

Abstract
In 1999, two independent groups identified plasmacytoid dendritic cells (PDC) as major type I interferon- (IFN-) producing cells in the blood. Since then, evidence is accumulating that PDC are a multifunctional cell population effectively coordinating innate and adaptive immune responses. This paper focuses on the role of different immune cells and their interactions in the surveillance of alpha herpes virus infections, summarizes current knowledge on PDC surface receptors and their role in direct cell-cell contacts, and develops a risk factor model for the clinical implications of herpes simplex and varicella zoster virus reactivation. Data from studies involving knockout mice and cell-depletion experiments as well as human studies converge into a “spider web”, in which the direct and indirect crosstalk between many cell populations tightly controls acute, latent, and recurrent alpha herpes virus infections. Notably, cells involved in innate immune regulations appear to shape adaptive immune responses more extensively than previously thought.
1. Introduction
The human alpha herpes viruses comprise three different viruses: herpes simplex virus type 1 (HSV-1), type 2 (HSV-2), and varicella zoster virus (VZV) [1]. These highly cytopathic viruses are characterized by a short replication cycle, a broad cell tropism, and an efficient spread in cell culture. Importantly, they exhibit a distinct neurotropism, which, after primary infection at mucocutaneous sites, guides the viral particles along the peripheral sensory nerves to the dorsal root ganglia or the trigeminal ganglion, where they establish latent infection. Under circumstances of local or systemic immune suppression, alpha herpes viruses are reactivated and transported the same way, but in the reverse direction, to the epithelial surfaces. Primary HSV-1 and HSV-2 infections can manifest as stomatitis aphthosa, herpetic whitlow, and neonatal herpes acquired by passage through the maternal birth canal [2]. Reactivations are well known as cold sores, corneal, and genital herpes. Primary and recurrent VZV infections manifest as chickenpox and shingles, respectively [3]. In rare cases, alpha herpes viruses cause severe diseases such as encephalitis, acute retinal necrosis, and life-threatening systemic infections. It still remains a mystery why only a few individuals in the large cohort of seropositives are so severely affected by these viruses.
The first animal model to investigate the pathogenesis of acute, latent, and recurrent herpes viral infections was described by von Szily, who, after inoculation of herpes simplex virus into the anterior eye chamber of rabbits, observed the spread of inflammation along the optic nerve and infiltration of the uvea of the contralateral eye [4]. These results were confirmed by Whittum and colleagues in a mouse model [5], which paved the way for further experiments in which herpes virus pathogenesis was analyzed in knockout mice or by depletion of individual cell populations.
The focus of this paper is the immune control of alpha herpes virus infections. In particular, we wanted to summarize the role of cells involved in innate and adaptive immune responses and to highlight their interactions in the efficient control of acute and recurrent herpes virus infections with respect to the current literature. The reader is also referred to excellent reviews by others which addressed similar or related aspects of alpha herpes virus infections [6–9]. The corresponding immune escape mechanisms of alpha herpes viruses were recently described by others [10].
2. The Spider Web—Immune Surveillance of Alpha Herpes Virus Infections
The current literature on the control of acute and latent herpes virus infections is summarized in Figure 1. There is not only evidence that single cell populations play a direct role in the suppression of alpha herpes virus replication, but cells interact with each other and across the innate-adaptive barrier in mediating efficient surveillance.
Figure 1.
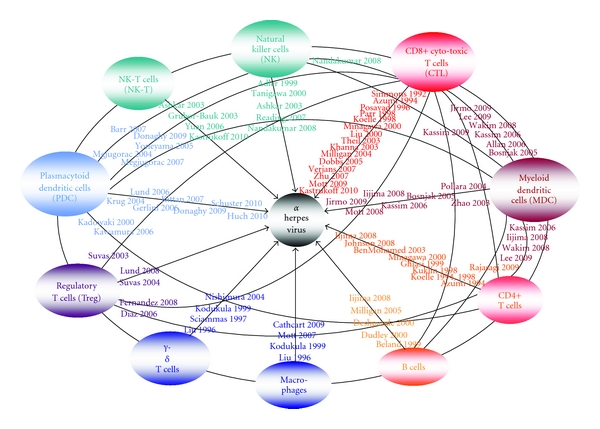
“Spider web” for the control of alpha herpes virus infections by cells of the innate and adaptive immune system (left and right side of the figure, resp.).
2.1. Innate Immune Control of Acute Herpes Virus Infections
The resolution of acute herpes simplex infections is mostly associated with macrophages and gamma delta T cells. Liu and colleagues detected both cell types infiltrating the trigeminal ganglion after corneal HSV-1 infection in 1996 [11]. Mice depleted of gamma delta T cells or macrophages suffered from a more severe HSV-1 infection after footpad or ocular inoculation [12, 13]. Similar findings were obtained when the macrophage-derived inducible nitric oxide synthase was inhibited, and tumor necrosis factor- (TNF-) alpha or interferon- (IFN-) gamma secreted by macrophages and gamma delta T cells, respectively, were neutralized [12]. Mice deficient in gamma delta T cells were shown to be susceptible to intravaginal HSV-2 infection [14]. Depletion of macrophages in immunized mice increased the replication of HSV-1 in the eye of infected mice at early timepoints [15]. After injection of HSV-1 into the anterior eye chamber of BALB/c mice, an early influx of macrophages prevented the spread to the ipsilateral retina [16].
A further case was made for the role of natural killer (NK) and NK-T cells in the control of acute herpes simplex virus infections. Thus, it was shown that mice lacking NK and T cells suffered from more severe encephalitis after intranasal HSV-1 inoculation compared to mice only lacking T cells [17]. When HSV-1 was inoculated into the anterior eye chamber of NK-depleted BALB/c mice, the virus rapidly spread to the ispilateral retina [18]. Interleukin (IL-)18 was shown to be involved in the rapid activation of NK cells and thus in the control of early HSV-1 replication in the lung after intranasal infection of C57BL/6 mice [19]. NK-deficient mice also displayed a reduced quality and quantity of CD8+ cytotoxic T lymphocytes (CTL), which defines a novel helper role for NK cells [20]. C57BL/6 mice deficient in NK and NK-T cells were more susceptible to vaginal HSV-2 infection than control mice; these cells were the early source of IFN-gamma in the vaginal fluids [21]. In C57BL/6 mice lacking CD1d or NK-T cells, the clearance of HSV-1 inoculated by skin scarification was impaired compared to control mice [22]. These data are confirmed by a study showing that HSV-1 evades NK-T cell recognition by suppressing CD1d recycling [23]. The presence of NK-T cells, however, appeared to be less critical, when a less virulent strain of HSV-1 was used for infection [24].
2.2. Control of Herpes Virus Latency by Adaptive Immune Responses
In a study published in 1994, the inoculation of HSV-1 into the anterior eye chamber of BALB/c mice was followed by the infiltration of the ipsi- and contralateral retina after depletion of CD4+ and CD8+ T cells [25]. These data were extended by others, reporting that CD4+ or CD8+ T cell functions were required for the clearance of HSV-1 from trigeminal ganglia and establishment of latent nonlytic infection in mice [26]. In recurrent human genital HSV-2 lesions, HSV-specific T cell clones were recovered, the majority of which was positive for CD4 [27]. In a followup study, the authors showed that the viral clearance from the lesions was associated with a high level of local CD4+ and CD8+ T cell cytolytic activities towards HSV-infected cells [28].
Several reports focus on CD4+ T cells as the major cell population in the immune control of alpha herpes virus infections. In a study using T cell subtype knockout mice and mice depleted with T cell subset-specific antibodies, immunity against vaginal HSV-2 challenge after vaccination with recombinant vectors expressing viral glycoproteins was dependent on CD4+ T cells [29]. Transfer of CD4+ T cells (and to a minor extent CD8+ T cells) from immunized BALB/c mice into severe combined immunodeficient (SCID) mice contributed to latent HSV-1 infection in the trigeminal ganglia [30]. Another group identified immunodominant epitopes from the HSV-1 glycoprotein D that conferred Th1 CD4+ T cell protective immunity against lethal ocular challenge in a mouse model [31]. In CD8+ T cell-depleted and -deficient mice, CD4+ T cells gradually cleared infectious HSV-1 from neural tissues, not dependent on perforin or Fas [32]. This mechanism was confirmed in another study, in which effector CD4+ T cells protected mice from HSV-2 infection after recurrent vaginal challenge, using a noncytolytic mechanism via local IFN-gamma secretion [33].
Various studies emphasize the role of CD8+ T cells in controlling alpha herpes virus infections. In this respect, CD8+ T cells were shown to be responsible for the clearance of infectious HSV-1 virions from murine ganglia [34]. Mice depleted of CD4+ and CD8+ T cells, but also those depleted of only CD8+ T cells, suffered from a more severe HSV-2 infection in the vaginal epithelium than nondepleted immune mice [35]. A high frequency of HSV-specific CD8+ cytotoxic T cell precursors has been detected in patients suffering from genital herpes [36]. Addition of CD8alpha antibodies to ex vivo cultures of trigeminal ganglia caused HSV-1 reactivation from latency as evident from the expression of late proteins and infectious virions [37]. These data suggested ongoing viral replication at the sites of herpes virus latency, which was confirmed by detection of viral transcripts of the ICP4 and thymidine kinase genes in mouse ganglia [38, 39]. Others quantified the amount of virus replication and reported about 500 neurons with latency-associated transcripts in each trigeminal ganglion compared to one neuron with early and late viral transcripts and viral protein per 10 trigeminal ganglia [40]. Latent HSV-1 infection in human trigeminal ganglia was shown to be accompanied by a chronic inflammatory process [41], and CD8+ T cells specific for an immunodominant HSV-1 glycoprotein B epitope could block HSV-1 reactivation from latency in explanted trigeminal ganglion cultures [42]. The mechanism was shown not to be dependent on perforin- or Fas/FasL-mediated cytolytic pathways but on IFN-gamma secretion [43, 44]. Herpes virus latency in human trigeminal and genital ganglia was reported to be associated with local, persistent T cell responses against HSV-1 and HSV-2, respectively [45, 46]. The level of HSV-1 latency correlated with higher levels of PD-1 mRNA, suggesting an exhaustion of CD8+ T cells in the trigeminal ganglia of latently infected mice [47]. Notably, recent data show a diminished CD8+ T cell response in the draining lymph nodes of CD4+ T cell-depleted mice [48]. Altogether, CD4+ and CD8+ T cells appear to play a major role in maintaining alpha herpes virus latency by suppression of lytic replication. In neuronal tissues, this control is mostly effected through noncytotoxic immune responses.
Beland and colleagues showed in 1999 that B cell deficient mice succumbed to HSV-1 encephalitis compared to control C57BL/6 mice and that the transfer of hyperimmune sera protected from death [49]. SCID mice which were given immune serum prior to corneal HSV-1 infection survived without latent infection [30]. In B cell-deficient mice, vaginal HSV-2 infection and spread was limited by the transfer of immune sera [50], and the susceptibility to cutaneous HSV infection was increased up to 1000-fold in BALB/c and C57BL/6 mice with a defect in the μ-immunoglobulin chain [51]. In addition, virus-specific plasma cells were found over an extended period in sensory ganglia following intravaginal HSV-2 inoculation, which may contribute to latency [52].
2.3. Modulation of Adaptive Immune Responses by Regulatory T Cells (Treg)
The role of Treg in the control of alpha herpes virus infections is controversially debated. Publications focus on the suppressive role of Treg on CD4+ and CD8+ T cells [53, 54]. Thus, enhanced CD8+ T cell responses and a lower viral load were observed in mice depleted for Treg prior to HSV-1 inoculation into the foot pad [54]. In a followup study, a more severe immunoinflammatory stromal keratitis was induced after HSV-1 inoculation into Treg-depleted mice [55]. Another study reported enhanced CD8+ cell cytotoxicity and IFN-gamma responses by CD4+ and CD8+ T cells as well as reduced viral titers in the draining lymph nodes of Treg-depleted mice [56]. Into a different direction goes a recent report on Treg-depleted mice, which suffered from increased viral load and accelerated fatal infection after vaginal HSV-2 inoculation, suggesting that Treg coordinate early protective responses by allowing a timely entry of NK cells, DC, and T cells into the infected tissue [57]. The discovery of distinct surface markers of Treg, for example, GARP, may facilitate further studies about the role of these cells in controlling HSV infection [58].
2.4. Coordination of Innate and Adaptive Immunity by Dendritic Cells (DC)
There is ample evidence that myeloid and plasmacytoid DC directly and indirectly contribute to the control of alpha herpes virus infections. Mice which were inoculated intravaginally with HSV-2 showed a rapid recruitment of submucosal myeloid dendritic cells (MDC) to the infected epithelium, which, presenting viral peptides in the MHC class II context, emerged in the draining lymph nodes and stimulated IFN-gamma secretion from HSV-specific CD4+ T cells [59]. The binding of HSV-1 glycoprotein D caused type I IFN secretion and maturation of MDC [60]. Others reported that HSV-infected apoptotic DC were phagocytosed by uninfected bystander DC, which, after cross-presentation of viral antigens, stimulated virus-specific CD8+ T cells [61]. The depletion of CD11c+ DC in the highly resistant C57BL/6 strain resulted in enhanced susceptibility to HSV-1 infection and spread into the nervous system; the authors also observed an impaired activation of NK cells, CD4+ and CD8+ T cells [62]. The viral antigen appears to be taken up initially by migratory DC and then transferred to lymphoid-resident DC for presentation and CTL priming [63]. Immunization of mice with Flt3-ligand DNA increased the number of CD11c+ CD8alpha+ DC and enhanced HSV-1 latency [64]. In extra-lymphoid tissues, HSV-specific memory CD8+ T cell responses were stimulated through a tripartite interaction with CD4+ T cells and DC recruited from the blood [65]. Dependent on the mode of infection, lymph node-resident or tissue-derived migratory DC pick up and present HSV-1 epitopes to CD4+ and CD8+ T cells [66]. Recently, it was shown that the uptake of virus released from target cells led to cross-presentation of viral antigens, which was important for CTL immunity in mice [67]. The cooperation of dendritic cells and B cells restimulated memory CD4+ T cells to secrete IFN-gamma [33], and the depletion of DC impaired the magnitude of IFN-gamma expression and cytotoxicity of NK cells [68].
The importance of type I interferons has been highlighted in a study in which the expression of IFN-alpha in astrocytes protected mice from acute and latent ocular HSV-1 infection [69]. The type I IFN response of plasmacytoid dendritic cells (PDC), the major producers of type I IFN, to HSV-1 is mediated via the Toll-like receptor (TLR) 9 /MyD88 pathway [70]. However, mice lacking MyD88 or TLR9 control HSV-1 replication, indicating that TLR9-independent pathways are also involved [70, 71]. HSV-1 stimulation of PDC, concomitant to IFN-alpha production, stimulated naïve CD4+ T cells to produce IFN-gamma and IL-10 [72]. It also induced migration of activated T and NK cells by production of the chemokines CCL4 and CXCL10 [73]. In a mouse model of cutaneous HSV-1 infection, PDC-depleted mice showed a reduced CD8+ T cell IFN-gamma production and HSV-specific lysis accompanied by an increase in viral load; PDC alone failed to induce CTL, however provided efficient help to lymph node dendritic cells [74]. In mice vaginally inoculated with HSV-2, PDC were recruited to the site of infection; PDC-depleted and TLR9-deficient mice had survival curves which were significantly worse than wild type mice [75]. In the human system, HSV-stimulated PDC were shown to stimulate the differentiation of naïve CD4+ T cells into cytotoxic “regulatory” T cells, which expressed perforin and granzymes, and produced IFN-gamma and IL-10 [76]. Another group reported that PDC-derived IL-18 rapidly activated NK cells in HSV-1 infection [77]. Human PDC were recruited to varicella skin lesions [78, 79], and infiltrated the dermis of recurrent genital herpes simplex lesions, closely associated with NK and T cells [80]. HSV-infected monocyte-derived DC induced IFN-alpha production in PDC through the exchange of cellular material [81]. We have reported reduced PDC counts and function together with reduced CD8+ T cell IFN-gamma production in patients with HSV- and VZV-associated acute retinal necrosis [82]. However, it remained unclear whether these deficits were cause or consequence of uncontrolled virus replication. Altogether, evidence is accumulating that PDC are directly and indirectly involved in the control of acute and latent alpha herpes virus infections.
3. The PDC “Life Cycle”—A Reflection of the Surface Receptor Repertoire
What is the evidence that PDC—in addition to secretion of type I IFN and proinflammatory cytokines—shape the immune response by direct cell-cell contact? The antigen-presenting properties of PDC, which we are just beginning to understand, were recently addressed in an excellent review [83]. The PDC surface receptors are increasingly coming into the focus of scientific research [84–88]. However, data about the function of PDC surface receptors in alpha herpes virus infections are scarce, and in most cases the function is deduced from the regulation of expression upon stimulation of PDC with HSV, other viruses or CpG, and from the function of these surface receptors on other cell populations.
Having these caveats in mind, we propose a model of the PDC “life cycle” (Figure 2), which was originally developed from chip and flow cytometry analyses of PDC in response to HSV-1 [87]. The timely coordinated regulation of PDC surface receptors suggests an inflammatory migration profile similar to gamma delta T cells [89], in which PDC are attracted to the site of infection, encounter their cognate antigen, reenter the blood stream, and then migrate to secondary lymphatic tissue. In more detail, immature PDC express three receptors for proinflammatory cytokines (CD183, CD184, and CD195), which were shown to be functional on these cells [90–92]. The presence of receptors involved in adhesion and rolling at the vascular endothelium (CD31, CD43, CD44, CD47, CD62L, CD99, and CD162) and those mediating firm adhesion and transmigration (CD11a, CD18, CD29, and CD49d) [93] suggest that PDC are recruited to inflamed tissue; explicit evidence is available for CD31 [92]. These data are corroborated by the detection of PDC in HSV and VZV lesions [75, 78–80]. At the site of infection, uptake of antigen may be mediated by three herpes virus entry mediators, namely CD111 (HVE-C), CD112 (HVE-B), and CD270 (HVE-A, HVEM) [80]. Other receptors relevant for antigen uptake may be CD4, the C-type lectin BDCA2 (CD303) [94, 95], Fc receptors [96–98], and CD170 (Siglec-5) [99]. Subsequent PDC activation and maturation can be seen from the enhanced expression of CD38, CD69, and CD83. Concomitant downregulation of adhesion molecules suggests that PDC can emigrate from the site of infection and reenter the blood stream. Homing to lymphatic tissue appears to be mediated by CD62L (L-selectin) [93, 100–102] and enhanced expression of the CC chemokine receptor 7 (CD197), which binds to CCL19 and CCL21 expressed by high endothelial venules in lymph nodes [90, 91, 103, 104]. Upon HSV-1 stimulation, CD197 was one of the few receptors which were upregulated with delay [87], suggesting that PDC indeed migrate from the site of infection to secondary lymphatic tissue. However, it has not been formally excluded that two different PDC populations migrate to different localizations.
Figure 2.
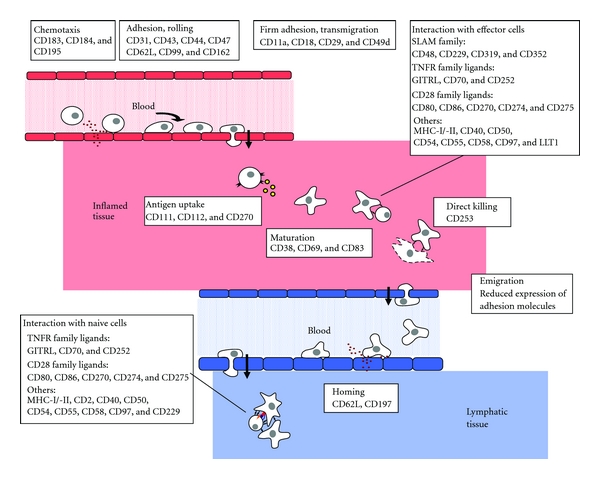
Proposed model for the plasmacytoid dendritic cell (PDC) “life cycle”, which is based on the timely coordinated expression of surface markers after contact with herpes simplex virus (HSV). The upper part of the figure depicts the attraction of PDC to inflamed tissue, where they take up their antigen and interact with local effector cells. Then, PDC reenter the blood stream and home to secondary lymphatic tissue via high endothelial venules (HEV), where they shape the adaptive immune response via interaction with naïve T cells, as outlined at the lower part of the figure.
The model of the PDC “life cycle” proposes two sites of interaction with other cells of the immune system: the inflamed tissue where PDC may encounter effector cells, and the lymphatic tissue, where PDC may interact with naïve cells and shape adaptive immune responses by the expression of coregulatory surface molecules. At the site of infection, expression of the SLAM family members CD48, CD229 (LY9), CD319 (CRACC), and CD352 (NTBA) may be particularly important for the interaction of PDC with NK and T cells [85, 87, 105–109]. CD319 may also induce proliferation of B cells [110]. This crosstalk appears to be mediated predominantly via homotypic receptor interactions [111].
Interaction of PDC with effector cells may also be influenced by the TNF receptor family [112]. PDC stimulated by virus or CpG express the ligand for the glucocorticoid-induced tumor necrosis factor receptor (GITRL), which may promote cytotoxicity and IFN-gamma production of CD357 (GITR)-expressing NK and T cells [113]. CpG-stimulated PDC fully activated NK-T cells involving direct contact of CD252 (OX40L) and CD134 (OX40) [114, 115]. CpG-activated PDC were also shown to promote B cell differentiation and immunoglobulin secretion via interaction of CD70 with CD27 [116].
A third family mediating coregulatory signals for naïve and memory T cells are the CD28 family ligands [112]. Of these, CD80, CD86, CD270, CD274 (B7-H1, PD-L1), and CD275 (B7-H2, ICOS-L) were described to be expressed on the PDC surface [80, 93, 95, 100, 117]. CpG- or HSV-stimulated PDC appear to drive CD4+ Treg immune responses via CD275 [117, 118], whereas CD40L-exposed PDC promoted the proliferation of CD8+ Treg [119].
CD50, CD54, and CD229 were reported to play an important role in the immunological synapse [120, 121], whereas the role of BDCA4 is still controversially discussed [122, 123]. PDC express the costimulatory molecules CD58 and CD97, which may costimulate NK and T cells [93, 124–126]. TLR9-activated PDC express the lectin-like transcript 1 (LLT1, CLEC2D), which interacts with CD161 on NK and effector T cells [127]. Recognition of virus-infected cells with subsequent induction of apoptotic and cytotoxic immune responses may be mediated by the enhanced expression of TRAIL (CD253) [128–130].
Several publications provide evidence for direct antigen presentation by PDC. After internalization and processing of influenza viruses, PDC cross-presented these antigens on MHC class I to CD8+ T cells [131]. Cross-priming of naïve CD8+ T cells with ovalbumin-exposed PDC was dependent on concomitant TLR activation [132]. Endocytosis of cytomegalovirus antigen together with CpG-B enhanced virus-specific memory CD4+ T cell responses [95]. In contrast, Yoneyama reported an indirect effect of PDC on the induction of CD8+ CTL by providing efficient help to lymph node DC via CD2 and CD40L in a model of cutaneous HSV-1 infection [74]. In addition, infiltrating CD8+ T cells may reciprocally influence PDC immune responses by secretion of IL-3 [133], which contributes to maturation and survival of PDC [100, 117].
4. The Risk Factor Model—Clinical Implications of Herpes Virus Reactivation
One of the best studied models of alpha herpes virus reactivation from latency is the acute retinal necrosis (ARN). Roughly one and two thirds of cases are caused by reactivation of HSV and VZV, respectively. Clinical course, treatment options and therapy outcome of this severe disease were recently reviewed [134–136]. The vasculitis, retinal necrosis, and intraocular inflammation start in the periphery and rapidly progress circumferentially, mostly affecting nonimmunocompromised patients at a frequency of 0.5 cases per 1 million. Current therapies combine antiviral and anti-inflammatory drugs with surgical intervention. An early vitrectomy with silicon oil instillation is associated with a lower incidence of retinal detachment compared to conservative treatment; however, the overall visual prognosis is poor [137, 138]. The cause appears to be, besides retinal detachment, herpes virus-associated retinal ischemia and atrophy of the optic nerve [139].
In patients suffering from herpes virus-associated ARN, several risk factors can be identified (Figure 3). As such, there are previous ARN or chorioretinal scars from other diseases, for example, toxoplasmosis; periocular trauma; ocular; or brain surgery; previous herpes simplex virus encephalitis and iatrogenic or acquired immune deficiency caused by corticoid and/or antineoplastic treatment and HIV-1 infection, respectively [140–142]. In a systemic survey of nine patients with ARN, we additionally identified meningitis/encephalitis and frequent infections in childhood, recurrent herpes virus affections at unusual localizations in patients and relatives, as well as (bacterial) infections and stress around ARN [82]. These factors point to a local break in the integrity of ocular tissues and a weakened immune system distracted by other challenges.
Figure 3.
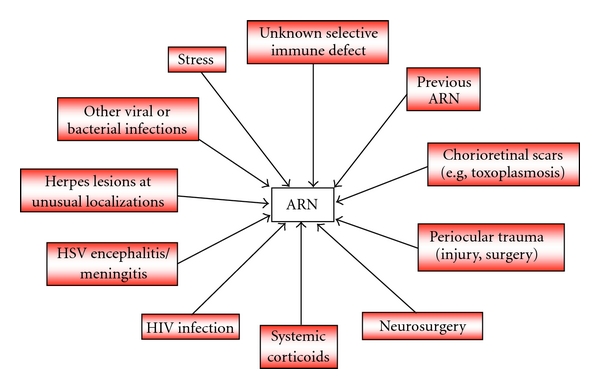
Risk factor model for the reactivation of latent herpes simplex (HSV) or varicella zoster virus infections leading to acute retinal necrosis (ARN).
In addition, one may propose an (unknown) selective defect in the innate or adaptive immunity, which predisposes to alpha herpes virus reactivation. In mice, TLR9 plays a major role for the recognition of HSV-1 and HSV-2 although TLR-independent pathways have been described [70, 143]. Knockout mice for TLR2 and TLR9 were shown to be more susceptible to HSV-1 encephalitis [144, 145]. Similarly, the interferon response factor 3, which is in the downstream signaling cascade of endosomal and cytosolic recognition factors for double-stranded RNA, plays an important role in the control of HSV-1 replication [146, 147]. In humans, TLR9 is expressed on PDC and B cells, but—unlike the murine system—not on myeloid DC, which may explain why severe alpha herpes virus infections have not been associated with TLR9 polymorphisms in humans. Instead, defects in STAT1 [148], TLR3 [149], and the endoplasmatic reticulum transmembrane protein UNC93B have been reported in patients suffering from herpes simplex virus-associated encephalitis [150, 151].
5. Conclusions
What have we learned from the published data ? General conclusions are shortened in terms of the variability of animal models used: different animal strains, different viruses, and different modes of infection may lead to different conclusions. Another caveat is that mouse data do not readily translate into the human system, for which the data are still limited. Yet, three important conclusions can be drawn. First, many cell populations are involved to keep primary and recurrent alpha herpes virus infections under control, which may reflect how seriously these cytopathic infections are taken by the immune system. A second conclusion is that the textbook knowledge of innate cells controlling primary infection and adaptive immunity supervising latent alpha herpes virus infections is no longer clearcut. Cells involved in innate immune regulations appear to shape adaptive immune responses more extensively than previously thought. Is there a master regulator in this system, for example, the two dendritic cell populations that coordinate each other and all other cells ? Or does each component of the immune system substitute for another in some respect ? A third conclusion is that interactions of different cell populations via soluble factors and in particular via surface receptors appear to be crucial to fight against alpha herpes virus infections [152, 153], which, if not controlled efficiently, will cause severe disabling diseases.
Acknowledgments
The authors thank Bernhard Fleckenstein for continuous support. P. Schuster, J. B. Boscheinen, and K. Tennert were supported by the graduate college 1071 (“Viruses of the Immune System”, Project B5). They acknowledge funding by the German Research Foundation (SCHM1702/2-1, SCHM1702/2-2, and SCHM1702/3-1) and the “Akademie der Wissenschaften und Literatur zu Mainz”. They apologize to all appreciated colleagues whose data they may have overlooked.
Conflict of Interests
The authors have no financial conflict of interests.
Abbreviations
- ARN:
Acute retinal necrosis
- CCR:
CC chemokine receptor
- CTL:
Cytotoxic T lymphocytes
- DC:
Dendritic cells
- HSV:
Herpes simplex virus
- IFN:
Interferon
- IL:
Interleukin
- L:
Ligand
- MDC:
Myeloid dendritic cells
- NK:
Natural killers
- PDC:
Plasmacytoid dendritic Cells
- SCID:
Severe combined immunodeficiency
- TLR:
Toll-like receptor
- TNF:
Tumor necrosis factor
- Treg:
Regulatory T cells
- VZV:
Varicella zoster virus.
References
- 1.Pellett PE, Roizman B. The family Herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins; 2007. pp. 2479–2499. [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins; 2007. pp. 2501–2601. [Google Scholar]
- 3.Cohen JI, Straus SE, Arvin AM. Varicella-zoster virus. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins; 2007. pp. 2773–2818. [Google Scholar]
- 4.von Szily AV. An experimental endogenous transmission of infection from bulbus to bulbus. Klinische Monatsblatter fur Augenheilkunde. 1924;75:593–602. [Google Scholar]
- 5.Whittum JA, McCulley JP, Niederkorn JY, Streilein JW. Ocular disease induced in mice by anterior chamber inoculation of herpes simplex virus. Investigative Ophthalmology and Visual Science. 1984;25(9):1065–1073. [PubMed] [Google Scholar]
- 6.Decman V, Freeman ML, Kinchington PR, Hendricks RL. Immune control of HSV-1 latency. Viral Immunology. 2005;18(3):466–473. doi: 10.1089/vim.2005.18.466. [DOI] [PubMed] [Google Scholar]
- 7.Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Current Opinion in Immunology. 2004;16(4):463–469. doi: 10.1016/j.coi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Van De Walle GR, Cox E, Nauwynck H, Favoreel HW. The role of dendritic cells in alphaherpesvirus infections: archetypes and paradigms. Reviews in Medical Virology. 2009;19(6):338–358. doi: 10.1002/rmv.628. [DOI] [PubMed] [Google Scholar]
- 9.Koelle DM, Corey L. Herpes simplex: insights on pathogenesis and possible vaccines. Annual Review of Medicine. 2008;59:381–395. doi: 10.1146/annurev.med.59.061606.095540. [DOI] [PubMed] [Google Scholar]
- 10.Griffin BD, Verweij MC, Wiertz EJHJ. Herpesviruses and immunity: the art of evasion. Veterinary Microbiology. 2010;143(1):89–100. doi: 10.1016/j.vetmic.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. Journal of Virology. 1996;70(1):264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kodukula P, Liu T, Van Rooijen N, Jager MJ, Hendricks RL. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. Journal of Immunology. 1999;162(5):2895–2905. [PubMed] [Google Scholar]
- 13.Sciammas R, Kodukula P, Tang Q, Hendricks RL, Bluestone JA. T cell receptor-γ/δ cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. Journal of Experimental Medicine. 1997;185(11):1969–1975. doi: 10.1084/jem.185.11.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura H, Yajima T, Kagimoto Y, et al. Intraepithelial γδ T cells may bridge a gap between innate immunity and acquired immunity to herpes simplex virus type 2. Journal of Virology. 2004;78(9):4927–4930. doi: 10.1128/JVI.78.9.4927-4930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mott K, Brick DJ, Van Rooijen N, Ghiasi H. Macrophages are important determinants of acute ocular HSV-1 infection in immunized mice. Investigative Ophthalmology and Visual Science. 2007;48(12):5605–5615. doi: 10.1167/iovs.07-0894. [DOI] [PubMed] [Google Scholar]
- 16.Cathcart HM, Fields MA, Zheng M, Marshall B, Atherton SS. Infiltrating cells and IFNγ production in the injected eye after uniocular anterior chamber inoculation of HSV-1. Investigative Ophthalmology and Visual Science. 2009;50(5):2269–2275. doi: 10.1167/iovs.08-2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler H, Beland JL, Del-Pan NC, Kobzik L, Sobel RA, Rimm IJ. In the absence of T cells, natural killer cells protect from mortality due to HSV-1 encephalitis. Journal of Neuroimmunology. 1999;93(1-2):208–213. doi: 10.1016/s0165-5728(98)00236-7. [DOI] [PubMed] [Google Scholar]
- 18.Tanigawa M, Bigger JE, Kanter MY, Atherton SS. Natural killer cells prevent direct anterior-to-posterior spread of herpes simplex virus type I in the eye. Investigative Ophthalmology and Visual Science. 2000;41(1):132–137. [PubMed] [Google Scholar]
- 19.Reading PC, Whitney PG, Barr DP, et al. IL-18, but not IL-12, regulates NK cell activity following intranasal herpes simplex virus type 1 infection. Journal of Immunology. 2007;179(5):3214–3221. doi: 10.4049/jimmunol.179.5.3214. [DOI] [PubMed] [Google Scholar]
- 20.Nandakumar S, Woolard SN, Yuan D, Rouse BT, Kumaraguru U. Natural killer cells as novel helpers in anti-herpes simplex virus immune response. Journal of Virology. 2008;82(21):10820–10831. doi: 10.1128/JVI.00365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. Journal of Virology. 2003;77(18):10168–10171. doi: 10.1128/JVI.77.18.10168-10171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grubor-Bauk B, Simmons A, Mayrhofer G, Speck PG. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant Vα14-Jα281 TCR. Journal of Immunology. 2003;170(3):1430–1434. doi: 10.4049/jimmunol.170.3.1430. [DOI] [PubMed] [Google Scholar]
- 23.Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nature Immunology. 2006;7(8):835–842. doi: 10.1038/ni1364. [DOI] [PubMed] [Google Scholar]
- 24.Cornish AL, Keating R, Kyparissoudis K, Smyth MJ, Carbone FR, Godfrey DI. NKT cells are not critical for HSV-1 disease resolution. Immunology and Cell Biology. 2006;84(1):13–19. doi: 10.1111/j.1440-1711.2005.01396.x. [DOI] [PubMed] [Google Scholar]
- 25.Azumi A, Atherton SS. Sparing of the ipsilateral retina after anterior chamber inoculation of HSV-1: requirement for either CD4+ or CD8+ T cells. Investigative Ophthalmology and Visual Science. 1994;35(8):3251–3259. [PubMed] [Google Scholar]
- 26.Ghiasi H, Perng GC, Nesburn AB, Wechsler SL. Either a CD4+ or CD8+ T cell function is sufficient for clearance of infectious virus from trigeminal ganglia and establishment of herpes simplex virus type 1 latency in mice. Microbial Pathogenesis. 1999;27(6):387–394. doi: 10.1006/mpat.1999.0314. [DOI] [PubMed] [Google Scholar]
- 27.Koelle DM, Abbo H, Peck A, Ziegweid K, Corey L. Direct recovery of herpes simplex virus (HSV)-specific T lymphocyte clones from recurrent genital HSV-2 lesions. Journal of Infectious Diseases. 1994;169(5):956–961. doi: 10.1093/infdis/169.5.956. [DOI] [PubMed] [Google Scholar]
- 28.Koelle DM, Posavad CM, Barnum GR, Johnson ML, Frank JM, Corey L. Clearance of HSV-2 from recurrent genital lesions correlates with infiltration of HSV-specific cytotoxic T lymphocytes. The Journal of Clinical Investigation. 1998;101(7):1500–1508. doi: 10.1172/JCI1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuklin NA, Daheshia M, Chun S, Rouse BT. Role of mucosal immunity in herpes simplex virus infection. Journal of Immunology. 1998;160(12):5998–6003. [PubMed] [Google Scholar]
- 30.Minagawa H, Yanagi Y. Latent herpes simplex virus-1 infection in SCID mice transferred with immune CD4+T cells: a new model for latency. Archives of Virology. 2000;145(11):2259–2272. doi: 10.1007/s007050070019. [DOI] [PubMed] [Google Scholar]
- 31.BenMohamed L, Bertrand G, McNamara CD, et al. Identification of novel immunodominant CD4+ Th1-type T-cell peptide epitopes from herpes simplex virus glycoprotein D that confer protective immunity. Journal of Virology. 2003;77(17):9463–9473. doi: 10.1128/JVI.77.17.9463-9473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson AJ, Chu CF, Milligan GN. Effector CD4+ T-cell involvement in clearance of infectious herpes simplex virus type 1 from sensory ganglia and spinal cords. Journal of Virology. 2008;82(19):9678–9688. doi: 10.1128/JVI.01159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iijima N, Linehan MM, Zamora M, et al. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. Journal of Experimental Medicine. 2008;205(13):3041–3052. doi: 10.1084/jem.20082039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmons A, Tscharke DC. Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: Implications for the fate of virally infected neurons. Journal of Experimental Medicine. 1992;175(5):1337–1344. doi: 10.1084/jem.175.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parr MB, Parr EL. Mucosal immunity to herpes simplex virus type 2 infection in the mouse vagina is impaired by in vivo depletion of T lymphocytes. Journal of Virology. 1998;72(4):2677–2685. doi: 10.1128/jvi.72.4.2677-2685.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posavad CM, Koelle DM, Corey L. High frequency of CD8 cytotoxic T-lymphocyte precursors specific for herpes simplex viruses in persons with genital herpes. Journal of Virology. 1996;70(11):8165–8168. doi: 10.1128/jvi.70.11.8165-8168.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. Journal of Experimental Medicine. 2000;191(9):1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. Journal of Virology. 1997;71(8):5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer MF, Coen DM. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. Journal of Virology. 1995;69(3):1389–1399. doi: 10.1128/jvi.69.3.1389-1399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theil D, Derfuss T, Paripovic I, et al. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. American Journal of Pathology. 2003;163(6):2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18(5):593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobbs ME, Strasser JE, Chu CF, Chalk C, Milligan GN. Clearance of herpes simplex virus type 2 by CD8+ T cells requires gamma interferon and either perform- or Fas-mediated cytolytic mechanisms. Journal of Virology. 2005;79(23):14546–14554. doi: 10.1128/JVI.79.23.14546-14554.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milligan GN, Dudley-McClain KL, Young CG, Chu CF. T-cell-mediated mechanisms involved in resolution of genital herpes simplex virus type 2 (HSV-2) infection of mice. Journal of Reproductive Immunology. 2004;61(2):115–127. doi: 10.1016/j.jri.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Verjans GMGM, Hintzen RQ, Van Dun JM, et al. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(9):3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu J, Koelle DM, Cao J, et al. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. Journal of Experimental Medicine. 2007;204(3):595–603. doi: 10.1084/jem.20061792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. Journal of Virology. 2009;83(5):2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajasagi NK, Kassim SH, Kollias CM, Zhao X, Chervenak R, Jennings SR. CD4+ T cells are required for the priming of CD8 T cells following infection with herpes simplex virus type 1. Journal of Virology. 2009;83(10):5256–5268. doi: 10.1128/JVI.01997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beland JL, Sobel RA, Adler H, Del-Pan NC, Rimm IJ. B cell-deficient mice have increased susceptibility to HSV-1 encephalomyelitis and mortality. Journal of Neuroimmunology. 1999;94(1-2):122–126. doi: 10.1016/s0165-5728(98)00238-0. [DOI] [PubMed] [Google Scholar]
- 50.Dudley KL, Bourne N, Milligan GN. Immune protection against HSV-2 in B-cell-deficient mice. Virology. 2000;270(2):454–463. doi: 10.1006/viro.2000.0298. [DOI] [PubMed] [Google Scholar]
- 51.Deshpande SP, Kumaraguru U, Rouse BT. Dual role of B cells in mediating innate and acquired immunity to herpes simplex virus infections. Cellular Immunology. 2000;202(2):79–87. doi: 10.1006/cimm.2000.1666. [DOI] [PubMed] [Google Scholar]
- 52.Milligan GN, Meador MG, Chu CF, Young CG, Martin TL, Bourne N. Long-term presence of virus-specific plasma cells in sensory ganglia and spinal cord following intravaginal inoculation of herpes simplex virus type 2. Journal of Virology. 2005;79(17):11537–11540. doi: 10.1128/JVI.79.17.11537-11540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diaz GA, Koelle DM. Human CD4+ CD25 high cells suppress proliferative memory lymphocyte responses to herpes simplex virus type 2. Journal of Virology. 2006;80(16):8271–8273. doi: 10.1128/JVI.00656-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. Journal of Experimental Medicine. 2003;198(6):889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. Journal of Immunology. 2004;172(7):4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez MA, Puttur FK, Wang YM, Howden W, Alexander SI, Jones CA. T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. Journal of Immunology. 2008;180(3):1556–1564. doi: 10.4049/jimmunol.180.3.1556. [DOI] [PubMed] [Google Scholar]
- 57.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320(5880):1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13439–13444. doi: 10.1073/pnas.0901965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao X, Deak E, Soderberg K, et al. Vaginal submucosal dendritic cells, but not langerhans cells, induce protective Th1 responses to herpes simplex virus-2. Journal of Experimental Medicine. 2003;197(2):153–162. doi: 10.1084/jem.20021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollara G, Jones M, Handley ME, et al. Herpes simplex virus type-1-induced activation of myeloid dendritic cells: the roles of virus cell interaction and paracrine type I IFN secretion. Journal of Immunology. 2004;173(6):4108–4119. doi: 10.4049/jimmunol.173.6.4108. [DOI] [PubMed] [Google Scholar]
- 61.Bosnjak L, Miranda-Saksena M, Koelle DM, Boadle RA, Jones CA, Cunningham AL. Herpes simplex virus infection of human dendritic cells induces apoptosis and allows cross-presentation via uninfected dendritic cells. Journal of Immunology. 2005;174(4):2220–2227. doi: 10.4049/jimmunol.174.4.2220. [DOI] [PubMed] [Google Scholar]
- 62.Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. Journal of Virology. 2006;80(8):3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allan RS, Waithman J, Bedoui S, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 64.Mott KR, Underhill D, Wechsler SL, Ghiasi H. Lymphoid-related CD11c+ CD8α + dendritic cells are involved in enhancing herpes simplex virus type 1 latency. Journal of Virology. 2008;82(20):9870–9879. doi: 10.1128/JVI.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wakim LM, Waithman J, Van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319(5860):198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- 66.Lee HK, Zamora M, Linehan MM, et al. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. Journal of Experimental Medicine. 2009;206(2):359–370. doi: 10.1084/jem.20080601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jirmo AC, Nagel CH, Bohnen C, Sodeik B, Behrens GMN. Contribution of direct and cross-presentation to CTL immunity against herpes simplex virus 1. Journal of Immunology. 2009;182(1):283–292. doi: 10.4049/jimmunol.182.1.283. [DOI] [PubMed] [Google Scholar]
- 68.Kassim SH, Rajasagi NK, Ritz BW, et al. Dendritic cells are required for optimal activation of natural killer functions following primary infection with herpes simplex virus type 1. Journal of Virology. 2009;83(7):3175–3186. doi: 10.1128/JVI.01907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carr DJJ, Veress LA, Noisakran S, Campbell IL. Astrocyte-targeted expression of IFN-α1 protects mice from acute ocular herpes simplex virus type 1 infection. Journal of Immunology. 1998;161(9):4859–4865. [PubMed] [Google Scholar]
- 70.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103(4):1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 71.Hochrein H, Schlatter B, O’Keeffe M, et al. Herpes simplex virus type-1 induces IFN-α production via Toll-like receptor 9-dependent and -independent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(31):11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadowaki N, Antonenko S, Lau JYN, Liu YJ. Natural interferon α/β-producing cells link innate and adaptive immunity. Journal of Experimental Medicine. 2000;192(2):219–225. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. Journal of Leukocyte Biology. 2004;75(3):504–514. doi: 10.1189/jlb.0603291. [DOI] [PubMed] [Google Scholar]
- 74.Yoneyama H, Matsuno K, Toda E, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. Journal of Experimental Medicine. 2005;202(3):425–435. doi: 10.1084/jem.20041961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting edge: plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. Journal of Immunology. 2006;177(11):7510–7514. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- 76.Kawamura K, Kadowaki N, Kitawaki T, Uchiyama T. Virus-stimulated plasmacytoid dendritic cells induce CD4+ cytotoxic regulatory T cells. Blood. 2006;107(3):1031–1038. doi: 10.1182/blood-2005-04-1737. [DOI] [PubMed] [Google Scholar]
- 77.Barr DP, Belz GT, Reading PC, et al. A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. European Journal of Immunology. 2007;37(5):1334–1342. doi: 10.1002/eji.200636362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gerlini G, Mariotti G, Bianchi B, Pimpinelli N. Massive recruitment of type I interferon producing plasmacytoid dendritic cells in varicella skin lesions. Journal of Investigative Dermatology. 2006;126(2):507–509. doi: 10.1038/sj.jid.5700052. [DOI] [PubMed] [Google Scholar]
- 79.Huch JH, Cunningham AL, Arvin AM, et al. Impact of varicella-zoster virus on dendritic cell subsets in human skin during natural infection. Journal of Virology. 2010;84(8):4060–4072. doi: 10.1128/JVI.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Donaghy H, Bosnjak L, Harman AN, et al. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. Journal of Virology. 2009;83(4):1952–1961. doi: 10.1128/JVI.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Megjugorac NJ, Jacobs ES, Izaguirre AG, George TC, Gupta G, Fitzgerald-Bocarsly P. Image-based study of interferongenic interactions between plasmacytoid dendritic cells and HSV-infected monocyte-derived dendritic cells. Immunological Investigations. 2007;36(5-6):739–761. doi: 10.1080/08820130701715845. [DOI] [PubMed] [Google Scholar]
- 82.Kittan NA, Bergua A, Haupt S, et al. Impaired plasmacytoid dendritic cell innate immune responses in patients with herpes virus-associated acute retinal necrosis. Journal of Immunology. 2007;179(6):4219–4230. doi: 10.4049/jimmunol.179.6.4219. [DOI] [PubMed] [Google Scholar]
- 83.Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29(3):352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells—virus experts of innate immunity. Seminars in Immunology. 2005;17(4):253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 85.Cabezón R, Sintes J, Llinàs L, Benitez-Ribas D. Analysis of HLDA9 mAbs on plasmacytoid dendritic cells. Immunology Letters. 2011;134(2):167–173. doi: 10.1016/j.imlet.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 86.Fitzgerald-Bocarsly P, Feng D. The role of type I interferon production by dendritic cells in host defense. Biochimie. 2007;89(6-7):843–855. doi: 10.1016/j.biochi.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schuster P, Donhauser N, Pritschet K, et al. Co-ordinated regulation of plasmacytoid dendritic cell surface receptors upon stimulation with herpes simplex virus type 1. Immunology. 2010;129(2):234–247. doi: 10.1111/j.1365-2567.2009.03176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunological Reviews. 2010;234(1):142–162. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moser B, Brandes M. γδ T cells: an alternative type of professional APC. Trends in Immunology. 2006;27(3):112–118. doi: 10.1016/j.it.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 90.Kohrgruber N, Gröger M, Meraner P, et al. Plasmacytoid dendritic cell recruitment by immobilized CXCR3 ligands. Journal of Immunology. 2004;173(11):6592–6602. doi: 10.4049/jimmunol.173.11.6592. [DOI] [PubMed] [Google Scholar]
- 91.Yoneyama H, Matsuno K, Zhang Y, et al. Evidence for recruitment of plasmacytoid dendritic cell precursors to inflamed lymph nodes through high endothelial venules. International Immunology. 2004;16(7):915–928. doi: 10.1093/intimm/dxh093. [DOI] [PubMed] [Google Scholar]
- 92.de la Rosa G, Longo N, Rodríguez-Fernández JL, et al. Migration of human blood dendritic cells across endothelial cell monolayers: adhesion molecules and chemokines involved in subset-specific transmigration. Journal of Leukocyte Biology. 2003;73(5):639–649. doi: 10.1189/jlb.1002516. [DOI] [PubMed] [Google Scholar]
- 93.Dzionek A, Fuchs A, Schmidt P, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. Journal of Immunology. 2000;165(11):6037–6046. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 94.Dzionek A, Sohma Y, Nagafune J, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon α/β induction. Journal of Experimental Medicine. 2001;194(12):1823–1834. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jaehn PS, Zaenker KS, Schmitz J, Dzionek A. Functional dichotomy of plasmacytoid dendritic cells: antigen-specific activation of T cells versus production of type I interferon. European Journal of Immunology. 2008;38(7):1822–1832. doi: 10.1002/eji.200737552. [DOI] [PubMed] [Google Scholar]
- 96.Fanning SL, George TC, Feng D, et al. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-α production. Journal of Immunology. 2006;177(9):5829–5839. doi: 10.4049/jimmunol.177.9.5829. [DOI] [PubMed] [Google Scholar]
- 97.Båve U, Magnusson M, Eloranta ML, Perers A, Alm GV, Rönnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with Lupus IgG. Journal of Immunology. 2003;171(6):3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 98.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. Journal of Clinical Investigation. 2005;115(2):407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lock K, Zhang J, Lu J, Lee SH, Crocker PR. Expression of CD33-related siglecs on human mononuclear phagocytes, monocyte-derived dendritic cells and plasmacytoid dendritic cells. Immunobiology. 2004;209(1-2):199–207. doi: 10.1016/j.imbio.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 100.Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent T1 polarization. Nature Immunology. 2000;1(4):305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- 101.Cella M, Jarrossay D, Faccheth F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature Medicine. 1999;5(8):919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 102.Venturi GM, Tu L, Kadono T, et al. Leukocyte migration is regulated by L-selectin endoproteolytic release. Immunity. 2003;19(5):713–724. doi: 10.1016/s1074-7613(03)00295-4. [DOI] [PubMed] [Google Scholar]
- 103.Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. Journal of Immunology. 2001;167(4):1862–1866. doi: 10.4049/jimmunol.167.4.1862. [DOI] [PubMed] [Google Scholar]
- 104.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nature Immunology. 2004;5(12):1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 105.Falco M, Marcenaro E, Romeo E, et al. Homophilic interaction of NTBA, a member of the CD2 molecular family: induction of cytotoxicity and cytokine release in human NK cells. European Journal of Immunology. 2004;34(6):1663–1672. doi: 10.1002/eji.200424886. [DOI] [PubMed] [Google Scholar]
- 106.Flaig RM, Stark S, Watzl C. Cutting edge: NTB-A activates NK cell via homophilic interaction. Journal of Immunology. 2004;172(11):6524–6527. doi: 10.4049/jimmunol.172.11.6524. [DOI] [PubMed] [Google Scholar]
- 107.Stark S, Watzl C. 2B4 (CD244), NTB-A and CRACC (CS1) stimulate cytotoxicity but no proliferation in human NK cells. International Immunology. 2006;18(2):241–247. doi: 10.1093/intimm/dxh358. [DOI] [PubMed] [Google Scholar]
- 108.Martín M, Del Valle JM, Saborit I, Engel P. Identification of Grb2 as a novel binding partner of the signaling lymphocytic activation molecule-associated protein binding receptor CD229. Journal of Immunology. 2005;174(10):5977–5986. doi: 10.4049/jimmunol.174.10.5977. [DOI] [PubMed] [Google Scholar]
- 109.Valdez PA, Wang H, Seshasayee D, et al. NTB-A, a new activating receptor in T cells that regulates autoimmune disease. Journal of Biological Chemistry. 2004;279(18):18662–18669. doi: 10.1074/jbc.M312313200. [DOI] [PubMed] [Google Scholar]
- 110.Jae KL, Mathew SO, Vaidya SV, Kumaresan PR, Mathew PA. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine expression on human B lymphocytes. Journal of Immunology. 2007;179(7):4672–4678. doi: 10.4049/jimmunol.179.7.4672. [DOI] [PubMed] [Google Scholar]
- 111.Calpe S, Wang N, Romero X, et al. The SLAM and SAP gene families control innate and adaptive immune responses. Advances in Immunology. 2008;97:177–250. doi: 10.1016/S0065-2776(08)00004-7. [DOI] [PubMed] [Google Scholar]
- 112.Duttagupta PA, Boesteanu AC, Katsikis PD. Costimulation signals for memory CD8+ T cells during viral infections. Critical Reviews in Immunology. 2009;29(6):469–486. doi: 10.1615/critrevimmunol.v29.i6.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hanabuchi S, Watanabe N, Wang Y-H, et al. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL) Blood. 2006;107(9):3617–3623. doi: 10.1182/blood-2005-08-3419. [DOI] [PubMed] [Google Scholar]
- 114.Marschner A, Rothenfusser S, Hornung V, et al. CpG ODN enhance antigen-specific NKT cell activation via plasmacytoid dendritic cells. European Journal of Immunology. 2005;35(8):2347–2357. doi: 10.1002/eji.200425721. [DOI] [PubMed] [Google Scholar]
- 115.Diana J, Griseri T, Lagaye S, et al. NKT cell-plasmacytoid dendritic cell cooperation via OX40 controls viral infection in a tissue-specific manner. Immunity. 2009;30(2):289–299. doi: 10.1016/j.immuni.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 116.Shaw J, Wang YH, Ito T, Arima K, Liu YJ. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood. 2010;115(15):3051–3057. doi: 10.1182/blood-2009-08-239145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ito T, Yang M, Wang YH, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. Journal of Experimental Medicine. 2007;204(1):105–115. doi: 10.1084/jem.20061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moseman EA, Liang X, Dawson AJ, et al. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. Journal of Immunology. 2004;173(7):4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- 119.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. Journal of Experimental Medicine. 2002;195(6):695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Romero X, Zapater N, Calvo M, et al. CD229 (Ly9) lymphocyte cell surface receptor interacts homophilically through its N-terminal domain and relocalizes to the immunological synapse. Journal of Immunology. 2005;174(11):7033–7042. doi: 10.4049/jimmunol.174.11.7033. [DOI] [PubMed] [Google Scholar]
- 121.Montoya MC, Sancho D, Bonello G, et al. Role of ICAM-3 in the initial interaction of T lymphocytes and APCs. Nature Immunology. 2002;3(2):159–168. doi: 10.1038/ni753. [DOI] [PubMed] [Google Scholar]
- 122.Dzionek A, Inagaki Y, Okawa K, et al. Plasmacytoid dendritic cells: from specific surface markers to specific cellular functions. Human Immunology. 2002;63(12):1133–1148. doi: 10.1016/s0198-8859(02)00752-8. [DOI] [PubMed] [Google Scholar]
- 123.Tordjman R, Lepelletier Y, Lemarchandel V, et al. A neuronal receptor, neuropilin-I, is essential for the initiation of the primary immune response. Nature Immunology. 2002;3(5):477–482. doi: 10.1038/ni789. [DOI] [PubMed] [Google Scholar]
- 124.Crawford K, Stark A, Kitchens B, et al. CD2 engagement induces dendritic cell activation: implications for immune surveillance and T-cell activation. Blood. 2003;102(5):1745–1752. doi: 10.1182/blood-2002-07-2206. [DOI] [PubMed] [Google Scholar]
- 125.Capasso M, Durrant LG, Stacey M, Gordon S, Ramage J, Spendlove I. Costimulation via CD55 on human CD4+ T cells mediated by CD97. Journal of Immunology. 2006;177(2):1070–1077. doi: 10.4049/jimmunol.177.2.1070. [DOI] [PubMed] [Google Scholar]
- 126.Matmati M, Pouwels W, Van Bruggen R, et al. The human EGF-TM7 receptor EMR3 is a marker for mature granulocytes. Journal of Leukocyte Biology. 2007;81(2):440–448. doi: 10.1189/jlb.0406276. [DOI] [PubMed] [Google Scholar]
- 127.Rosen DB, Cao W, Avery DT, et al. Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. Journal of Immunology. 2008;180(10):6508–6517. doi: 10.4049/jimmunol.180.10.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chaperot L, Blum A, Manches O, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. Journal of Immunology. 2006;176(1):248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 129.Hardy AW, Graham DR, Shearer GM, Herbeuval JP. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-α . Proceedings of the National Academy of Sciences of the United States of America. 2007;104(44):17453–17458. doi: 10.1073/pnas.0707244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stary G, Klein I, Kohlhofer S, et al. Plasmacytoid dendritic cells express TRAIL and induce CD4 T-cell apoptosis in HIV-1 viremic patients. Blood. 2009;114(18):3854–3863. doi: 10.1182/blood-2009-04-217927. [DOI] [PubMed] [Google Scholar]
- 131.Di Pucchio T, Chatterjee B, Smed-Sörensen A, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nature Immunology. 2008;9(5):551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mouriès J, Moron G, Schlecht G, Escriou N, Dadaglio G, Lederc C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood. 2008;112(9):3713–3722. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fuchs A, Cella M, Kondo T, Colonna M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood. 2005;106(6):2076–2082. doi: 10.1182/blood-2004-12-4802. [DOI] [PubMed] [Google Scholar]
- 134.Rautenberg P, Grančičova L, Hillenkamp J, Nölle B, Roider JB, Fickenscher H. Acute retinal necrosis from the virologist’s perspective. Ophthalmologe. 2009;106(12):1065–1073. doi: 10.1007/s00347-009-2048-4. [DOI] [PubMed] [Google Scholar]
- 135.Gandorfer A, Thurau S. Acute retinal necrosis. Ophthalmologe. 2009;106(8):751–760. doi: 10.1007/s00347-009-1986-1. [DOI] [PubMed] [Google Scholar]
- 136.Pleyer U, Winterhalter S. Diagnosic and therapeutic aspects of herpes virus associated uveitis. Klinische Monatsblatter fur Augenheilkunde. 2010;227(5):407–412. doi: 10.1055/s-0029-1245338. [DOI] [PubMed] [Google Scholar]
- 137.Hillenkamp J, Nölle B, Bruns C, Rautenberg P, Fickenscher H, Roider J. Acute retinal necrosis: clinical features, early vitrectomy, and outcomes. Ophthalmology. 2009;116(10):1971–1975.e2. doi: 10.1016/j.ophtha.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 138.Hillenkamp J, Nolle B, Rautenberg P, Fickenscher H, Roider J. Acute retinal necrosis: clinical features and therapy options. Ophthalmologe. 2009;106(12):1058–1064. doi: 10.1007/s00347-009-2047-5. [DOI] [PubMed] [Google Scholar]
- 139.Witmer MT, Pavan PR, Fouraker BD, Levy-Clarke GA. Acute retinal necrosis associated optic neuropathy. doi: 10.1111/j.1755-3768.2010.01911.x. Acta Ophthalmologica. In press. [DOI] [PubMed] [Google Scholar]
- 140.Van Gelder RN, Willig JL, Holland GN, Kaplan HJ. Herpes simplex virus type 2 as a cause of acute retinal necrosis syndrome in young patients. Ophthalmology. 2001;108(5):869–876. doi: 10.1016/s0161-6420(01)00556-5. [DOI] [PubMed] [Google Scholar]
- 141.Tran THC, Stanescu D, Caspers-Velu L, et al. Clinical characteristics of acute HSV-2 retinal necrosis. American Journal of Ophthalmology. 2004;137(5):872–879. doi: 10.1016/j.ajo.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 142.Ganatra JB, Chandler D, Santos C, Kuppermann B, Margolis TP. Viral causes of the acute retinal necrosis syndrome. American Journal of Ophthalmology. 2000;129(2):166–172. doi: 10.1016/s0002-9394(99)00316-5. [DOI] [PubMed] [Google Scholar]
- 143.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. Journal of Experimental Medicine. 2003;198(3):513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lima GK, Zolini GP, Mansur DS, et al. Toll-Like Receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. American Journal of Pathology. 2010;177(5):2433–2445. doi: 10.2353/ajpath.2010.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sørensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. Journal of Immunology. 2008;181(12):8604–8612. doi: 10.4049/jimmunol.181.12.8604. [DOI] [PubMed] [Google Scholar]
- 146.Menachery VD, Leib DA. Control of herpes simplex virus replication is mediated through an interferon regulatory factor 3-dependent pathway. Journal of Virology. 2009;83(23):12399–12406. doi: 10.1128/JVI.00888-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Menachery VD, Pasieka TJ, Leib DA. Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. Journal of Virology. 2010;84(19):9685–9694. doi: 10.1128/JVI.00706-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nature Genetics. 2003;33(3):388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 149.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 150.Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 151.Tabeta K, Hoebe K, Janssen EM, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nature Immunology. 2006;7(2):156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 152.Gowrishankar K, Steain M, Cunningham AL, et al. Characterization of the host immune response in human ganglia after herpes zoster. Journal of Virology. 2010;84(17):8861–8870. doi: 10.1128/JVI.01020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kastrukoff LF, Lau AS, Takei F, et al. Redundancy in the immune system restricts the spread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice. Virology. 2010;400(2):248–258. doi: 10.1016/j.virol.2010.02.013. [DOI] [PubMed] [Google Scholar]