

Abstract
Hereditary etiologies of pancreatic and hepatobiliary cancers are increasingly recognized. An estimated >10% of pancreatic and increasing number of hepatobiliary cancers are hereditary. The cumulative risk of hereditary pancreatic cancer ranges from measurable but negligible in cystic fibrosis to a sobering 70% in cases of hereditary pancreatitis. Candidates for pancreatic cancer surveillance are those with a risk pancreatic cancer estimated to be >10-fold that of the normal population. Screening for pancreatic cancer in high-risk individuals is typically performed by endoscopic ultrasound and should begin at least 10 years prior to the age of the youngest affected relative. Disease states known to be associated with increased risk of hepatocellular cancer include hereditary hemochromatosis, autoimmune hepatitis, porphyria, and α1-antitrypsin deficiency, with relative risks as high as 36-fold. Although much less is known about hereditary bile-duct cancers, Muir-Torre syndrome and bile salt export pump deficiency are diseases whose association with hereditary carcinogenesis is under investigation.
1. Introduction
Hereditary etiologies of carcinogenesis have been increasingly recognized over the past century. In 1889, Billroth was one of the first to recognize the occurrence of multiple primary malignant neoplasms [1] and Lynch et al. in 1967 one of the first to draw attention to the etiologic role of hereditary factors in their occurrence [2]. Since then, at least 78 identified genetic syndromes of hereditary cancer have been described [3] and many more syndromes undoubtedly remain to be identified.
Recent molecular and genetic advancements have brought international attention to pancreatic cancer (PC), with an estimated >10% of cases being hereditary in etiology. Although less common then hereditary PC, hereditary hepatobiliary cancers are also increasingly recognized.
2. Pancreas Cancer
PC is a lethal disease, with 277,000 new cases diagnosed per year globally [4]; in the USA, it was estimated to be 43,140 new cases of pancreatic cancer and 36,800 deaths from the disease in 2010 [5]. The definition of familial, or hereditary PC (HPC), the vast majority of which is pancreatic ductal adenocarcinoma (PDAC), is generally accepted to be defined as PC occurring in families with ≥2 first-degree relatives (FDRs) [6].
Six years after the landmark 1967 publication by Lynch et al. on the role of hereditary factors in the occurrence of multiple primary malignant neoplasms [2], a kindred of four siblings with pancreas adenocarcinoma was described [7]. Over the next two decades, several more publications highlighted the importance of the hereditary form of pancreatic adenocarcinoma, not only within a single generation but also across multiple generations and families [8, 9] (Table 1).
Table 1.
Epidemiological studies of HPC.
| Study, year [ref] | Type | Location | No. of patients | Findings |
|---|---|---|---|---|
| Falk, 1988 [9] | Case control | Louisiana, USA | 363 | OR 5.25 if FH |
|
| ||||
| Lynch, 1990 [10] | Descriptive | Nebraska, USA | 47 | 10-fold increased occurrence of PC if FH of PC (7.8% versus 0.6%) |
|
| ||||
| Ghadirian, 1991 [11] | Case control | Montréal, Canada | 179 | 13-fold increased occurrence of PC if FH of PC (6.7% versus 0.7%) |
|
| ||||
| Fernandez, 1994 [12] | Case control | Italy | 362 | 4-fold increased occurrence of PC if FH of PC (3.9% versus 1.1%); RR 3 if FH |
|
| ||||
| Dergham, 1997 [13] | Case series | Detroit, USA | 81 | 9% occurrence of PC if FH of PC. |
|
| ||||
| Coughlin, 2000 [14] | Cohort | USA | 3751 | RR 1.5 (men)–1.7 (women) if FH |
|
| ||||
| Hemminki, 2003 [15] | Cohort | Sweden | 21,000 | SIR 2.4 if FH (parent) |
|
| ||||
| Klein, 2004 [16] | Registry | Baltimore, USA | 5179 | SIR 32 if 3 FDR with PCSIR 6.4 if 2 FDR with PCSIR 4.5 if 1 FDR with PC |
|
| ||||
| Permuth-Wey, 2008 [17] | Meta-analysis | USA, Italy, Canada, Japan | 6568 | RR 3.4 if FH of PC |
OR: odds ratio; RR: relative risk; SIR: standardized incidence ratio; FH: family history; FDR: first-degree relative.
Population-based case-control studies have quantified the increase in risk for individuals from families with convincing family histories of PDAC. For example, a study in 1990 from the Hereditary Cancer Institute at Creighton University identified familial clustering among 47 cases of pancreatic cancer in 18 nuclear families with two or more first-degree relatives affected [10]. In this study, while only 0.7% of controls had positive family histories of the disease, 6.7% of PDAC cases had positive family histories. Ghadirian et al. [11] conducted a similar study on 179 French-speaking patients in Montréal and found that 7.8% of the patients who had PDAC had a family history of the disease compared with only 0.6% of the control population representing a 13-fold increase. Dergham et al. [13] reported a similar figure of 9% of pancreatic cancer patients having a family history. In a Northern Italy study including >300 patients with PDAC, a family history of pancreatic cancer was associated with an increased relative risk of 3 for the development of pancreatic cancer (RR = 3) [12].
Larger, national studies [14, 15] and a meta-analysis of studies [17] of thousands to tens of thousands of patients with PC similarly have revealed an increased relative risk ranging from 1.5 to 3.4 based on family history of PC. The risk of PC in family members of those who have PC is likely best estimated by analysis of the National Familial Pancreas Tumor Registry at Johns Hopkins Hospital [16]: among 5179 individuals in 838 kindreds, the risk (standardized incidence ratios) of persons with 1, 2, or 3 FDRs with PC was 4.6, 6.4, and 32, respectively (Table 1).
In addition to familial PC, among other genetic defect(s) which remain to be discovered, there are at least five well-described inherited profiles with known genetic defects that substantially increase the carrier's cumulative lifetime risk for developing HPC (Table 2).
Table 2.
Known inherited syndromes associated with increased risk of HPC.
| Syndrome | Genes | Gene function | Rate of PC in syndrome | O/E → risk of PC | Cumulative risk of PC | References |
|---|---|---|---|---|---|---|
| HP |
PRSS1; SPINK1 |
Trypsinogen; Protease Inhibitor | 8/246 = 3.2% 10/200 = 5.0% 26/418 = 6.2% |
8/0.15 → 53 10/0.115 → 87 26/NR → 67 |
25–70% | [18–20] |
| FAMMM | CDKN2/P16 | Tumor suppressor | 9/200 = 4.5% 66/466 = 14% 15/656 = 2.3% |
6/0.16 → 38a
2/0.03 → 65b 8/0.6 → 13 |
13–17% | [21–23] |
| HBOC |
BRCA1;
BRCA2; (PALB2) |
Tumor suppressor | 14/1181 = 1.2% | 14/4.4 → 5.9 7/1.3 → 8.9c |
1.2–6.9% | [24–26] |
| PJ | STK11/LKB1 | Tumor suppressor | 6/240 = 2.5% 4/31 = 13% |
NR/NR → 132 | 5–36% | [27–29] |
| HNPCC |
MLH1;
MSH2; MSH6; PMS2 |
DNA mismatch repair | 47 cases in 31 families | O/E→ 8.6 | 3.7% | [30] |
| CF | CFTR | Transmembrane conductance regulator | 1/28, 842 = 0.0035%d | 1/0.4 → 2.6 7/1.7 → 5.3 |
“Negligible” | [31, 32] |
| FPC | Unk. | Unk. | 2/1253 = 0.16% 4/634 = 0.63% 5/106 = 4.7% |
2/0.44 → 4.5 4/0.62 → 6.4 5/0.16 → 32 |
NR | [33] |
NR: not reported; HP: hereditary pancreatitis; FAMMM: familial atypical multiple mole melanoma; HBOC: hereditary breast and ovarian cancer; PJ: Peutz-Jeghers; HNPCC: hereditary nonpolyposis colorectal cancer; FPC: familial pancreas cancer; CF: cystic fibrosis.
aFemales ≥56 years old.
bFemales <55 years old.
cIf outside the ovarian cancer cluster region.
Families, not individuals.
2.1. Specific Diseases with Inherited Predisposition for Pancreatic Cancer
2.1.1. Hereditary Pancreatitis
Hereditary pancreatitis (HP) is an autosomal dominant disease that presents with episodes of acute pancreatitis in early childhood, as early as 5 years of age. As a result of such early onset of acute pancreatitis, most of these patients develop chronic pancreatitis in their second decade of life, unlike chronic alcoholic pancreatitis which presents later in life. Chronic HP is further distinguished from chronic alcoholic pancreatitis by the equal gender ratio, but is otherwise largely similar to nonhereditary pancreatitis (Table 3) [34].
Table 3.
Comparison of chronic alcoholic and chronic hereditary pancreatitis.
| Pancreatitis type | Chronic alcoholic pancreatitis | Chronic hereditary pancreatitis | P |
|---|---|---|---|
| Male to female ratio | 12.5 : 1 | 1 : 1 | |
| Age of onset (years) | 40 | 10.5 | <.05 |
| Delay in diagnosis (years) | 3 | 14.3 | <.05 |
| Presence of pseudocysts | 10% | 33% | <.05 |
| Presence of pancreatic calcifications | 57% | 58% | NS |
| Endocrine insufficiency | 70% | 50% | NS |
| Exocrine insufficiency | 42% | 38% | NS |
| Need for surgery | 41% | 50% | NS |
Modified from [34].
Any difficulty differentiating HP from the nonhereditary form on the basis of biochemical and laboratory differences was mitigated by the discovery of a genetic difference: a 1996 study of five kindreds with hereditary pancreatitis revealed that an arginine-histamine substitution at residue 117 of the cationic trypsinogen gene, PRSS1, was associated with the phenotypic expression of acute pancreatitis due to failure of the affected trypsinogen, leading to autodigestion of the pancreas [35]. A second mutation was later described—a single A to T mutation—to be associated with a less severe form of hereditary pancreatitis [36].
Hereditary pancreatitis is associated with a markedly increased risk (53-fold) for developing PC (Table 2), which usually develops after several decades of pancreatitis especially in those with paternal mode of inheritance [18]. Smoking, a known risk factor for PC, was found to have an even more pronounced detrimental effect on those with hereditary pancreatitis: in a cohort of 497 patients, smoking was found to double the risk for PC and to be associated with a 2-decade-earlier onset of PC compared with nonsmoking status [37].
2.1.2. Familial Atypical Multiple Mole Melanoma Syndrome
The familial atypical multiple mole melanoma syndrome (FAMMM) is an autosomal dominant syndrome associated with an increased risk of cutaneous melanoma, dysplastic nevi, and PC, among other neoplasms. The association of the FAMMM syndrome with PC was described in 1990 by Bergman et al. [21], who studied nine families (200 individuals) with FAMMM in the Netherlands, reporting nine cases of PC, which has a 13.4-fold increased risk compared with the general population.
The CDKN2A gene, also known as P16, is a tumor-suppressor gene with various mutations implicated in the development of FAMMM as well as other systemic cancers. Studies of FAMMM individuals whose P16/CDKN2A was rendered dysfunctional by mutations have noted that the risk of PC was increased 15- to 38-fold [22, 38].
2.1.3. Hereditary Breast and Ovarian Cancer Syndrome and PALB2 Loss
Mutations of BRCA1 and BRCA2, common in hereditary breast and ovarian cancer (HBOC) syndrome, predispose carriers to developing breast, ovarian, and a variety of other cancers including PC. Sequencing of BRCA2 in individuals with HPC from North American [39] and European [40] HPC registries have revealed deleterious mutations in nearly one-fifth of these individuals, making BRCA2 the most common genetic defect in HPC. Loss of BRCA2 in HPC has been observed to confer an increased risk of PC as high as nearly 9-fold [24]. The role of BRCA1 seems to be less pronounced, but still measureable at a 2- to 3-fold increased risk of PC in carriers of BRCA1 mutations [25, 26]. Goggins et al. at Johns Hopkins University [41] studied 41 unselected adenocarcinomas of the pancreas, 15 of which had allelic loss at the BRCA2 locus and 4 of those (9.8% overall) had a second-allele abnormality. Three of these four cancers (7.3% overall) were considered germ-line mutations (with confirmation in normal tissue) suggesting that the rate of germ-line BRCA2 mutations in apparently sporadic pancreatic cancer may be as high as in breast or ovarian cancer [41].
The recently recognized partner and localizer of BRCA2, PALB2, is also frequently lost in HPC, at a rate of approximately 3% in recent North American [42] and European [43] sequencing studies of HPC kindreds, making it the second most commonly lost gene in HPC. Knowledge of such mutations as BRCA2 and PALB2 has major therapeutic implications; PC in this group of patients has been shown to be exquisitely sensitive to DNA cross-linking agents such as mitomycin C (MMC) [44].
2.1.4. Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) is an autosomal dominant syndrome associated with loss of STK11/LKB1 gene function and is characterized by hamartomatous polyps in the gastrointestinal tract and pigmented skin lesions on the lips, oral mucosa, and digits. In a large retrospective cohort study of 34 PJS patients identified over 50 years of Mayo Clinic records, the overall risk for developing cancer in affected individuals was 9.9. The relative risk of developing gastrointestinal cancers was 50.5, and the risk was 5-fold higher in women (RR 151) than men (RR 30) [45]. The risk for developing PC was reported to be 5% at the age of 40 and 8% at the age of 60 in a study of 240 international PJS patients possessing the STK11 mutation [27]. Of note, all pancreatic cancers in that study were diagnosed between the age of 34 and 49 years [27]. That multi-institutional effort was recently extended to include 419 PJS patients, 297 with an identified STK11/LKB1 germ-line mutation [46], with similar results in the risk of PC (3- and 7-fold risk at 40 and 60 years of age, respectively.
2.1.5. Lynch Syndrome
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (HNPCC), is an autosomal dominant condition associated with mutations in DNA mismatch repair (MMR) genes including MLH1, MSH2, MSH6, PMS2, and others. The resulting compromise of DNA maintenance and repair leads to the accumulation of errors in the genome manifested in microsatellite instability and loss of normal tumor-suppressor function. Whereas Lynch I syndrome is comprised only of colorectal cancers, Lynch II has been characterized to include a number of extracolonic cancers including PC.
Kastrinos et al. [30] analyzed the data on 6,342 individuals from 147 families with MMR gene mutations from two major US cancer centers and found that 21.1% of the families reported a case of pancreatic cancer. The cumulative risk of pancreatic cancer in these individuals was 1.3% at age 50 and 3.7% at age 70, which corresponds to a an 8.6-fold increase compared to the general population [30].
2.2. Surveillance and Screening for PC
PC, specifically PDAC, is generally a lethal disease, and even at high-volume institutions, the median survival following resection of PDAC is less than 20 months and the 5-year survival is only 20% [47]. However, when very small, very favorable cases of PC are selected, long-term survival is possible, with 4- and 5-year survivals of 78% and 59% reported [48, 49]. Usually, such cases are incidentally and fortuitously discovered at an early stage. While screening the low-risk general population for PC would be associated with an unfavorable risk/benefit ratio (due the low overall incidence of PC and to the lack of a screening test that is readily available, noninvasive, and accurate), screening a population at very high (>10-fold [50]) risk may offer an opportunity to cure an otherwise uncurable cancer if discovered early.
Investigators at the University of Washington were among the first to describe the use of prospective screening and surveillance for high-risk individuals [51]: 7 of 14 individuals from 3 high-risk families who were screened with endoscopic ultrasonography (EUS), endoscopic retrograde cholangiopancreatography (ERCP), computed tomography (CT), and serum CEA and CA19-9 were found to have high-risk lesions based on concerning features on EUS and ERCP features. Pancreatectomy was therefore recommended and performed in all 7 patients, and all 7 patients had widespread dysplasia (PanINs), but no cancer or normal pancreas parenchyma was found in any of the specimens [51]. In a follow-up study at the same institution, Kimmey et al. screened 46 high-risk patients with EUS [52]: 13 patients had abnormal findings, 12 of whom underwent pancreatectomy with all 12 specimens showing widespread dysplasia (PanIN).
Canto et al. [53] at Johns Hopkins similarly reported on 38 high-risk (most with ≥3 relatives with PC) individuals who underwent screening with EUS and, if abnormal, then biopsy, ERCP, and CT. Resection was offered to and performed on 6 patients with a mass seen on EUS. On final pathology, 4 patients had a benign lesion, one patient had an IPMN, and one patient had PDAC [53]. Other centers around the world, including Germany, the Netherlands, and the US [54–56] have similarly begun screening programs.
Recommendations regarding screening and surveillance are in evolution. The University of Washington currently recommends surveillance to the following: (1) individuals with 2 or more first-degree relatives with PC, (2) individuals with one first-degree relative with PC diagnosed under the age of 50, (3) individuals with 2 or more relatives with pancreatic cancer, one of whom had PC at an early age, and (4) individuals with a genetic disorders, such as PJS and FAMMM [57]. Screening recommendations of the Fourth International Symposium of Inherited Diseases of the Pancreas [50], including both University of Washington and Johns Hopkins investigators, are slightly more stringent (Table 4) and include anyone deemed to have a risk of PC ≥10-fold the general population. As such, candidates for screening and surveillance include those with FAMMM, PJS, HP, or ≥3 first-degree relatives with PC, individuals with ≥3 first-, second-, or third-degree relatives with PC (at least one of whom is a first-degree relative), any member of a PJS family, those carrying mutations of BRCA1, BRCA2, or an MMR gene, and with at least one first- or second-degree relative with PC, and candidates with 2 relatives with PC in the same lineage (directly connected), at least one of whom is a first-degree relative of the candidate [50].
Table 4.
Candidates for pancreatic cancer surveillance.
| Candidates for PC surveillance (with >10-fold increased risk of PC) |
|---|
| Anyone with ≥3 first-degree relatives with PC |
| Individuals with ≥3 first-, second-, or third-degree relatives with PC, at least one of whom is a first-degree relative |
| Anyone with FAMMM, PJS, or HP |
| Any member of a PJS family |
| Carriers of mutations of BRCA1, BRCA2, or an MMR gene and with at least one first- or second-degree relative with PC |
| A person with 2 relatives in the same lineage (directly connected) with PC, at least one of whom is a first-degree relative of the candidate |
| Some people with two first-degree relatives with PC and favorable expert opinion |
Modified from [50].
After deciding which patients to screen, the questions of how and when to screen remain. In addition to EUS and ERCP, magnetic resonance imaging with cholangiopancreatography (MRI/MRCP) has more recently gained increasing interest as a screening modality. Vasen et al. [58] used MRI/MRCP to screen high-risk individuals with P16-leiden mutations. After a 4-year median follow-up period, out of 79 individuals screened, pancreatic cancer was diagnosed in 9% and precursor lesions in 11% [58]. Whichever screening tool is employed, a screening program should take place only in the setting of a high-volume center and with full informed consent. Patients who are not willing to undergo pancreatectomy for suspicious lesions identified on screening should not undergo screening. Care must also be taken to exclude patients with a recent history of pancreatitis or heavy alcohol intake, since EUS findings are similar in that population [59].
Normal EUS findings include homogenous parenchyma and a thin-walled, anechoic main pancreatic duct. Abnormal EUS features that are considered to warrant ERCP followup (Figure 1) include hypoechoic nodules and cysts, echogenic foci, parenchymal heterogeneity, narrowing or dilation of the pancreatic duct, and duct-wall echogenicity [52, 53, 59–61]. Unfortunately, many of these changes are also present in chronic pancreatitis and in recent heavy alcohol intake, as such patients must be stratified accordingly. Concerning ERCP features include saccular deformities or other irregularities of the pancreatic ducts [60, 61].
Figure 1.
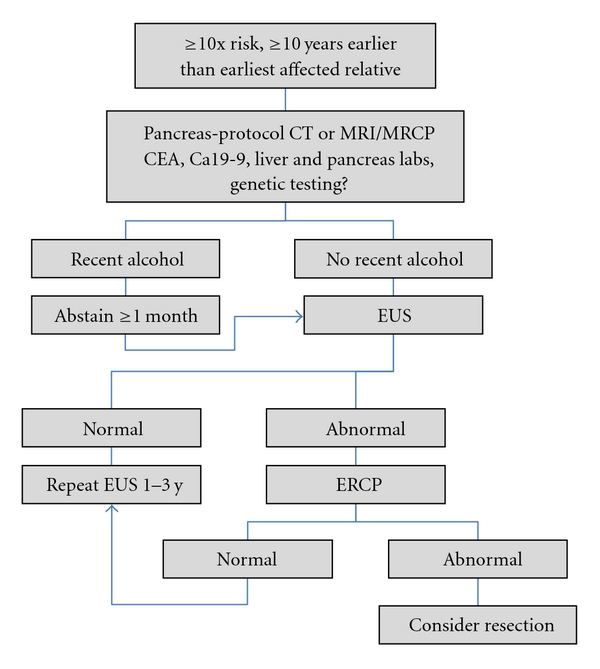
Flow chart for pancreatic cancer screening in high-risk individuals.
When to start screening is similarly not universally defined [50]. Applying the screening principles of colorectal cancer by beginning screening for pancreatic cancer 10 years earlier than the youngest affected member in the family is a reasonable starting point. Taking into account, however, the long time between initiation of a PDAC tumor cell and the presence of a PDAC tumor beginning to have metastatic capability (11.7 ± 3.1 years [62]), and taking into account evidence that consecutive generations with FPC die of PC a median of 10 years sooner each subsequent generation [63], and finally taking into account that smokers with FPC develop cancer a decade before nonsmokers, it is reasonable to use judgment in screening selected individuals much earlier. A reasonable screening algorithm is presented in Figure 1.
The treatment of patients who are deemed to have highrisk for cancer or a precancer lesion and who are found to have an abnormality by screening is pancreatectomy. Some recommend routine total pancretectomy, citing multifocality of the disease [59], whereas others recommended partial pancreatectomy [53]. Inadequate data exist to determine which option is associated with the most favorable risk/benefit ratio, but each patient's ability to manage the severe diabetes following total pancreatectomy must be weighed carefully on a case-by-case basis with the risk of leaving behind at-risk pancreas.
3. Hepatobiliary Cancers
3.1. Hepatocellular Carcinoma
Compared with PC, much less is known about hereditary hepatobiliary cancers. Although the great majority of hepatocellular carcinoma (HCC) cases are sporadic, some data exist to suggest an inherited component of risk. In a study of nearly 5000 HBV carriers from the Liver Unit of Chang-Gung Memorial Hospital and the Government Employee Central Clinics in Taipei, those who had a family history of HCC had a 2.4-fold risk of HCC compared with HBV carriers without a family history of HCC and this risk increased to 5.6-fold if two or more relatives were affected [64]. Similarly, analysis of the Swedish Cancer Registry [65], covering >10 million individuals, revealed a 4.7-fold increased risk of HCC in offspring of patients with HCC. In addition to general familial risk, several specific, known inherited liver diseases have been associated with increased risk for the development of HCC (Table 5).
Table 5.
Inherited diseases of the liver associated with HCC.
| Inherited diseases of the liver associated with HCC | ||
|---|---|---|
| Disease | RR | References |
| Hereditary hemochromatosis | 2–20 | [66–68] |
| Autoimmune hepatitis | 23 | [69] |
| Porphyria | 5–36 | [70, 71] |
| α1-antitrypsin deficiency | 5 | [72] |
| Progressive familial intrahepatic cholestasis | Unk. | [73, 74] |
| Glycogen storage disease type 1 (von Gierke disease) | Unk. | [75] |
| Hereditary tyrosinemia type I | Unk. | [76–78] |
| Wilson's disease | Unk. | [79] |
| Niemann-Pick disease | Unk. | [80] |
| Gaucher disease | Unk | [81] |
| Hereditary telangiectasias | Unk. | [82, 83] |
3.1.1. Specific Diseases with Inherited Predisposition for HCC
Hereditary hemochromatosis (HH) is an autosomal recessive disease associated with various mutations in the HFE gene resulting in progressive iron overload in the liver and elsewhere and is associated with an increased risk of HCC. To study whether HH, per se, and not chronic liver disease, is responsible for the increased cancer risk, Fracanzani et al. [66] analyzed the rate HCC in 230 patients with HH and 230 others with noniron-related chronic liver disease, finding a 1.9-fold increased risk of HCC in HH patients after controlling for alcohol abuse, smoking, and family history of cancer. In a modeling study using published life tables, age-specific cancer rates, and DNA studies of archived liver biopsy specimens, Haddow et al. [67] calculated the lifetime risk of HCC in a cohort of 5000 men with the common homozygous C282Y mutation in the HFE gene to be 23-fold compared with 1,000,000 normal men. Elmberg et al. [68] studied 1847 Swedish patients with HH and 5973 of their first-degree relatives. Patients with HH had a 20-fold increased risk of HCC, but their first-degree relatives had no increased risk of overall cancers and an only 1.5-fold increased risk of hepatobiliary cancers such as HCC [68]. A recent meta-analysis of 9 studies including 1102 HCC cases and 3766 controls in Europe revealed that the C282Y mutation but not the H63D mutation was associated with HCC in patients with alcoholic cirrhosis [84].
Other hereditary liver diseases have been associated with increased risk of HCC development, such as autoimmune hepatitis (RR 23) [69], porphyria (RR 5–36) [70, 71], α1-antitrypsin deficiency (RR 5) [72], progressive familial intrahepatic cholestasis (RR 3.7) [73, 74], glycogen storage disease type 1 (von Gierke disease) (RR unk.) [75], hereditary tyrosinemia type I (RR unk.) [76–78], Wilson's disease (RR unk.) [79], Niemann-Pick disease (RR unk.) [80], Gaucher disease (RR unk.) [81], and hereditary telangieatasias (RR unk.) [82, 83], but these associations are poorly studied due to the rarity of the disease processes.
3.2. Bile-Duct and Gallbladder Cancer
In a large, cooperative, case-control series from Milan, Fernandez et al. [12] prospectively followed 740 patients with pancreatic and hepatobiliary cancers compared with 1408 matched control patients and found a family history of gallbladder cancer in 1 of 58 patients with gallbladder cancer and in 2 of 1408 controls, yielding a relative risk of 13.9 but with a wide confidence interval (95% CI 1.2–163.9). Interestingly, a family history of stomach cancer was associated with a nearly 2-fold relative risk of gallbladder cancer [12].
Analysis of the >10-million-person-large Swedish Cancer Registry revealed a 5.2-fold increased risk of gallbladder cancer in the offspring of patients with gallbladder cancer [65]. There was a similar (3.8-fold), but only borderline significant, risk of extrahepatic bile-duct cancer when a family history of maternal ovarian cancer was present [65].
Several studies have reported an association between a family history of gallstones and gallbladder cancer. In a recent study from China's Shanghai Cancer Institute, Hsing et al. [85] confirmed earlier reports [86] that simply a family history of gallstones conferred an increased risk of gallbladder cancer (2.1-fold [85] to 3.6-fold [86]), even after adjustment for age, gender, marital status, education, smoking, alcohol drinking body mass index, and importantly, the presence of gallstones, which themselves further increase the risk.
3.2.1. Specific Diseases with Inherited Predisposition for Bile-Duct Cancer
In a cohort of 472 patients from 15 different families with HNPCC, cancer of the biliopancreatic tract was seen in 18 patients, 11 (79%) of which were confirmed as arising in the biliary tree or ampulla of Vater [87]. Despite a >9-fold increased risk of bile-duct cancer in patients with HNPCC [88], routine screening for bile-duct cancer has not been recommended [89], owing in large part to the difficulty in detecting these cancers and their rarity.
Muir-Torre syndrome (MTS) is an autosomal dominant syndrome described in the 1960s [90, 91] that predisposes to sebaceous skin lesions or keratoacanthomas and visceral tumors. MTS is a variant of HNPCC with the majority of germ-line mutations occurring in the MSH2 gene [92]. Several cases of bile-duct and ampullary cancers have been reported in association with MTS [93–95], including a report of a novel missense mutation in the MSH2 gene [93]. Although screening for biliary cancers is not currently practical, it has been suggested that screening for ampullary cancers in MTS patients would have a favorable risk/benefit ratio [95].
Bile salt export pump deficiency (BSEP), caused by mutations in ABCB11 [96], has been associated with bile-duct cancer [97]. In a study of 82 different ABCB11 mutations in 109 families [74], 19 of 128 patients (15%) with BSEP mutations developed hepatobiliary mutations, but only 2 of the 19 were bile-duct cancers, the remaining being HCC.
4. Summary
Among all hereditary cancers of the pancreas, liver, and biliary tree, only those of the pancreas have been studied well enough to allow for recommendations regarding screening and surveillance. While several known but rare forms of hereditary hepatobiliary cancer exist, screening recommendation cannot be made at this time due to the scarcity of available data. In contrast, any individual with a risk of PC estimated to be ≥10-fold should be screened with EUS by an experienced endoscopist in an experienced center after genetic counseling and informed consent, provided that the individual is willing to undergo pancreatectomy. Screening should begin at least 10 years prior to the age of the youngest affected relative and perhaps even earlier for select patients, such as smokers.
References
- 1.Billroth H. Die Allgemeine Chirurgie, Pathologie, und Therapie. Georg Reimer; 1889. [Google Scholar]
- 2.Lynch HT, Krush AJ, Larsen AL. Heredity and multiple primary malignant neoplasms: six cancer families. The American Journal of the Medical Sciences. 1967;254(3):322–329. doi: 10.1097/00000441-196709000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Garber JE, Offit K. Hereditary cancer predisposition syndromes. Journal of Clinical Oncology. 2005;23(2):276–292. doi: 10.1200/JCO.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International Journal of Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 6.Hruban RH, Petersen GM, Ha PK, Kern SE. Genetics of pancreatic cancer: from genes to families. Surgical Oncology Clinics of North America. 1998;7(1):1–23. [PubMed] [Google Scholar]
- 7.MacDermott RP, Kramer P. Adenocarcinoma of the pancreas in four siblings. Gastroenterology. 1973;65(1):137–139. [PubMed] [Google Scholar]
- 8.Ehrenthal D, Haeger L, Griffin T, Compton C. Familial pancreatic adenocarcinoma in three generations. A case report and a review of the literature. Cancer. 1987;59(9):1661–1664. doi: 10.1002/1097-0142(19870501)59:9<1661::aid-cncr2820590923>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 9.Falk RT, Pickle LW, Fontham ET, Correa P, Fraumeni JF. Life-style risk factors for pancreatic cancer in Louisiana: a case-control study. The American Journal of Epidemiology. 1988;128(2):324–336. doi: 10.1093/oxfordjournals.aje.a114972. [DOI] [PubMed] [Google Scholar]
- 10.Lynch HT, Fitzsimmons ML, Smyrk TC, et al. Familial pancreatic cancer: clinicopathologic study of 18 nuclear families. The American Journal of Gastroenterology. 1990;85(1):54–60. [PubMed] [Google Scholar]
- 11.Ghadirian P, Boyle P, Simard A, Baillargeon J, Maisonneuve P, Perret C. Reported family aggregation of pancreatic cancer within a population-based case-control study in the Francophone Community in Montreal, Canada. International Journal of Pancreatology. 1991;10(3-4):183–196. doi: 10.1007/BF02924156. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez E, La Vecchia C, D’Avanzo B, Negri E, Franceschi S. Family history and the risk of liver, gallbladder, and pancreatic cancer. Cancer Epidemiology Biomarkers and Prevention. 1994;3(3):209–212. [PubMed] [Google Scholar]
- 13.Dergham ST, Dugan MC, Arlauskas P, et al. Relationship of family cancer history to the expression of p53, p21WAF-1, HER-2/neu, and K-ras mutation in pancreatic adenocarcinoma . International Journal of Pancreatology. 1997;21(3):225–234. doi: 10.1007/BF02821608. [DOI] [PubMed] [Google Scholar]
- 14.Coughlin SS, Calle EE, Patel AV, Thun MJ. Predictors of pancreatic cancer mortality among a large cohort of United States adults. Cancer Causes and Control. 2000;11(10):915–923. doi: 10.1023/a:1026580131793. [DOI] [PubMed] [Google Scholar]
- 15.Hemminki K, Li X. Familial and second primary pancreatic cancers: a nationwide epidemiologic study from Sweden. International Journal of Cancer. 2003;103(4):525–530. doi: 10.1002/ijc.10863. [DOI] [PubMed] [Google Scholar]
- 16.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Research. 2004;64(7):2634–2638. doi: 10.1158/0008-5472.can-03-3823. [DOI] [PubMed] [Google Scholar]
- 17.Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Familial Cancer. 2009;8(2):109–117. doi: 10.1007/s10689-008-9214-8. [DOI] [PubMed] [Google Scholar]
- 18.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. Journal of the National Cancer Institute. 1997;89(6):442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 19.Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. The American Journal of Gastroenterology. 2008;103(1):111–119. doi: 10.1111/j.1572-0241.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- 20.Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clinical Gastroenterology and Hepatology. 2004;2(3):252–261. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 21.Bergman W, Watson P, de Jong J, Lynch HT, Fusaro RM. Systemic cancer and the FAMMM syndrome. The British Journal of Cancer. 1990;61(6):932–936. doi: 10.1038/bjc.1990.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borg A, Sandberg T, Nilsson K, et al. High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. Journal of the National Cancer Institute. 2000;92(15):1260–1266. doi: 10.1093/jnci/92.15.1260. [DOI] [PubMed] [Google Scholar]
- 23.Vasen HF, Gruis NA, Frants RR, Van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) International Journal of Cancer. 2000;87(6):809–811. [PubMed] [Google Scholar]
- 24.van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. Journal of Medical Genetics. 2005;42(9):711–719. doi: 10.1136/jmg.2004.028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BCRA1 mutation carriers identified in a risk evaluation program. Journal of the National Cancer Institute. 2002;94(18):1365–1372. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- 26.Thompson D, Easton DF. Cancer incidence in BRCA1 mutation carriers. Journal of the National Cancer Institute. 2002;94(18):1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 27.Lim W, Olschwang S, Keller JJ, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology. 2004;126(7):1788–1794. doi: 10.1053/j.gastro.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. The New England Journal of Medicine. 1987;316(24):1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 29.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 30.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. Journal of the American Medical Association. 2009;302(16):1790–1795. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maisonneuve P, FitzSimmons SC, Neglia JP, Campbell PW, Lowenfels AB. Cancer risk in nontransplanted and transplanted cystic fibrosis patients: a 10-year study. Journal of the National Cancer Institute. 2003;95(5):381–387. doi: 10.1093/jnci/95.5.381. [DOI] [PubMed] [Google Scholar]
- 32.Maisonneuve P, Marshall BC, Lowenfels AB. Risk of pancreatic cancer in patients with cystic fibrosis. Gut. 2007;56(9):1327–1328. doi: 10.1136/gut.2007.125278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersen GM, de Andrade M, Goggins M, et al. Pancreatic cancer genetic epidemiology consortium. Cancer Epidemiology Biomarkers and Prevention. 2006;15(4):704–710. doi: 10.1158/1055-9965.EPI-05-0734. [DOI] [PubMed] [Google Scholar]
- 34.Paolini O, Hastier P, Buckley M, et al. The natural history of hereditary chronic pancreatitis: a study of 12 cases compared to chronic alcoholic pancreatitis. Pancreas. 1998;17(3):266–271. doi: 10.1097/00006676-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nature Genetics. 1996;14(2):141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 36.Gorry MC, Gabbaizedeh D, Furey W, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113(4):1063–1068. doi: 10.1053/gast.1997.v113.pm9322498. [DOI] [PubMed] [Google Scholar]
- 37.Lowenfels AB, Maisonneuve P, Whitcomb DC, Lerch MM, DiMagno EP. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. Journal of the American Medical Association. 2001;286(2):169–170. doi: 10.1001/jama.286.2.169. [DOI] [PubMed] [Google Scholar]
- 38.Goldstein AM, Fraser MC, Struewing JP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. The New England Journal of Medicine. 1995;333(15):970–974. doi: 10.1056/NEJM199510123331504. [DOI] [PubMed] [Google Scholar]
- 39.Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17% Cancer Research. 2002;62(13):3789–3793. [PubMed] [Google Scholar]
- 40.Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutations in familial pancreatic carcinoma. Journal of the National Cancer Institute. 2003;95(3):214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- 41.Goggins M, Sehutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Research. 1996;56(23):5360–5364. [PubMed] [Google Scholar]
- 42.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):p. 217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slater EP, Langer P, Niemczyk E, et al. PALB2 mutations in European familial pancreatic cancer families. Clinical Genetics. 2010;78(5):490–494. doi: 10.1111/j.1399-0004.2010.01425.x. [DOI] [PubMed] [Google Scholar]
- 44.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Molecular Cancer Therapeutics. 2011;10(1):3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boardman LA, Thibodeau SN, Schaid DJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Annals of Internal Medicine. 1998;128(11):896–899. doi: 10.7326/0003-4819-128-11-199806010-00004. [DOI] [PubMed] [Google Scholar]
- 46.Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clinical Cancer Research. 2006;12(10):3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 47.Winter JM, Cameron JL, Campbell KA, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. Journal of Gastrointestinal Surgery. 2006;10(9):1199–1210. doi: 10.1016/j.gassur.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 48.Furukawa H, Okada S, Saisho H, et al. Clinicopathologic features of small pancreatic adenocarcinoma: a collective study. Cancer. 1996;78(5):986–990. doi: 10.1002/(SICI)1097-0142(19960901)78:5<986::AID-CNCR7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu Y, Yasui K, Matsueda K, Yanagisawa A, Yamao K. Small carcinoma of the pancreas is curable: new computed tomography finding, pathological study and postoperative results from a single institute. Journal of Gastroenterology and Hepatology. 2005;20(10):1591–1594. doi: 10.1111/j.1440-1746.2005.03895.x. [DOI] [PubMed] [Google Scholar]
- 50.Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460–1469. doi: 10.1136/gut.2006.108456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Annals of Internal Medicine. 1999;131(4):247–255. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 52.Kimmey MB, Bronner MP, Byrd DR, Brentnall TA. Screening and surveillance for hereditary pancreatic cancer. Gastrointestinal Endoscopy. 2002;56(4):S82–S86. doi: 10.1016/s0016-5107(02)70092-8. [DOI] [PubMed] [Google Scholar]
- 53.Canto MI, Goggins M, Yeo CJ, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clinical Gastroenterology and Hepatology. 2004;2(7):606–621. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 54.Langer P, Kann PH, Fendrich V, et al. Five years of prospective screening of high-risk individuals from families with familial pancreatic cancer. Gut. 2009;58(10):1410–1418. doi: 10.1136/gut.2008.171611. [DOI] [PubMed] [Google Scholar]
- 55.Poley JW, Kluijt I, Gouma DJ, et al. The yield of first-time endoscopic ultrasonography in screening individuals at a high risk of developing pancreatic cancer. The American Journal of Gastroenterology. 2009;104(9):2175–2181. doi: 10.1038/ajg.2009.276. [DOI] [PubMed] [Google Scholar]
- 56.Verna EC, Hwang C, Stevens PD, et al. Pancreatic cancer screening in a prospective cohort of high-risk patients: a comprehensive strategy of imaging and genetics. Clinical Cancer Research. 2010;16(20):5028–5037. doi: 10.1158/1078-0432.CCR-09-3209. [DOI] [PubMed] [Google Scholar]
- 57.UWSOMDOG. Pancreatic Cancer Care, Families with Pancreatic Cancer, Who is at Risk? 2011. http://www.uwgi.org/pancreaticcancer/Family.Risk.aspx?i=4.
- 58.Vasen HF, Wasser M, van Mil A, et al. Magnetic resonance imaging surveillance detects early-stage pancreatic cancer in carriers of a p16-Leiden mutation. Gastroenterology. 2011;140(3):850–856. doi: 10.1053/j.gastro.2010.11.048. [DOI] [PubMed] [Google Scholar]
- 59.Brentnall TA. Cancer surveillance of patients from familial pancreatic cancer kindreds. Medical Clinics of North America. 2000;84(3):707–718. doi: 10.1016/s0025-7125(05)70253-4. [DOI] [PubMed] [Google Scholar]
- 60.Canto MI. Screening and surveillance approaches in familial pancreatic cancer. Gastrointestinal Endoscopy Clinics of North America. 2008;18(3):535–553. doi: 10.1016/j.giec.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Brentnall TA. Management strategies for patients with hereditary pancreatic cancer. Current Treatment Options in Oncology. 2005;6(5):437–445. doi: 10.1007/s11864-005-0046-6. [DOI] [PubMed] [Google Scholar]
- 62.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McFaul CD, Greenhalf W, Earl J, et al. Anticipation in familial pancreatic cancer. Gut. 2006;55(2):252–258. doi: 10.1136/gut.2005.065045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu MW, Chang HC, Liaw YF, et al. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. Journal of the National Cancer Institute. 2000;92(14):1159–1164. doi: 10.1093/jnci/92.14.1159. [DOI] [PubMed] [Google Scholar]
- 65.Hemminki K, Li X. Familial liver and gall bladder cancer: a nationwide epidemiological study from Sweden. Gut. 2003;52(4):592–596. doi: 10.1136/gut.52.4.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fracanzani AL, Conte D, Fraquelli M, et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33(3):647–651. doi: 10.1053/jhep.2001.22506. [DOI] [PubMed] [Google Scholar]
- 67.Haddow JE, Palomaki GE, McClain M, Craig W. Hereditary haemochromatosis and hepatocellular carcinoma in males: a strategy for estimating the potential for primary prevention. Journal of Medical Screening. 2003;10(1):11–13. doi: 10.1258/096914103321610743. [DOI] [PubMed] [Google Scholar]
- 68.Elmberg M, Hultcrantz R, Ekbom A, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125(6):1733–1741. doi: 10.1053/j.gastro.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 69.Werner M, Almer S, Prytz H, et al. Hepatic and extrahepatic malignancies in autoimmune hepatitis. A long-term follow-up in 473 Swedish patients. Journal of Hepatology. 2009;50(2):388–393. doi: 10.1016/j.jhep.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 70.Andant C, Puy H, Bogard C, et al. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. Journal of Hepatology. 2000;32(6):933–939. doi: 10.1016/s0168-8278(00)80097-5. [DOI] [PubMed] [Google Scholar]
- 71.Fracanzani AL, Taioli E, Sampietro M, et al. Liver cancer risk is increased in patients with porphyria cutanea tarda in comparison to matched control patients with chronic liver disease. Journal of Hepatology. 2001;35(4):498–503. doi: 10.1016/s0168-8278(01)00160-x. [DOI] [PubMed] [Google Scholar]
- 72.Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? The European Journal of Gastroenterology and Hepatology. 1996;8(10):989–994. doi: 10.1097/00042737-199610000-00010. [DOI] [PubMed] [Google Scholar]
- 73.Knisely AS, Strautnieks SS, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44(2):478–486. doi: 10.1002/hep.21287. [DOI] [PubMed] [Google Scholar]
- 74.Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 75.Taddei T, Mistry P, Schilsky ML. Inherited metabolic disease of the liver. Current Opinion in Gastroenterology. 2008;24(3):278–286. doi: 10.1097/MOG.0b013e3282fcbc0f. [DOI] [PubMed] [Google Scholar]
- 76.Weinberg AG, Mize CE, Worthen HG. The occurence of hepatoma in the chronic form of hereditary tyrosinemia. Journal of Pediatrics. 1976;88(3):434–438. doi: 10.1016/s0022-3476(76)80259-4. [DOI] [PubMed] [Google Scholar]
- 77.Fisch RO, McCabe ER, Doeden D. Homotransplantation of the liver in a patient with hepatoma and hereditary tyrosinemia. Journal of Pediatrics. 1978;93(4):592–596. doi: 10.1016/s0022-3476(78)80893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scott CR. The genetic tyrosinemias. The American Journal of Medical Genetics Part C. 2006;142(2):121–126. doi: 10.1002/ajmg.c.30092. [DOI] [PubMed] [Google Scholar]
- 79.Iwadate H, Ohira H, Suzuki T, et al. Hepatocellular carcinoma associated with Wilson’s disease. Internal Medicine. 2004;43(11):1042–1045. doi: 10.2169/internalmedicine.43.1042. [DOI] [PubMed] [Google Scholar]
- 80.Birch NC, Radio S, Horslen S. Metastatic hepatocellular carcinoma in a patient with niemann-pick disease, type C. Journal of Pediatric Gastroenterology and Nutrition. 2003;37(5):624–626. doi: 10.1097/00005176-200311000-00023. [DOI] [PubMed] [Google Scholar]
- 81.Xu R, Mistry P, Mckenna G, et al. Hepatocellular carcinoma in type 1 Gaucher disease: a case report with review of the literature. Seminars in Liver Disease. 2005;25(2):226–229. doi: 10.1055/s-2005-871201. [DOI] [PubMed] [Google Scholar]
- 82.Jameson CF. Primary hepatocellular carcinoma in hereditary haemorrhagic telangiectasia: a case report and literature review. Histopathology. 1989;15(5):550–552. doi: 10.1111/j.1365-2559.1989.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 83.Sussman EB, Sternberg SS. Hereditary hemorrhagic telangiectasia. A case with hepatocellular carcinoma and acquired hepatocerebral degeneration. Archives of Pathology and Laboratory Medicine. 1975;99(2):95–100. [PubMed] [Google Scholar]
- 84.Jin F, Qu LS, Shen XZ. Association between C282Y and H63D mutations of the HFE gene with hepatocellular carcinoma in European populations: a meta-analysis. Journal of Experimental and Clinical Cancer Research. 2010;29(18) doi: 10.1186/1756-9966-29-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hsing AW, Bai Y, Andreotti G, et al. Family history of gallstones and the risk of biliary tract cancer and gallstones: a population-based study in Shanghai, China. International Journal of Cancer. 2007;121(4):832–838. doi: 10.1002/ijc.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Strom BL, Soloway RD, Rios-Dalenz JL, et al. Risk factors for gallbladder cancer. An international collaborative case-control study. Cancer. 1995;76(10):1747–1756. doi: 10.1002/1097-0142(19951115)76:10<1747::aid-cncr2820761011>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 87.Mecklin JP, Jarvinen HJ, Virolainen M. The association between cholangiocarcinoma and hereditary nonpolyposis colorectal carcinoma. Cancer. 1992;69(5):1112–1114. doi: 10.1002/cncr.2820690508. [DOI] [PubMed] [Google Scholar]
- 88.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. International Journal of Cancer. 1999;81(2):214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 89.Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with Lynch syndrome. The Lancet Oncology. 2009;10(4):400–408. doi: 10.1016/S1470-2045(09)70041-5. [DOI] [PubMed] [Google Scholar]
- 90.Muir EG, Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. The British Journal of Surgery. 1967;54(3):191–195. doi: 10.1002/bjs.1800540309. [DOI] [PubMed] [Google Scholar]
- 91.Torre D. Multiple sebaceous tumors. Archives of Dermatology. 1968;98(5):549–551. doi: 10.1001/archderm.98.5.549. [DOI] [PubMed] [Google Scholar]
- 92.Mangold E, Pagenstecher C, Leister M, et al. A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. Journal of Medical Genetics. 2004;41(7):567–572. doi: 10.1136/jmg.2003.012997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vernez M, Hutter P, Monnerat C, Halkic N, Gugerli O, Bouzourene H. A case of Muir-Torre syndrome associated with mucinous hepatic cholangiocarcinoma and a novel germline mutation of the MSH2 gene. Familial Cancer. 2007;6(1):141–145. doi: 10.1007/s10689-006-9105-9. [DOI] [PubMed] [Google Scholar]
- 94.Akhtar S, Oza KK, Khan SA, Wright J. Muir-Torre syndrome: case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. Journal of the American Academy of Dermatology. 1999;41(5):681–686. doi: 10.1016/s0190-9622(99)70001-0. [DOI] [PubMed] [Google Scholar]
- 95.Matthews JJ, Roberts R, O’Reilly DA, Schick S, Kingsnorth AN. Muir-Torre syndrome: a case for surveillance of the ampulla of Vater. Digestive Surgery. 2002;19(1):65–66. doi: 10.1159/000052010. [DOI] [PubMed] [Google Scholar]
- 96.Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nature Genetics. 1998;20(3):233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 97.Scheimann AO, Strautnieks SS, Knisely AS, Byrne JA, Thompson RJ, Finegold MJ. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. Journal of Pediatrics. 2007;150(5):556–559. doi: 10.1016/j.jpeds.2007.02.030. [DOI] [PubMed] [Google Scholar]