act as central nervous system (CNS) stimulants and are likely culprits in this interaction. Tramadol (Ultram), an analgesic gaining in popularity, has actions resembling tricyclic antidepressants and should also be avoided in patients receiving MAO inhibitors.
Opioids demonstrate a synergistic CNS depressant effect with all classes of sedatives, including the benzodiazepines and propofol used in sedation and general anesthesia regimens.21 Their combination is most certainly beneficial for this indication, but this synergistic influence mandates that dosages are titrated carefully. When opioids and sedatives are combined for other indications, generally by oral administration, dosages must be conservative. A summary of important drug interactions regarding analgesics is presented in Table 4.
Table 4.
Interactions of Analgesic Drugs Commonly Used in Dental Practice*

Sedatives and Antiemetics
Midazolam (Versed), triazolam (Halcion), and alprazolam (Xanax) are substrates for CYP3A4, which provide their elimination. Drugs that inhibit these enzymes can elevate serum concentrations and prolong the elimination of these benzodiazepines. The risk for this potentiation is most significant with the HIV protease inhibitors and azole antifungals. Large amounts of grapefruit juice have also been implicated, but in amounts under 1 quart per day any influence is probably insignificant.22 This interaction is not likely to influence the amount of sedative required to achieve satisfactory sedation but will most certainly delay the elimination of the drug and prolong patient recovery. If large amounts of these sedatives are required for effective sedation, the dentist must carefully reconsider normal discharge criteria and patient instructions when patients are taking any medications that inhibit CYP3A (Table 3). These issues are also a concern for newer nonbenzodiazepine sedatives, zolpidem (Ambien), and zaleplon (Sonata), which also are eliminated by CYP3A4.
Promethazine (Phenergan), prochlorperazine (Compazine), and droperidol (Inapsine) are used as antiemetics and as sedatives. A portion of their action is predicated on blocking dopamine receptors within the central nervous system. They should be avoided in all patients with Parkinson's, but especially those medicated with dopamine agonists such as levodopa.
All sedatives can at the very least potentiate the effect of any other CNS depressant including opioids, psychotropics, and anticonvulsants. The intensity of any interaction is impossible to predict due to varied dosages and tolerance the patient has developed over time. This risk is obviated when sedatives are administered by careful intravenous titration, but selecting oral dosages is difficult. Higher doses may be required due to tolerance, but lower doses may be required due to continued sensitivity. When making any decision the dentist should always err on the side of caution.
Local Anesthetics
The systemic effects produced by combinations of local anesthetics follow principles of summation.23 When adhering to dosage limits, guidelines for various agents should be regarded as additive. For example, if you have administered half the maximum dose for lidocaine and wish to add bupivacaine, reduce its maximum dose by half.
It is also essential that local anesthetics be respected as CNS depressants, and they potentiate any respiratory depression associated with sedatives and opioids. Furthermore, serum concentrations required to produce anesthetic‐induced seizures are lower if hypercarbia (elevated carbon dioxide) is present. This is the case when respiratory depression is produced by concurrent administration of sedatives and opioids. Goodson and Moore24 have documented catastrophic consequences of this drug interaction in pediatric patients receiving preoperative sedation along with excessive dosages of local anesthetics.
At times, relatively large amounts or repeated doses of local anesthetics are required for extensive and prolonged procedures. When this occurs, issues regarding hepatic clearance of local anesthetics should be considered. Keep in mind that both liver and renal functions decline 50% by age 65.25 Also, beta blockers reduce hepatic blood flow, and this may prolong the elimination of amide local anesthetics. Articaine is the exception because it has an ester side chain and is inactivated in serum by plasma cholinesterases.
Vasopressors
There is continued concern regarding pharmacokinetic interactions between vasopressors and antidepressants, but most of this worry relates to misconceptions regarding clearance of catecholamines. Neuronal uptake is the principal method for termination of endogenous adrenergic neurotransmitters, but adrenergic drugs are terminated primarily by hepatic biotransformation. Those that are not catecholamine in structure are metabolized in the liver by MAO, but catecholamines are inactivated by catechol‐o‐methyltransferase (COMT). Epinephrine and levonordefrin are catecholamines and are metabolized primarily by COMT, not MAO. MAO inhibitors do not delay their elimination. Novel agents that enhance dopaminergic activity are being introduced in the management of Parkinson's disease. One of these, entacapone (Comtan), is a COMT inhibitor that may delay metabolism of both epinephrine and levonordefrin. Should this interaction occur, the normally transient duration of cardiovascular stimulation caused by these drugs may last somewhat longer.
Tricyclic antidepressants inhibit neuronal uptake of norepinephrine. This poses no concern for epinephrine, but levonordefrin closely resembles norepinephrine in structure and may undergo some degree of neuronal uptake. Yagiela et al26 found the pressor effect of both norepinephrine and levonordefrin was increased sixfold with concurrent administration of desipramine, a tricyclic antidepressant. Based on this information some caution is warranted regarding tricyclic antidepressants and levonordefrin.
Risks for pharmacodynamic interactions with MAO inhibitors and tricyclic antidepressants cannot be entirely dismissed, however. These antidepressants do produce some degree of cardiac excitation, especially with higher dosages. The added cardiotonic effects produced by epinephrine or levonordefrin must always be considered for patients taking any medication capable of producing sympathomimetic effects. In addition to the antidepressants mentioned, these include digoxin, anorexics, decongestants, and agents for attention deficit disorders such as amphetamines and methylphenidate (Ritalin). Vasopressors are not contraindicated in these patients, but they should be administered with caution. For example, when using an epinephrine‐containing product, a reasonable protocol is to record baseline heart rate and blood pressure preoperatively and again following every 20–40 µg administered. This would equate to 1–2 cartridges containing a 1 ∶ 100,000 epinephrine concentration.
Although a minor consideration, the hemostasis and duration of anesthesia attributed to vasoconstriction may be attenuated by medications that act as antagonists for alpha adrenergic receptors on blood vessels.26 This includes many of the antipsychotic agents, tricyclic antidepressants, and alpha blockers prescribed for hypertension such as doxazosin (Cardura).
The potential pharmacodynamic interaction between any vasopressor and nonselective beta blockers is a significant concern. The interaction occurs within minutes of injecting the anesthetic solution and presents as a sudden elevation in blood pressure with a reflex slowing of heart rate. Consequences of this interaction are well‐documented for both epinephrine and levonordefrin.27–29 Its significance requires a thorough understanding by those administering local anesthetic formulations. A thorough explanation and illustration are provided in Figure 2.
Figure 2.

Epinephrine‐beta blocker interaction. In this illustration the following cardiovascular changes follow the administration of epinephrine in a dosage of 10 µg/min. (A) First, it is essential to understand the precise cardiovascular influences of epinephrine in a normal (control) patient. The cardiotonic effects of epinephrine are most familiar. It activates beta‐1 receptors on the sinoatrial node to increase heart rate (HR) and also activates beta‐1 receptors on myocardial cells increasing their force of contraction. This provides an increase in systolic blood pressure (SBP). In addition to its cardiotonic effects, epinephrine has the ability to activate both alpha and beta‐2 receptors on blood vessels producing constriction or dilation, respectively. Epinephrine is commonly viewed only as a vasoconstrictor by many clinicians because this is the effect it produces when injected subcutaneously or submucosally. This is because the tiny vessels found in these locations contain only alpha receptors and are constricted by epinephrine. In contrast, larger systemic arteries that determine vascular resistance and diastolic blood pressure contain both alpha and beta‐2 receptors, with the latter most prevalent. Following absorption, low doses of epinephrine found in local anesthetic formulations (eg, 20–100 µg) preferentially activate beta‐2 receptors which dilate the arteries, and diastolic blood pressure (DBP) actually declines. (B) In the presence of a nonselective beta blocker (eg, propranolol) the cardiovascular influences of epinephrine are strikingly different. This is primarily due to the blockade of beta‐2 receptors on systemic arteries. Epinephrine will now activate the remaining alpha receptors leading to vasoconstriction and an increase in diastolic blood pressure. To meet this increase in resistance, intrinsic mechanisms within myocardial cells respond with greater force and elevate systolic blood pressure as well. Together this will increase mean arterial pressure (MAP). The sudden elevation in MAP is sensed by baroreceptors within the carotid sinuses triggering a reflex slowing in heart rate, which is further accentuated by the fact that the beta‐1 receptors in the sinoatrial node are blocked.
The time course for this interaction is identical to that observed for normal cardiovascular responses to epinephrine. It commences following absorption from the injection site which generally peaks within 5 minutes. Catecholamines are cleared rapidly by hepatic COMT and any cardiovascular effects diminish as serum levels decline over the subsequent 15–20 minutes. It is significant that the mechanism of this interaction involves the action of beta blockers on vascular beta‐2 receptors. Selective beta blockers that act only on cardiac beta‐1 receptors do not generally introduce a risk for this interaction. However, some caution is advised when these selective agents are prescribed at high doses because some of their specificity is lost. The most commonly prescribed beta blockers for consideration are listed in Table 5. Vasopressors are not contraindicated in patients taking beta blockers, but doses should be conservative and blood pressure monitored periodically during administration as described above.
Table 5.
Commonly Prescribed Beta Blockers*

Antimicrobials
Antimicrobial drugs have been implicated in numerous drug interactions. Fortunately for the dentist, both periodontal and odontogenic infections are managed using a fairly limited number of antibiotic classes. Nevertheless, a few interactions are significant, and this section will first address issues regarding antibiotics in general, followed by a few more specific considerations.
Since the early 1980s, numerous articles have suggested that a majority of antibiotics may reduce the efficacy of oral contraceptives (OCs). The putative mechanism for this interaction relates to the ability of antibiotics to reduce normal intestinal flora that enhance bioavailability. Estrogens and progestins normally undergo enterohepatic recirculation, whereby absorbed steroids are conjugated in bile and excreted into the duodenum. Intestinal flora hydrolyze these conjugates allowing the steroids to be reabsorbed. Most publications claiming this interaction have been either anecdotal reports or elaborate theories based on these reports. Furthermore, many of these are found in poorly refereed literature and have included anonymous case reports of dentists who were sued for child support following an unwanted pregnancy attributed to an antibiotic‐OC interaction. None of these have cited legal proceedings that can be researched, and they should be viewed with suspicion. To date, rifampin (an antituberculosis agent) is the only antibiotic having a confirmed interaction with OCs.30 Despite the equivocal status of this issue, the Physicians' Desk Reference and other drug compendia mention the possibility of this interaction.
In a thorough review of oral contraceptives, Sondheimer31 stated that the generally accepted rate of unwanted pregnancy among OC users is 0.5%–1% (8% among teens), and the most common reason for failure is nonuse. Furthermore, he added that most antibiotics do not decrease the effectiveness of the OC. It is entirely within reason to suspect that a woman might also be taking an antibiotic during the month of OC failure, most likely penicillin or tetracycline. To date, all human studies measuring the influence of antibiotics on estrogen and progestin serum concentrations have found no interaction with antibiotics other than rifampin.32–34
Although alternative methods of contraception are easy to suggest, it is naive to assume a patient will consider them acceptable. If antibiotics are indicated, their benefit certainly outweighs anecdotal suggestions of OC ineffectiveness. To imply that the interaction has been established, or is even reasonably possible, is an irresponsible posture. Future quotes and publications based on conjecture should be discouraged. They merely serve as fuel for legal reprise. There is no sound evidence to support the contention that antibiotics, other than rifampin, reduce the effectiveness of oral contraceptives.35,36
Additional interactions related to antibiotic‐induced reduction in gut flora deserve mention, in particular those related to the anticoagulant warfarin (Coumadin). This anticoagulant acts by inhibiting synthesis of vitamin K which is required for activation of several coagulation factors. Further reduction of vitamin K levels can accentuate the anticoagulant effects of warfarin. Gut bacteria are an important source of vitamin K, and antibiotics may reduce this important source. Again, it can be said that short‐term use of antibiotics is probably not a concern, but it would be prudent to order international normalized ratio (INR) monitoring weekly during prolonged use. This is particularly true for cephalosporins, macrolides, and tetracycline. Though unrelated to gut flora in mechanism, two antibiotics must be avoided entirely in patients taking warfarin. Both erythromycin and metronidazole inhibit CYP2C9 microsomal enzymes responsible for metabolism of warfarin.
It is estimated that roughly 10% of the general population harbors a species of gut flora that contribute to the metabolism of digoxin.37 This is probably of little concern for short‐term use of antibiotics (5–7 days), but prolonged coverage may introduce concern for digoxin toxicity.
Finally, erythromycin and clarithromycin are substrates for CYP3A4 and also inhibit the activity of these enzymes. This potential for interaction has been established for increasing the toxicity of HMG‐CoA reductase inhibitors (“statins”) and HIV protease inhibitors. A summary of antibiotic interactions is presented in Table 6.
Table 6.
Interactions of Antibiotic Drugs Commonly Used in Dental Practice1

CONTINUING EDUCATION QUESTIONS
-
When administered concurrently, various local anesthetics demonstrate which of the following interactions?
A. Antagonism
B. Potentiation
C. Summation
D. Synergism
-
Which of the following should never be administered through an intravenous infusion until all other medications have been cleared?
A. Diazepam
B. Midazolam
C. Nalbuphine
D. Propofol
-
Most pharmacokinetic drug interactions involve an alteration in which of the following?
A. CYP450 enzyme activity
B. Drug absorption
C. Plasma protein binding
D. Renal clearance
-
The interaction between epinephrine and nonselective beta blockers manifests clinically as which of the following?
A. Decrease in diastolic pressure and increase in heart rate
B. Decrease in systolic pressure and decrease in heart rate
C. Increase in mean arterial pressure and decrease in heart rate
D. Increase in systolic pressure and increase in heart rate
REFERENCES
- 1.Chicago: Wolters Kluwer Health; 2010. Clin‐eguide. [Google Scholar]
- 2.Abramowicz M, editor. Drug interactions. Med Lett Drugs Ther. 2003;45:46–48. editor. [PubMed] [Google Scholar]
- 3.Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine; 2007. Available at: http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed April 15, 2010. [Google Scholar]
- 4.Horn JR. Important drug interactions and their mechanisms. In: Katzung BG, Masters SB, Trevor AJ, editors. Basic and Clinical Pharmacology. 11th ed. City, State: McGraw‐Hill Companies Inc; 2009. [Google Scholar]
- 5.Burke A, Smyth E, FitzGerald GA. Analgesic‐antipyretic agents; pharmacotherapy of gout. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw‐Hill; 2006. [Google Scholar]
- 6.Catella‐Lawson F, Reilly MP, Kapoor SC. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573–574. doi: 10.1016/s0140-6736(03)12509-3. [DOI] [PubMed] [Google Scholar]
- 8.Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287–1294. doi: 10.1056/NEJM199405053301808. [DOI] [PubMed] [Google Scholar]
- 9.Abramowicz M, editor. Do NSAIDs interfere with the cardioprotective effects of aspirin? Med Lett Drugs Ther. 2004;46:61–62. ed. [PubMed] [Google Scholar]
- 10.Cryer B, Berlin RG, Cooper SA, Hsu C, Wason S. Double‐blind, randomized, parallel, placebo‐controlled study of ibuprofen effects on thromboxane B2 concentrations in aspirin‐treated healthy adult volunteers. Clin Ther. 2005;27:185–191. doi: 10.1016/j.clinthera.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Patel TN, Goldberg KC. Use of aspirin and ibuprofen compared with aspirin alone and the risk of myocardial infarction. Arch Intern Med. 2004;164:852–856. doi: 10.1001/archinte.164.8.852. [DOI] [PubMed] [Google Scholar]
- 12.Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug‐drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ. 2007;177:347–351. doi: 10.1503/cmaj.070186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Y, Tsoi K, Hunt RH. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119:719–727. doi: 10.1016/j.amjmed.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Pinto A, Farrar JT, Hersh EV. Prescribing NSAIDs to patients on SSRIs: possible adverse drug interaction of importance to dental practitioners. Compend Contin Educ Dent. 2009;30:142–151. [PubMed] [Google Scholar]
- 15.Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol. JAMA. 1994;272:1845–1850. doi: 10.1001/jama.1994.03520230055038. [DOI] [PubMed] [Google Scholar]
- 16.Abramowicz M, editor. Acetaminophen safety—deja vu. Med Lett Drugs Ther. 2009;51:53–54. editor. [PubMed] [Google Scholar]
- 17.Abramowicz M, editor. Warfarin‐acetaminophen interaction. Med Lett Drugs Ther. 2008;50:45. editor. [PubMed] [Google Scholar]
- 18.Gutstein HB, Akil H. Opioid analgesics. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw‐Hill; 2006. [Google Scholar]
- 19.Abramowicz M, editor. Drug interactions. Med Lett Drugs Ther. 1999;41:61–62. editor. [PubMed] [Google Scholar]
- 20.Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613–624. doi: 10.1016/S0025-6196(11)60750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinik HR, Bradley EL, Jr, Kissin I. Triple anesthetic combination: propofol‐midazolam‐alfentanil. Anesth Analg. 1994;78:354–358. doi: 10.1213/00000539-199402000-00026. [DOI] [PubMed] [Google Scholar]
- 22.Abramowicz M, editor. CYPA and drug interactions. Med Lett Drugs Ther. 2005;47:54–55. ed. [PubMed] [Google Scholar]
- 23.Berde CB, Strichartz GR. Local anesthetics. In: Miller RD, Eriksson LI, Fleisher LA, Wiener‐Kronish JP, Young WL, editors. Miller's Anesthesia. 7th ed. Philadelphia, Pa: Elsevier, Churchhill Livingstone; 2009. [Google Scholar]
- 24.Goodson JM, Moore PA. Life‐threatening reactions after pedodontic sedation: an assessment of narcotic, local anesthetic and antiemetic drug interactions. J Am Dent Assoc. 1983;107:239–245. doi: 10.14219/jada.archive.1983.0225. [DOI] [PubMed] [Google Scholar]
- 25.Montamat SC, Cusack BJ, Vestal RE. Management of drug therapy in the elderly. N Engl J Med. 1989;321:303–309. doi: 10.1056/NEJM198908033210507. [DOI] [PubMed] [Google Scholar]
- 26.Yagiela JA, Duffin SR, Hunt LM. Drug interactions and vasoconstrictors used in local anesthetic solutions. Oral Surg Oral Med Oral Pathol. 1985;59:565–571. doi: 10.1016/0030-4220(85)90181-1. [DOI] [PubMed] [Google Scholar]
- 27.Foster CA, Aston SJ. Propranolol‐epinephrine interaction: a potential disaster. Plast Reconstr Surg. 1983;72:74–78. doi: 10.1097/00006534-198307000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Gandy W. Severe epinephrine‐propranolol interaction. Ann Emerg Med. 1989;18:98–99. doi: 10.1016/s0196-0644(89)80324-5. [DOI] [PubMed] [Google Scholar]
- 29.Mito RS, Yagiela JA. Hypertensive response to levonordefrin in a patient receiving propranolol: report of a case. J Am Dent Assoc. 1988;116:55–57. doi: 10.14219/jada.archive.1988.0155. [DOI] [PubMed] [Google Scholar]
- 30.Reimers D, Jezek A. The simultaneous use of rifampicin and other antitubercular agents with oral contraceptives. Prax Klin Pneumol. 1971;25:255–262. [PubMed] [Google Scholar]
- 31.Sondheimer SJ. Update on oral contraceptive pills and postcoital contraception. Curr Opin Obstet Gynecol. 1992;4:502–505. [PubMed] [Google Scholar]
- 32.Back DJ, Orme ML. Pharmacokinetic drug interactions with oral contraceptives. Clin Pharmacokinet. 1990;18:472–484. doi: 10.2165/00003088-199018060-00004. [DOI] [PubMed] [Google Scholar]
- 33.Archer JS, Archer DF. Oral contraceptive efficacy and antibiotic interaction: a myth debunked. J Am Acad Dermatol. 2002;46:917–923. doi: 10.1067/mjd.2002.120448. [DOI] [PubMed] [Google Scholar]
- 34.Dogterom P, van den Heuvel MW, Thomsen T. Absence of pharmacokinetic interactions of the combined contraceptive vaginal ring NuvaRing with oral amoxicillin or doxycycline in two randomised trials. Clin Pharmacokinet. 2005;44:429–438. doi: 10.2165/00003088-200544040-00007. [DOI] [PubMed] [Google Scholar]
- 35.Becker DE. Drug interactions in dental practice: a summary of facts and controversies. Compendium. 1994;15:1228–1242. [PubMed] [Google Scholar]
- 36.DeRossi SS, Hersh EV. Antibiotics and oral contraceptives. Dent Clin North Am. 2002;46:653–664. doi: 10.1016/s0011-8532(02)00017-4. [DOI] [PubMed] [Google Scholar]
- 37.Rocco TP, Fang JC. Pharmacological treatment of heart failure. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw‐Hill; 2006. [Google Scholar]