

Abstract
Advances in modern medicine have led to an increase in the median life span and an expansion of the world’s population over the age of 65. With increasing numbers of the population surviving to the extreme of age, those at risk for the development of pneumonia will approach 2 billion by the year 2050. Numerous age-related changes in the lung likely contribute to the enhanced occurrence of pneumonia in the elderly. Inflammation in the elderly has been shown to increase risk prior to infection; age-associated inflammation enhances bacterial ligand expression in the lungs which increases the ability of bacteria to attach and invade host cells. Conversely, the elaboration of the acute inflammatory response during early infection has been found to decrease with age resulting in a delayed immune response and diminished bacterial killing. Finally, the resolution of the inflammatory response during the convalescent stage back to “baseline” is often prolonged in the elderly and associated with negative outcomes, such as adverse cardiac events. The focus of this review will be to discuss our current understanding of the potential mechanisms by which dysregulated inflammation (both prior to and following an infectious insult) enhances susceptibility to and severity of community acquired pneumonia (CAP) in the elderly with an emphasis on pneumococcal pneumonia, the leading cause of CAP.
Keywords: Aging, Pneumonia, Inflammation, Toll-like Receptors, Statins
Aging is associated with an increased susceptibility to infectious disease. Despite improved clinical diagnosis and treatment guidelines, pneumonia remains the leading cause of infectious death for the elderly (those greater than 65 years of age) [1]. According to the United States Census Bureau by the year 2050 greater than 2 billion individuals will be over the age of 65 world-wide [1]. In the United States, the elderly population is estimated to double by the year 2050 reaching 88.5 million or approximately 20% of the population [2]. Thus, as the population ages, understanding the etiology and pathogenesis of pneumonia remain an important area of research.
The lower airways are sterile sites containing few neutrophils and lymphocytes under homeostatic conditions. However, the cellularity of the lung increases with age in humans and laboratory animals with studies documenting increased numbers of neutrophils and lymphocytes even in those with no apparent comorbidities [3]. The most abundant immune cell in the normal lung is the alveolar macrophage whose function is to rapidly clear inhaled particulates or infectious agents and, if necessary, initiate the inflammatory response. Resident alveolar macrophages and recruited neutrophils and monocytes serve to keep lung infections under control until the adaptive immune response develops and clearance of the pathogen occurs.
Numerous age-related changes in both the innate and adaptive immune system likely contribute to the enhanced occurrence of pneumonia in the elderly and are thoroughly discussed in other reviews [4–6]. Herein, we summarize and discuss our current understanding of the potential mechanisms by which dysregulated inflammation (both prior to and following an infectious insult) enhances susceptibility to and severity of community acquired pneumonia (CAP) in the elderly with a focus on pneumococcal pneumonia, the leading cause of infectious death in the elderly.
PNEUMONIA IN THE ELDERLY
It is estimated that the annual incidence of pneumonia in the community-dwelling elderly population is between 25 and 44 per 1000 individuals [5]. For those in nursing homes or institutionalized the incidence increases to 33 to 114 cases per 1000 individuals [5]. Mortality rates for those with CAP approach 30% and are higher in those with underlying comorbidities. In addition to higher incidence and mortality, elderly patients presenting with pneumonia often have a higher rate and length of hospitalization leading to higher cost of care. According to a 2002 study by Kaplan and colleagues the annual incidence of hospitalization for CAP in the United States was 18.3 cases per 1,000 elderly persons compared to 4 per 1000 cases overall [7]. Moreover, prior hospitalization for CAP is associated with increased rates of mortality within 1 year following discharge [8, 9] often due to cardiovascular failure [10], which may be the result of cardiomyocyte damage from bacterial cell wall components [11].
Many age-related changes collectively contribute to the enhanced susceptibility of the elderly to pneumonia including, but not limited to, decreased normal lung function, reduced mucociliary clearance, and a decline in both innate and adaptive immunity (immunosenescence) [1, 12, 13]. Immunosenescence is also a driving force for the reactivation of latent infectious diseases such as Herpes Zoster (shingles) and tuberculosis [14–16]. By far, a major risk factor for CAP in the elderly is the increased presence of comorbid conditions such as COPD and cardiovascular disease.
Although the preponderance of pneumonias are of bacterial origin, Influenza and Respiratory Syncytial Virus (RSV) account for approximately 5–30% of pneumonia cases with an average annual rate of influenza-associated deaths among the elderly of 66.1 deaths per 100,000 [17]. Secondary bacterial pneumonia following viral infection is a common complication and cause of mortality in the elderly [18]. Streptococcus pneumoniae (S. pneumoniae), Hemophilus influenzae and atypical bacteria such as Chlamydia pneumoniae and Mycoplasma pneumoniae account for the majority of community-acquired bacterial pneumonias in the elderly [4, 6]. Staphylococcus aureus and Gram-negative bacilli are also common among nursing home residents and hospitalized patients [4, 6]. Due to the difficulty in obtaining sufficient sputum samples in the elderly the etiology of approximately 40% of pneumonias remain unidentified.
S. pneumoniae is the leading cause of CAP among the elderly [19]. S. pneumoniae is an encapsulated Gram-positive diplococcus that normally asymptomatically colonizes the nasopharynx of healthy humans. Although the incidence of pneumococcal diseases is greatest in those less than 2 years of age, case-fatality rates following infection increase for those > 55 years of age [20, 21]. Worldwide, it is estimated that 1.6 million people die of pneumococcal disease annually [22]. The World Health Organization estimates that the mortality rate of adults with pneumococcal pneumonia averages 10–20%, and may exceed 50% in high-risk groups [19, 22]. Immunocompetent healthy adults rarely succumb to pneumococcal pneumonia suggesting that age-related changes are required for S. pneumoniae to overcome host defenses and cause disease. In particular, those with underlying comorbidities such as diabetes or coronary artery disease are at increased risk for the development of life-threatening invasive pneumococcal disease [19, 23].
Protective immunity against pneumococcal disease is mediated by antibodies against the anti-phagocytic capsular polysaccharide. Currently, the CDC recommends that those over the age of 65 be vaccinated with Pneumovax® 23, which contains 23 of the most common polysaccharide capsular serotypes. Importantly, the protective efficacy of this vaccine among the elderly is estimated to be 55–70% against bacteremia and meningitis but has not reduced the incidence of pneumonia [22, 24, 25]. This is likely due in part to the fact that older individuals often produce antibodies with lower opsonic avidity [26–28]. Thus, antibody responses to vaccination are less robust and decline over time. In lieu of identifying better antigens for vaccination, boosting the immunogenicity of the capsular polysaccharide, or including adjuvants, studies identifying host immune factors that are altered with age are crucial to allow for better design of immunomodulatory therapeutics to reduce the severity of disease in the elderly.
AGE-ASSOCIATED NFLAMMATION ENHANCES SUSCEPTIBILITY TO CAP
Epidemiologic studies by multiple investigators indicate that individuals >65 years experience higher levels of pro-inflammatory cytokines in blood and tissues when compared to healthy young adults [29–32]. This age-related increase of circulating pro-inflammatory cytokines within serum and tissues was coined “inflamm-aging” by Franceschi et al. in 2000 [33], however, several studies had already been published describing the observation of increased Interleukin (IL)-6 and C- reactive protein (CRP) in serum from aged humans [34–36]. In the lungs, studies by Meyer et al. have shown that healthy elderly patients have increased IL-6, IL-8, immunoglobulin and neutrophil elastase, as well as higher numbers of neutrophils and lymphocytes in bronchoalveolar lavage when compared to younger subjects [13]. Inflammaging or age-associated inflammation (AAI) has also been described in aged rodents. Examination of tissues from aged mice revealed higher levels of Nuclear Factor kappa B (NFκB) activation in the brain, lungs, liver, spleen and lymphoid tissues as well as higher levels of TNFα, IL-6, IL-12, and COX-2 when compared to young adult mice [37].
Not surprisingly, those with pre-existing lung diseases such as chronic obstructive pulmonary disease are at increased risk for development of pneumonia. However, other chronic diseases associated with inflammation such as atherosclerosis and type II diabetes mellitus are also established risk factors for the development of CAP [12, 38]. For example, in a prospective study of 3,075 individuals, aged 70 to 79 years, elevated levels of IL-6 and TNFα in the blood were associated with increased risk for the development of CAP with adjusted odds ratio of 1.6 and 1.7, respectively [38]. While the frequency of CAP for those without comorbid conditions was 2.9%, the presence of one or more comorbid condition increased the frequency of CAP to 7% and 10.7%, respectively [38]. Likewise, studies by Glynn et al. and Antunes et al. found that increased levels of IL-6 correlated best with both disease-specific and generic severity scores for pneumonia [39, 40]. Taken together these findings highlight that even when there are no apparent signs of infection or disease increased levels of inflammation are present in the elderly and may impact susceptibility to and severity of pneumonia.
Pre-existing inflammation leads to the upregulation of host receptors that mediate adhesion and invasion in the lung
Bacterial attachment to host cells is an essential first step in the establishment and development of disease. S. pneumoniae possesses multiple strategies to mediate attachment and invasion of host cells including the bacterial protein choline binding protein A (CbpA) and phosphorylcholine found on the cell wall [41]. In agreement with inflammation as a risk factor for CAP, it is well documented that S. pneumoniae adhesion to host cells is enhanced 100-fold when cells are pre-treated with pro-inflammatory cytokines such as TNFα and IL-1β in vitro [42]. Thus, S. pneumoniae attachment and invasion is, in part, dependent on the activation of NFκB and the upregulation of host cell surface proteins that act as bacterial ligands including platelet-activating factor receptor (PAFr), polymeric immunoglobulin receptor (pIgR) and laminin receptor (LR).
S. pneumoniae attaches to PAFr, the chemokine receptor for platelet activating factor found on epithelial and endothelial cells [41, 42]. PAFr binding is mediated by phosphorylcholine, which is present on the bacterial cell wall and lipoteichoic residues that extend from the cell membrane [41, 42]. This host-pathogen interaction results in pneumococcal uptake through a β-arrestin-dependent mechanism which prevents lysosomal fusion and instead results in bacterial transcytosis [43]. S. pneumoniae also uses surface exposed CbpA to facilitate attachment to both pIgR and LR. pIgR normally functions to transport IgA across mucosal epithelial cells, however, during pneumococcal infection it mediates translocation of the bacteria to the basolateral surface during receptor recycling [41, 44]. Transfection of human pIgR into MDCK cells resulted in 10-fold more pneumococcal invasion compared to the vector alone control [41]. It should be noted that while CbpA binds to human pIgR, studies have found that this interaction does not occur with mouse or rat pIgR [45]. More recently, CbpA has also been shown to mediate adherence through interactions with human and mouse LR which is present in the lungs and vasculature [46, 47]. The combined interactions of the pneumococcus to LR and PAFr are thought to be a principle mechanism by which the bacteria translocate across the alveolar-capillary barrier to cause bacteremia during early pneumonia and the blood-brain barrier to cause meningitis.
In a recent study, we determined that healthy aged mice express increased levels of pIgR, PAFr and LR in the lungs compared to their younger counterparts [47, 48]. We further demonstrated that continuous administration of low levels of TNFα by implanted osmotic pumps for 5 days (to mimic age-associated inflammation) was sufficient to increase the protein levels of pIgR and PAFr within the lungs of young mice [48]. Importantly, increased pIgR and PAFr in young mice were associated with an increased ability of the bacteria to persist and replicate within the lungs early during infection, with 100-fold more bacteria in the lungs 48 hours post-infection compared to young mice receiving PBS [48]. Thus inflammation was found to be positively correlated with both increased receptor expression and enhanced susceptibility to pneumococcal challenge. More recently we were able to demonstrate that PAFr and LR were also significantly increased in lung biopsy specimens obtained from aged humans (64–82 years) when compared to younger individuals aged 43–50 years. Importantly, H. influenzae and N. meningitidis have also been shown to bind to PAFr and LR [49, 50]. Therefore, age-related enhanced ligand expression in the lungs of both mice and humans is one mechanism for the increased severity of pneumonia in the elderly not only for the pneumococcus, but potentially for other respiratory pathogens as well.
Potential sources of inflammatory mediators in the lungs during aging
AAI in the lungs is likely the result of multiple factors including systemic chronic underlying diseases such as obesity and type II diabetes mellitus (T2DM), as well as the aging process itself, a side-effect of cellular senescence, and an increased production of reactive oxygen species all of which can activate NFκB and drive inflammatory processes [32, 51]. In the case of obesity, adipose tissue has been shown to be a significant source of inflammatory cytokines, termed adipokines [52, 53]. Increased circulating cytokines are thought to be a driving force for beta cell dysfunction leading to obesity-induced T2DM [54]. Additionally, fatty acid levels are often elevated during aging, obesity and T2DM and have been shown to enhance cytokine production from macrophages [55]. Thus it is likely that systemic inflammatory mediators brought to the lung via the vasculature could have profound effects on the surrounding lung tissue, however, several lines of evidence point to intrinsic age-related changes within the lung as additional sources of inflammation during aging.
Mucosal epithelial cells not only function as a barrier to infectious pathogens and inhaled particulates but also actively participate in host defense by the production of mucins, beta defensins, and inflammatory mediators [56–59]. Cellular senescence is an age-associated phenomenon by which cells with DNA damage or shortened telomeres lose the ability to further divide but do not undergo apoptosis. Senescent epithelial cells have been reported to accumulate in skin, liver, muscle, fatty tissue and most recently in the lungs of aged mice [60, 61]. Studies have also identified increased cellular senescence in the lungs of individuals with COPD, an established risk factor for CAP [62]. Although senescent cells have lost the ability to divide they are metabolically active and produce profuse amounts of pro-inflammatory mediators, a phenomenon that has been termed senescence associated secretory phenotype or SASP [63]. For example, senescent fibroblasts and epithelial cells have been found to secrete the pro-inflammatory cytokines IL-1, IL-6 and IL-8, as well as proteases and growth factors [63].
We recently confirmed earlier findings of increased inflammatory cytokines in the lungs of healthy aged mice and went on to provide evidence of age-related cellular senescence as a potential source of inflammatory cytokines within the lungs [47]. In vitro induction of cellular senescence of a human type II pneumocyte cell (A549) line resulted in increased secretion of IL-6 and IL-8 and senescent lung cells were found to express elevated levels of LR and cytokeratin 10 (K10). We previously identified that S. pneumoniae binds to K10 on lung cells through the pneumococcal adhesin PsrP [64]. Importantly, exposure of A549 cells to conditioned media from senescent lung cells was able to increase the expression of PAFr and increase their permissiveness to pneumococcal adhesion [47]. Thus, we propose a model by which the accumulation of senescent lung cells due to aging or exposure to genotoxic agents such as cigarette smoke could enhance LR, PAFr and K10 in the lungs thereby promoting bacterial attachment and susceptibility to pneumococcal pneumonia (Figure 1).
Figure 1.
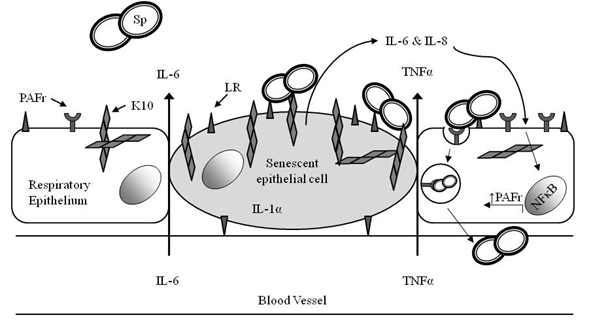
Systemic inflammation and cellular senescence in the lungs promote pneumococcal adhesion during early infection. Age-associated inflammation in the lungs may come from inflammatory cytokines brought to the lungs via the vasculature and from increased accumulation of pro-inflammatory senescent cells in the lungs. These cytokines (IL-6, IL-8, and TNFα) act on neighboring cells to activate NFκB and enhance the expression of platelet activating factor receptor (PAFr). PAFr is then able to bind to phosphorylcholine a component of the bacterial cell wall, and mediate endocytosis and invasion of activated epithelial cells by S. pneumoniae. Senescent cells also express elevated levels of Laminin Receptor (LR) and cytokeratin 10 (K10) which leads to greater bacterial adhesion.
AGE-RELATED CHANGES IN THE INFLAMMATORY RESPONSE MAY ENHANCE SUSCEPTIBILITY TO PNEUMONIA
While pre-existing inflammation enhances susceptibility to infection, inflammation is necessary to activate resident immune cells, recruit effector cells necessary for bacterial clearance, and aid in the activation of the adaptive response. Mice deficient in TNFα receptor, Interleukin-6 (IL-6) or IL-1β show enhanced mortality following pneumococcal infection [65–67]. Initiation and coordination of the inflammatory response occurs through the recognition of conserved pathogen associated molecular patterns (PAMPs) such as bacterial cell wall by pattern-recognition receptors (PRRs), with which include Toll-like receptors (TLRs), NOD-like receptors and RNA Helicases. TLRs are the most extensively studied PRRs and are expressed on a variety of cells including epithelial and endothelial cells, as well as cells of the innate and adaptive immune system.
TLRs detect microorganisms and initiate the inflammatory response
To date there have been 11 TLRs identified in humans and 13 TLRs identified in mice which recognize various microbial components such as Gram-positive lipoteichoic acid (LTA), Gram-negative lipopolysaccharide (LPS), and yeast zymosan. However, it is also important to note that TLRs can also become activated following recognition of host factors released during tissue injury; so-called damage-associated molecular patterns (DAMPs) [68]. Following TLR engagement, an intracellular signaling cascade ensues through cytoplasmic intermediates such as myeloid differentiation factor-88 (MyD88) and IL-1 Receptor associated kinase 1 (IRAK1) that ultimately result in Nuclear Factor kappa B (NFκB) and Mitogen Activated Protein Kinase (MAPK) activation and the production of pro- and anti-inflammatory cytokines, chemokines and anti-microbial peptides [69].
In particular, TLRs 2, 4 and 9 recognize components of the pneumococcus, however, only TLRs 2 and 4 are involved in the elaboration of inflammatory cytokines. TLR2 heterodimerizes with TLR1 to detect LTA, a component of the pneumococcal bacterial cell wall [70, 71], while TLR4 has been shown to respond to the pneumococcal toxin pneumolysin [72]. While TLRs 2 and 4 recognize extracellular PAMPs, TLR9 resides within endosomes where it recognizes unmethylated CpG DNA following bacterial internalization [73]. While the single loss of TLR2 or TLR4 does not significantly influence mouse survival following infection, TLR9 and Myd88-gene deficient mice are exquisitely susceptible to pneumococcal infection, emphasizing not only the redundant roles for TLRs, but also an essential role for Toll/IL-1 Receptor signaling in host survival [71]. In agreement with studies using gene-deficient mice, humans with loss of function mutations in key TLR signaling molecules such as MyD88, NEMO and IRAK4 (for a more comprehensive review see [74]) show enhanced susceptibility to pneumococcal infection, often resulting in severe invasive pneumococcal diseases such as bacteremia and meningitis.
TLR expression and function in the aging lung
TLRs are highly expressed on innate immune cells but have also been shown to be expressed on numerous cells within the lungs including endothelial cells, bronchial epithelial cells, and Type II pneumocytes [58]. TLR expression on these cells is low under steady state conditions but is significantly upregulated 12–24 hours following stimulation with bacterial products such as LPS [75]. Due to the risk of complications, assessment of age-related changes in the acute inflammatory response within the lung following a live infection in humans has not been examined. In a study of 15 elderly patients and 22 younger patients with pneumonia serum levels of GM-CSF, IL-1 beta, TNF-alpha, IL-8 and MIP-1α were lower in the elderly [76].
We have recently reported that protein levels of TLRs 1, 2 and 4 are reduced in the lungs of aged BALB/C mice compared to young adult control mice prior to infection [48]. Moreover, NFκB activation was reduced in the lungs 24 hours following infection with S. pneumoniae. In agreement with decreased NFκB activation and TLR levels in the lungs, the cytokine response during early pneumococcal pneumonia was also found to be significantly attenuated following intratracheal instillation of pneumococcal components. Thus we identified that the age-related susceptibility to pneumococcal pneumonia was associated with decreased TLR levels and function in the aging murine lung, however, it remains to be determined the precise cell types in the lungs that are experiencing TLR dysfunction as a consequence of age. In a study examining age-related changes in the antioxidant heme-oxygenase 1 (HO-1), intratracheal delivery of LPS demonstrated decreased upregulation of HO-1 in both the lungs and alveolar macrophages from aged male ICR mice (65–66 weeks) compared to young (9–22 weeks) [77]. While studies indicate that aged macrophages maintain the ability to phagocytose invading pathogens [78, 79], studies in humans and mice suggest that peripheral macrophages and dendritic cells are defective in their elaboration of inflammatory mediators following Toll-like receptor (TLR) stimulation (see below) and thus may be contributing to the reduced response we observed in the lungs.
TLR function declines with age
The impact of aging on human TLR function has been examined primarily in the context of LPS-induced TLR4 signaling of human peripheral monocyte and dendritic cell populations and has been reviewed by van Duin and Shaw [80]. Table 1 summarizes selected past and recent studies that have investigated the impact of age on the cytokine response of human and mice monocytes/macrophages and dendritic cells. Several studies found decreased production of inflammatory cytokines following LPS stimulation [51, 76, 81, 82], while others found no differences [83] or enhanced production [84, 85]. Recently, van Duin et al. examined the expression and function of TLRs 1–9 by flow cytometry using human peripheral monocytes from 81 individuals aged ≥ 65 years compared to 80 young adults aged 21–30. The only deficiency observed in TLR stimulation was in response to Pam3CSK4, a TLR1/2 specific agonist [81]. They further determined that the reduced cytokine response was associated with reduced TLR1 surface expression while TLR2 surface expression was unaffected by age. A more recent study confirmed and extended these findings by identifying that human TLR1 expression was reduced on monocytic subsets expressing CD14 and CD16, but not on CD14+ CD16− monocytes [82]. TLR1/2 recognizes triacylated lipoproteins that extend through Gram-positive cell walls and thus reductions in alveolar macrophage responses to TLR1/2 stimulation may explain the reduced cytokine response we previously observed in the aged mouse lungs [48]. Importantly all four monocyte subsets examined exhibited impaired production of TNFα and IL-6. Interestingly, CD14+ CD16+ monocytes have recently been characterized as an activated senescent monocyte subpopulation, which are present in higher numbers in atherosclerotic plaques and have been suggested to be a source of inflammatory mediators during aging [86, 87].
Table 1.
Select studies in humans and mice evaluating the age-related differences in TLR-induced cytokine production of monocyte/macrophages and dendritic cells
| Study | Species | Stimulus | Cell Type | Cytokine Production |
|---|---|---|---|---|
| Born et al. (1995) | Human | LPS | Whole blood | Increased TNFα and IL-1β |
| Gon et al. (1996) | Human | LPS | Monocytes | Decreased TNFα and IL-1β |
| Roubenoff et al. (1998) | Human | LPS | PMNC | No difference in TNFα and IL-1β |
| Bruunsgaard et al. (1999) | Human | LPS | Whole blood | Decreased TNFα and IL-1β |
| Gabriel et al. (2002) | Human | LPS | Whole blood | Increased IL-6 and IL-1β |
| Van Duin et al. (2007) | Human* | Pam3CSK4 | Monocytes | Decreased TNFα and IL-6 |
| Panda et al. (2010) | Human | LPS, Pam3CSK4 | Myeloid Dendritic cells | Decreased TNFα and IL-6 |
| Nyugen et al. (2010) | Human | Pam3CSK4 | Monocytes | Decreased TNFα and IL-6 |
| Renshaw et al. (2002) | C57BL/6¶ mice | LPS | Splenic Macrophages | Decreased TNFα and IL-6 |
| Boehmer et al. (2004) | BALB/c mice | LPS | Peritoneal Macrophages | Decreased TNFα and IL-6 |
| Chelvarajan et al. (2006) | BALB/c mice | LPS | Splenic Macrophages | Decreased TNFα and IL-6 |
This study assessed the function of multiple TLRs, however, significant differences were only identified in response to Pam3CSK4.
This study reported lower mRNA expression levels of TLRs 1–9 and decreased cytokine production in response to stimulation of TLRs1/2, TLRs2/6, TLR3, TLR4, TLR5, and TLR9.
Early studies by Renshaw et al. were the first to report that TLR-induced cytokine production was reduced in splenic macrophages from aged C57BL/6 mice [88]. This diminished response was associated with lower TLR expression as well as decreased TLR4 surface expression. In contrast, a later study found that aged thioglycolate-elicited peritoneal macrophages from BALB/c mice did not exhibit age-related changes in TLR4 surface expression although they exhibited reduced cytokine response. They concluded that age-related decreases in levels of mitogen-activated protein kinases such as p38 and JNK resulted in reduced phosphorylation following stimulation with LPS [89]. In order to further clarify these differences, another study utilized microarray to examine BALB/c splenic macrophages for age-related changes in gene expression following stimulation with LPS [90]. Multiple age-related differences in TLR signaling molecules were identified, including reductions in the expression of key signaling molecules (Myd88, TRAF6 and NFκB subunits) and increased expression of a negative regulator of TLR signaling, IRAKM [90]. However, this study found that activation of p38 MAPK was found to be significantly increased with age, while levels and phosphorylation of ERK were significantly reduced. Therefore, studies from mice and humans have not reached a consensus on the precise mechanism operative in reduced TLR responsiveness.
There are many factors that can account for the differences observed between studies. Differences in mouse strains (C57BL/6 vs. BALB/c), isolation and purification of macrophage populations, and animal housing may all effect experimental outcomes. TLR expression has been shown to be upregulated following NFκB activation [75]. Moreover, studies have demonstrated that macrophage TLR expression can be modulated by microenvironmental changes [91, 92]. Therefore, differences observed between studies may reflect the relative inflammatory state of the aged tissue microenvironment. Nonetheless, it is clear from these studies that macrophages isolated from aged mice and humans are impaired in the early production of pro-inflammatory cytokines following stimulation with certain purified PAMPs. It is tempting to speculate that these age-related defects in cytokine response substantially contribute to the increased severity of disease, however, the multitude of age-related changes that occur with age may make this phenotype difficult to assess in vivo. Additionally, a recent study determined that aged pulmonary CD11c+ cells (macrophages and dendritic cells) were able to produce TNFα and INFγ in the absence of TLR2, while pulmonary CD11c+ cells from young mice were not [93]. Thus it is possible that TLR dysfunction in the context of aging may have different effects in response to various microorganisms.
Age-related TLR dysfunction is not unique to macrophages as human and murine dendritic cell TLR function has also been shown to decline with age [94, 95]. Importantly, reduced TLR function by dendritic cells from elderly humans was correlated with decreased antibody response following influenza immunization indicating that TLR dysfunction contributes to the age-related decline in vaccine efficacy [94]. The findings that both macrophages and dendritic cells exhibit impaired TLR function suggest that this age-related phenotype may occur at an earlier stage of differentiation such as within the bone marrow. Age-related decreased TLR function may also reflect a state of tolerization, which has been demonstrated to occur using in vitro models of endotoxin tolerance and in vivo following resolution of respiratory influenza infection [96, 97].
ELDERLY PATIENTS HAVE A PROLONGED INFLAMMATORY RESPONSE FOLLOWING CAP
Following infection of the elderly, the resolution of the inflammatory response has been shown to be prolonged and associated with worsened outcomes. In a study of 22 hospitalized patients with confirmed pneumococcal pneumonia in 19/22 patients, at 1 week post-admission levels of TNFα, soluble TNF Receptor I (sTNFR) and the anti-inflammatory cytokine IL-10 were significantly higher in the elderly (68–91 years) compared to younger patients (37–55 years) [51]. In a model of endotoxemia, elderly volunteers also demonstrated a prolonged inflammatory response as indicated by longer circulationg levels of sTNFRs and CRP [98]. The mechanisms underlying this prolonged inflammatory state are unclear and these findings are to some extent contradictory with age-related hyporesponsiveness of innate immune cells. Possible explanations for this include greater disease severity and tissue injury in the elderly and/or defective recruitment of exudates macrophages to the site of tissue injury. Additionally, other age-related defects including decreased accumulation or function of immunosuppressive CD4+ T regulatory cells and altered production of TGF-β production by respiratory epithelial cells may also hinder the dampening of the acute proinflammatory response. Nonetheless, a prolonged inflammatory state likely contributes to the exacerbation of underlying pathologies and increased mortality of the elderly within 1 year following admission for CAP. A recent retrospective study of 50,119 male subjects with a mean age of 77.5 years found an increased incidence of cardiovascular events such as congestive heart failure and arrhythmias within 90 days of hospital admission for CAP [10].
THERAPIES THAT COUNTER AGE-ASSOCIATED INFLAMMATION DECREASE MORTALITY FOLLOWING CAP
Given the established negative consequences of pre-existing inflammation but the necessity for a robust pro-inflammatory response during acute infection it is not surprising that the pre-infection use of potent anti-inflammatory therapeutics such as steroids and anti-TNFα therapy are associated with severe pneumococcal infections. Conversely, milder anti-inflammatory drugs such as aspirin or NSAIDs which have been shown to reduce the incidence of colon cancer [99], have not been found to reduce the severity of CAP [100]. As it is not possible to stop the aging process, identifying drugs or treatments that could potentially reverse age-associated inflammation without exacerbating the age-related immune dysfunction would be beneficial to reduce the incidence and severity of pneumococcal pneumonia.
Statins have pleiotropic effects that have been found to reduce mortality following community acquired pneumonia
Statins, or 3-hydroxy-3 methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are among the most widely prescribed drugs in the United States. While statins can reduce plasma cholesterol by as much as 30–55%, it is increasingly evident that statins also have potent anti-inflammatory properties that are independent of their lipid-lowering ability. Several retrospective studies have identified statins to be beneficial in reducing the incidence of death associated with pneumonia and sepsis [101–103]. Mortensen et al. found that prior statin use was associated with a reduced 30 day mortality [102]. Importantly, prior statin use has also been found to reduce the risk of CAP in patients with diabetes [101] Thus, statin therapy offers potential prophylaxis for individuals who are at high-risk for CAP. Given that statins have reported pleiotropic effects, it is likely that statins may be reducing mortality following pneumococcal pneumonia by several cooperative mechanisms. Understanding these mechanisms may aid in the design of better therapeutics for CAP.
We recently found that statins reduce the severity of invasive pneumococcal disease in a mouse model of sickle-cell disease [104]. Sickle cell patients have a 400-fold increased risk for the development of lethal pneumococcal sepsis in part due to heightened inflammation in the lungs and vasculature [105]. Specifically, we found that intraperitoneal administration of statins for five days was able to reduce the levels of expression of PAFr and reduced the ability of pneumolysin, a pore-forming toxin that binds cholesterol, to damage endothelial cells in vitro and in vivo. Moreover, we found that administration of an oral statin diet for 4 weeks significantly decreased bacterial burden in the lungs and blood of aged BALB/c mice (20 months) [106]. Although statins have been found to reduce LPS-induced lung inflammation in human volunteers and pro-inflammatory responses of monocytes stimulated ex vivo [107–109], other studies, as well as our own observations, have found that during a live infection, the anti-inflammatory properties of statins are less apparent [104, 110, 111]. A recent study identified that statins increase bacterial killing of a wide range of Gram-positive organisms by enhancing the formation of phagocyte extracellular traps [112]. However, in a mouse model of Klebsiella pneumoniae, statin administration resulted in increased bacterial outgrowth due to reduced neutrophil accumulation within the lungs and a defect in neutrophil-dependent intracellular killing [110]. Thus, statin prophylaxis may be beneficial to protect against some pathogens but may not protect against all infectious diseases. Additionally, although statins are generally well tolerated, neuromuscular and hepatic complications may limit their use.
SUMMARY AND FUTURE PERSPECTIVES
Pneumonia in the elderly remains a serious health problem with significant morbidity and mortality rates with increasing age. Understanding the age-related changes that occur in the lungs may allow for better treatment or prevention of CAP in the elderly. Figure 2 provides a summary of how age-related dysregulated inflammation prior to, during acute infection, and during the convalescent stage potentially contributes to the enhanced susceptibility of the elderly to infectious disease and increased mortality.
Figure 2.
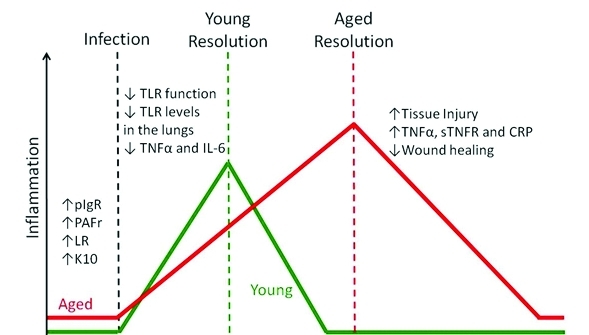
Schematic of dysregulated inflammation in the elderly. Chronic-low grade inflammation enhances bacterial ligand expression in the lungs which increases the ability of bacteria to attach to and invade host cells. Following infection, TLR dependent production of inflammatory cytokines in macrophages and dendritic cells has been found to decrease with age, leading to a delay in the activation of host defense mechanisms. Finally, the resolution of the inflammatory response during the convalescent stage is often prolonged in the elderly likely as a result of increased tissue injury and an age-related decrease in repair mechanisms.
Importantly, to date, studies trying to identify mechanisms for the diminished inflammatory response have yielded conflicting results. One explanation for this is that investigators have not taken into consideration the temporal differences between young and aged animals in regards to how they respond to infectious stimuli. For example, studies comparing aged convalescent individuals, which most likely show enhanced inflammation, are not comparable to those during acute infection, which may show diminished inflammatory markers. This window, where aged animals show a diminished capacity to respond with a robust pro-inflammatory response may in particular be variable or small, as an inability to adequately respond to infection will result in greater infectious burden that ultimately results in a greater disease severity and elevated immune response versus young individuals.
Finally, the use of anti-inflammatory drugs as a prophylactic therapy against S. pneumoniae may not only protect against disease but also might ameliorate the muted response during acute infection. There is an increasing body of evidence showing that repeated antigenic stimulation or chronic inflammation serves to tolerize immune cells and prevent their robust response. Thus by inhibiting pre-infection inflammation statins may also restore the acute response and prevent severe disease. Given the estimate that >2 billion individuals who are susceptible to pneumonia this important topic warrants future investigation.
ACKNOWLEDGEMENTS
CJO is supported by NIH AG033274
REFERENCES
- 1.Janssens JP. Pneumonia in the elderly (geriatric) population. Curr Opin Pulm Med. 2005;11:226–230. doi: 10.1097/01.mcp.0000158254.90483.1f. [DOI] [PubMed] [Google Scholar]
- 2.Vincent GKaVAV. Current Population Reports. U.S. Census Bureau; Washington, DC: 2010. THE NEXT FOUR DECADES, The Older Population in the United States: 2010 to 2050; pp. P25–1138. [Google Scholar]
- 3.Meyer KC, Rosenthal NS, Soergel P, Peterson K. Neutrophils and low-grade inflammation in the seemingly normal aging human lung. Mech Ageing Dev. 1998;104:169–181. doi: 10.1016/s0047-6374(98)00065-7. [DOI] [PubMed] [Google Scholar]
- 4.Loeb M. Pneumonia in the elderly. Curr Opin Infect Dis. 2004;17:127–130. doi: 10.1097/00001432-200404000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Janssens JP, Krause KH. Pneumonia in the very old. Lancet Infect Dis. 2004;4:112–124. doi: 10.1016/S1473-3099(04)00931-4. [DOI] [PubMed] [Google Scholar]
- 6.Chong CP, Street PR. Pneumonia in the elderly: a review of the epidemiology, pathogenesis, microbiology, and clinical features. South Med J. 2008;101:1141–1145. doi: 10.1097/SMJ.0b013e318181d5b5. quiz 1132, 1179. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan V, Angus DC, Griffin MF, Clermont G, Scott Watson R, Linde-Zwirble WT. Hospitalized community-acquired pneumonia in the elderly: age- and sex-related patterns of care and outcome in the United States. Am J Respir Crit Care Med. 2002;165:766–772. doi: 10.1164/ajrccm.165.6.2103038. [DOI] [PubMed] [Google Scholar]
- 8.Torres OH, Munoz J, Ruiz D, Ris J, Gich I, Coma E, Gurgui M, Vazquez G. Outcome predictors of pneumonia in elderly patients: importance of functional assessment. J Am Geriatr Soc. 2004;52:1603–1609. doi: 10.1111/j.1532-5415.2004.52492.x. [DOI] [PubMed] [Google Scholar]
- 9.Bohannon RW, Maljanian RD. Hospital readmissions of elderly patients hospitalized with pneumonia. Conn Med. 2003;67:599–603. [PubMed] [Google Scholar]
- 10.Perry TW, Pugh MJ, Waterer GW, Nakashima B, Orihuela CJ, Copeland LA, Restrepo MI, Anzueto A, Mortensen EM. Incidence of cardiovascular events after hospital admission for pneumonia. Am J Med. 2011;124:244–251. doi: 10.1016/j.amjmed.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fillon S, Soulis K, Rajasekaran S, Benedict-Hamilton H, Radin JN, Orihuela CJ, El Kasmi KC, Murti G, Kaushal D, Gaber MW, Weber JR, Murray PJ, Tuomanen EI. Platelet-activating factor receptor and innate immunity: uptake of gram-positive bacterial cell wall into host cells and cell-specific pathophysiology. J Immunol. 2006;177:6182–6191. doi: 10.4049/jimmunol.177.9.6182. [DOI] [PubMed] [Google Scholar]
- 12.Koivula I, Sten M, Makela PH. Risk factors for pneumonia in the elderly. Am J Med. 1994;96:313–320. doi: 10.1016/0002-9343(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 13.Meyer KC, Ershler W, Rosenthal NS, Lu XG, Peterson K. Immune dysregulation in the aging human lung. Am J Respir Crit Care Med. 1996;153:1072–1079. doi: 10.1164/ajrccm.153.3.8630547. [DOI] [PubMed] [Google Scholar]
- 14.Rimland D, Moanna A. Increasing incidence of herpes zoster among Veterans. Clin Infect Dis. 2010;50:1000–1005. doi: 10.1086/651078. [DOI] [PubMed] [Google Scholar]
- 15.Sambhara S, McElhaney JE. Immunosenescence and influenza vaccine efficacy. Curr Top Microbiol Immunol. 2009;333:413–429. doi: 10.1007/978-3-540-92165-3_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McElhaney JE, Effros RB. Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol. 2009;21:418–424. doi: 10.1016/j.coi.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson MG, P, Shay DK, MD, Zhou H, MSc,MPH, Bridges CB, MD, Cheng PY, PhD, Burns E, MA, Bresee JS, MD, Cox NJ., PhD Estimates of Deaths Associated with Seasonal Influenza --- United States, 1976--2007. Morbidity and Mortality Weekly Report (MMWR) 2010;59:1057–1062. [PubMed] [Google Scholar]
- 18.Mandell LA, Bartlett JG, Dowell SF, File TM, Jr, Musher DM, Whitney C. Update of practice guidelines for the management of community-acquired pneumonia in immunocompetent adults. Clin Infect Dis. 2003;37:1405–1433. doi: 10.1086/380488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson KA, Baughman W, Rothrock G, Barrett NL, Pass M, Lexau C, Damaske B, Stefonek K, Barnes B, Patterson J, Zell ER, Schuchat A, Whitney CG. Epidemiology of invasive Streptococcus pneumoniae infections in the United States, 1995–1998: Opportunities for prevention in the conjugate vaccine era. Jama. 2001;285:1729–1735. doi: 10.1001/jama.285.13.1729. [DOI] [PubMed] [Google Scholar]
- 20.Lexau CA, Lynfield R, Danila R, Pilishvili T, Facklam R, Farley MM, Harrison LH, Schaffner W, Reingold A, Bennett NM, Hadler J, Cieslak PR, Whitney CG. Changing epidemiology of invasive pneumococcal disease among older adults in the era of pediatric pneumococcal conjugate vaccine. Jama. 2005;294:2043–2051. doi: 10.1001/jama.294.16.2043. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson W, Hamborsky J, McIntyre L, Wolfe S. Centers for Disease Control and Prevention. Epidemiology and Prevention of Vaccine-Preventable Diseases. Public Health Foundation; Washington DC: 2007. Pneumococcal Disease. [Google Scholar]
- 22.Pneumococcal vaccines. WHO position paper. Wkly Epidemiol Rec. 1999;74:177–183. [PubMed] [Google Scholar]
- 23.Lynch JP, 3rd, Zhanel GG. Streptococcus pneumoniae: epidemiology and risk factors, evolution of antimicrobial resistance, and impact of vaccines. Curr Opin Pulm Med. 2010;16:217–225. doi: 10.1097/MCP.0b013e3283385653. [DOI] [PubMed] [Google Scholar]
- 24.Whitney CG, Farley MM, Hadler J, Harrison LH, Bennett NM, Lynfield R, Reingold A, Cieslak PR, Pilishvili T, Jackson D, Facklam RR, Jorgensen JH, Schuchat A. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med. 2003;348:1737–1746. doi: 10.1056/NEJMoa022823. [DOI] [PubMed] [Google Scholar]
- 25.Jackson LA, Neuzil KM, Yu O, Benson P, Barlow WE, Adams AL, Hanson CA, Mahoney LD, Shay DK, Thompson WW. Effectiveness of pneumococcal polysaccharide vaccine in older adults. N Engl J Med. 2003;348:1747–1755. doi: 10.1056/NEJMoa022678. [DOI] [PubMed] [Google Scholar]
- 26.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–499. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubins JB, Puri AK, Loch J, Charboneau D, MacDonald R, Opstad N, Janoff EN. Magnitude, duration, quality, and function of pneumococcal vaccine responses in elderly adults. J Infect Dis. 1998;178:431–440. doi: 10.1086/515644. [DOI] [PubMed] [Google Scholar]
- 28.Romero-Steiner S, Musher DM, Cetron MS, Pais LB, Groover JE, Fiore AE, Plikaytis BD, Carlone GM. Reduction in functional antibody activity against Streptococcus pneumoniae in vaccinated elderly individuals highly correlates with decreased IgG antibody avidity. Clin Infect Dis. 1999;29:281–288. doi: 10.1086/520200. [DOI] [PubMed] [Google Scholar]
- 29.Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol. 2001;8:131–136. doi: 10.1097/00062752-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Ershler WB. Interleukin-6: a cytokine for gerontologists. J Am Geriatr Soc. 1993;41:176–181. doi: 10.1111/j.1532-5415.1993.tb02054.x. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar D, Fisher PB. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–699. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 34.Ferrucci L, Harris TB, Guralnik JM, Tracy RP, Corti MC, Cohen HJ, Penninx B, Pahor M, Wallace R, Havlik RJ. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc. 1999;47:639–646. doi: 10.1111/j.1532-5415.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- 35.Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH, Jr, Heimovitz H, Cohen HJ, Wallace R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–512. doi: 10.1016/s0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- 36.Hager K, Machein U, Krieger S, Platt D, Seefried G, Bauer J. Interleukin-6 and selected plasma proteins in healthy persons of different ages. Neurobiol Aging. 1994;15:771–772. doi: 10.1016/0197-4580(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 37.Spencer NF, Poynter ME, Im SY, Daynes RA. Constitutive activation of NF-kappa B in an animal model of aging. Int Immunol. 1997;9:1581–1588. doi: 10.1093/intimm/9.10.1581. [DOI] [PubMed] [Google Scholar]
- 38.Yende S, Tuomanen EI, Wunderink R, Kanaya A, Newman AB, Harris T, de Rekeneire N, Kritchevsky SB. Preinfection systemic inflammatory markers and risk of hospitalization due to pneumonia. Am J Respir Crit Care Med. 2005;172:1440–1446. doi: 10.1164/rccm.200506-888OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antunes G, Evans SA, Lordan JL, Frew AJ. Systemic cytokine levels in community-acquired pneumonia and their association with disease severity. Eur Respir J. 2002;20:990–995. doi: 10.1183/09031936.02.00295102. [DOI] [PubMed] [Google Scholar]
- 40.Glynn P, Coakley R, Kilgallen I, Murphy N, O'Neill S. Circulating interleukin 6 and interleukin 10 in community acquired pneumonia. Thorax. 1999;54:51–55. doi: 10.1136/thx.54.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell. 2000;102:827–837. doi: 10.1016/s0092-8674(00)00071-4. [DOI] [PubMed] [Google Scholar]
- 42.Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- 43.Radin JN, Orihuela CJ, Murti G, Guglielmo C, Murray PJ, Tuomanen E. B-arrestin 1 determines the traffic pattern of PAFr-mediated enodocytosis of Streptococcus pneumoniae. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaetzel CS. Polymeric Ig receptor: defender of the fort or Trojan horse? Curr Biol. 2001;11:R35–38. doi: 10.1016/s0960-9822(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 45.Elm C, Braathen R, Bergmann S, Frank R, Vaerman JP, Kaetzel CS, Chhatwal GS, Johansen FE, Hammerschmidt S. Ectodomains 3 and 4 of human polymeric Immunoglobulin receptor (hpIgR) mediate invasion of Streptococcus pneumoniae into the epithelium. J Biol Chem. 2004;279:6296–6304. doi: 10.1074/jbc.M310528200. [DOI] [PubMed] [Google Scholar]
- 46.Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala'Aldeen DA, Tuomanen EI. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J Clin Invest. 2009;119:1638–1646. doi: 10.1172/JCI36759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shivshankar P, Boyd AR, Le Saux CJ, Yeh IT, Orihuela CJ. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell. 2011 doi: 10.1111/j.1474-9726.2011.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinojosa E, Boyd AR, Orihuela CJ. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J Infect Dis. 2009;200:546–554. doi: 10.1086/600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiser JN, Goldberg JB, Pan N, Wilson L, Virji M. The phosphorylcholine epitope undergoes phase variation on a 43-kilodalton protein in Pseudomonas aeruginosa and on pili of Neisseria meningitidis and Neisseria gonorrhoeae. Infect Immun. 1998;66:4263–4267. doi: 10.1128/iai.66.9.4263-4267.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swords WE, Buscher BA, Ver Steeg Ii K, Preston A, Nichols WA, Weiser JN, Gibson BW, Apicella MA. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol. 2000;37:13–27. doi: 10.1046/j.1365-2958.2000.01952.x. [DOI] [PubMed] [Google Scholar]
- 51.Bruunsgaard H, Skinhoj P, Qvist J, Pedersen BK. Elderly humans show prolonged in vivo inflammatory activity during pneumococcal infections. J Infect Dis. 1999;180:551–554. doi: 10.1086/314873. [DOI] [PubMed] [Google Scholar]
- 52.Arai Y, Takayama M, Abe Y, Hirose N. Adipokines and Aging. J Atheroscler Thromb. 2011 doi: 10.5551/jat.7039. [DOI] [PubMed] [Google Scholar]
- 53.Meijer RI, Serne EH, Smulders YM, van Hinsbergh VW, Yudkin JS, Eringa EC. Perivascular adipose tissue and its role in type 2 diabetes and cardiovascular disease. Curr Diab Rep. 2011;11:211–217. doi: 10.1007/s11892-011-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathur SK, Jain P, Mathur P. Microarray evidences the role of pathologic adipose tissue in insulin resistance and their clinical implications. J Obes. 2011;2011:587495. doi: 10.1155/2011/587495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dasu MR, Jialal I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am J Physiol Endocrinol Metab. 2011;300:E145–154. doi: 10.1152/ajpendo.00490.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem. 2006;281:12994–12998. doi: 10.1074/jbc.M511431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartlett JA, Fischer AJ, McCray PB., Jr Innate immune functions of the airway epithelium. Contrib Microbiol. 2008;15:147–163. doi: 10.1159/000136349. [DOI] [PubMed] [Google Scholar]
- 58.Armstrong L, Medford AR, Uppington KM, Robertson J, Witherden IR, Tetley TD, Millar AB. Expression of functional toll-like receptor-2 and -4 on alveolar epithelial cells. Am J Respir Cell Mol Biol. 2004;31:241–245. doi: 10.1165/rcmb.2004-0078OC. [DOI] [PubMed] [Google Scholar]
- 59.Ha U, Lim JH, Jono H, Koga T, Srivastava A, Malley R, Pages G, Pouyssegur J, Li JD. A novel role for IkappaB kinase (IKK) alpha and IKKbeta in ERK-dependent up-regulation of MUC5AC mucin transcription by Streptococcus pneumoniae. J Immunol. 2007;178:1736–1747. doi: 10.4049/jimmunol.178.3.1736. [DOI] [PubMed] [Google Scholar]
- 60.Kreiling JA, Tamamori-Adachi M, Sexton AN, Jeyapalan JC, Munoz-Najar U, Peterson AL, Manivannan J, Rogers ES, Pchelintsev NA, Adams PD, Sedivy JM. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell. 10:292–304. doi: 10.1111/j.1474-9726.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 62.Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:596–601. doi: 10.1513/pats.200904-017RM. [DOI] [PubMed] [Google Scholar]
- 63.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shivshankar P, Sanchez C, Rose LF, Orihuela CJ. The Streptococcus pneumoniae adhesin PsrP binds to Keratin 10 on lung cells. Mol Microbiol. 2009;73:663–679. doi: 10.1111/j.1365-2958.2009.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis. 1997;176:439–444. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
- 66.van der Poll T, Keogh CV, Buurman WA, Lowry SF. Passive immunization against tumor necrosis factor-alpha impairs host defense during pneumococcal pneumonia in mice. Am J Respir Crit Care Med. 1997;155:603–608. doi: 10.1164/ajrccm.155.2.9032201. [DOI] [PubMed] [Google Scholar]
- 67.Takashima K, Tateda K, Matsumoto T, Iizawa Y, Nakao M, Yamaguchi K. Role of tumor necrosis factor alpha in pathogenesis of pneumococcal pneumonia in mice. Infect Immun. 1997;65:257–260. doi: 10.1128/iai.65.1.257-260.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 69.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 70.Knapp S, Wieland CW, van 't Veer C, Takeuchi O, Akira S, Florquin S, van der Poll T. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- 71.Dessing MC, Schouten M, Draing C, Levi M, von Aulock S, van der Poll T. Role played by Toll-like receptors 2 and 4 in lipoteichoic acid-induced lung inflammation and coagulation. J Infect Dis. 2008;197:245–252. doi: 10.1086/524873. [DOI] [PubMed] [Google Scholar]
- 72.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by toll-like receptor 4 confers resistance to pneumococcal infection. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol. 2007;9:633–644. doi: 10.1111/j.1462-5822.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- 74.Chapman SJ. Can your genes make you more prone to pneumococcal disease? Expert Rev Anti Infect Ther. 2010;8:967–972. doi: 10.1586/eri.10.84. [DOI] [PubMed] [Google Scholar]
- 75.Saito T, Yamamoto T, Kazawa T, Gejyo H, Naito M. Expression of toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouse. Cell Tissue Res. 2005;321:75–88. doi: 10.1007/s00441-005-1113-9. [DOI] [PubMed] [Google Scholar]
- 76.Gon Y, Hashimoto S, Hayashi S, Koura T, Matsumoto K, Horie T. Lower serum concentrations of cytokines in elderly patients with pneumonia and the impaired production of cytokines by peripheral blood monocytes in the elderly. Clin Exp Immunol. 1996;106:120–126. [PubMed] [Google Scholar]
- 77.Ito Y, Betsuyaku T, Moriyama C, Nasuhara Y, Nishimura M. Aging affects lipopolysaccharide-induced upregulation of heme oxygenase-1 in the lungs and alveolar macrophages. Biogerontology. 2009;10:173–180. doi: 10.1007/s10522-008-9164-4. [DOI] [PubMed] [Google Scholar]
- 78.Murciano C, Yanez A, O'Connor JE, Gozalbo D, Gil ML. Influence of aging on murine neutrophil and macrophage function against Candida albicans. FEMS Immunol Med Microbiol. 2008;53:214–221. doi: 10.1111/j.1574-695X.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- 79.Esposito AL, Clark CA, Poirier WJ. An assessment of the respiratory burst and bactericidal activity of alveolar macrophages from adult and senescent mice. J Leukoc Biol. 1988;43:445–454. doi: 10.1002/jlb.43.5.445. [DOI] [PubMed] [Google Scholar]
- 80.van Duin D, Shaw AC. Toll-like receptors in older adults. J Am Geriatr Soc. 2007;55:1438–1444. doi: 10.1111/j.1532-5415.2007.01300.x. [DOI] [PubMed] [Google Scholar]
- 81.van Duin D, Mohanty S, Thomas V, Ginter S, Montgomery RR, Fikrig E, Allore HG, Medzhitov R, Shaw AC. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178:970–975. doi: 10.4049/jimmunol.178.2.970. [DOI] [PubMed] [Google Scholar]
- 82.Nyugen J, Agrawal S, Gollapudi S, Gupta S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol. 2010;30:806–813. doi: 10.1007/s10875-010-9448-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roubenoff R, Harris TB, Abad LW, Wilson PW, Dallal GE, Dinarello CA. Monocyte cytokine production in an elderly population: effect of age and inflammation. J Gerontol A Biol Sci Med Sci. 1998;53:M20–26. doi: 10.1093/gerona/53a.1.m20. [DOI] [PubMed] [Google Scholar]
- 84.Gabriel P, Cakman I, Rink L. Overproduction of monokines by leukocytes after stimulation with lipopolysaccharide in the elderly. Exp Gerontol. 2002;37:235–247. doi: 10.1016/s0531-5565(01)00189-9. [DOI] [PubMed] [Google Scholar]
- 85.Born J, Uthgenannt D, Dodt C, Nunninghoff D, Ringvolt E, Wagner T, Fehm HL. Cytokine production and lymphocyte subpopulations in aged humans. An assessment during nocturnal sleep. Mech Ageing Dev. 1995;84:113–126. doi: 10.1016/0047-6374(95)01638-4. [DOI] [PubMed] [Google Scholar]
- 86.Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. 2011;186:1809–1815. doi: 10.4049/jimmunol.1001866. [DOI] [PubMed] [Google Scholar]
- 87.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 88.Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–4701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 89.Boehmer ED, Goral J, Faunce DE, Kovacs EJ. Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol. 2004;75:342–349. doi: 10.1189/jlb.0803389. [DOI] [PubMed] [Google Scholar]
- 90.Chelvarajan RL, Liu Y, Popa D, Getchell ML, Getchell TV, Stromberg AJ, Bondada S. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol. 2006;79:1314–1327. doi: 10.1189/jlb.0106024. [DOI] [PubMed] [Google Scholar]
- 91.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 93.Rottinghaus EK, Vesosky B, Turner J. TLR-2 independent recognition of Mycobacterium tuberculosis by CD11c+ pulmonary cells from old mice. Mech Ageing Dev. 2010;131:405–414. doi: 10.1016/j.mad.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, Chen S, Towle V, Belshe RB, Fikrig E, Allore HG, Montgomery RR, Shaw AC. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol. 2010;184:2518–2527. doi: 10.4049/jimmunol.0901022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol. 2008;181:6747–6756. doi: 10.4049/jimmunol.181.10.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiong Y, Qiu F, Piao W, Song C, Wahl LM, Medvedev AE. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J Biol Chem. 2011;286:7905–7916. doi: 10.1074/jbc.M110.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krabbe KS, Bruunsgaard H, Hansen CM, Moller K, Fonsmark L, Qvist J, Madsen PL, Kronborg G, Andersen HO, Skinhoj P, Pedersen BK. Ageing is associated with a prolonged fever response in human endotoxemia. Clin Diagn Lab Immunol. 2001;8:333–338. doi: 10.1128/CDLI.8.2.333-338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruder EH, Laiyemo AO, Graubard BI, Hollenbeck AR, Schatzkin A, Cross AJ. Non-Steroidal Anti-Inflammatory Drugs and Colorectal Cancer Risk in a Large, Prospective Cohort. Am J Gastroenterol. 2011 doi: 10.1038/ajg.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Voiriot G, Dury S, Parrot A, Mayaud C, Fartoukh M. Nonsteroidal antiinflammatory drugs may affect the presentation and course of community-acquired pneumonia. Chest. 2011;139:387–394. doi: 10.1378/chest.09-3102. [DOI] [PubMed] [Google Scholar]
- 101.van de Garde EM, Hak E, Souverein PC, Hoes AW, van den Bosch JM, Leufkens HG. Statin therapy and reduced risk of pneumonia in patients with diabetes. Thorax. 2006 doi: 10.1136/thx.2006.062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mortensen EM, Restrepo MI, Anzueto A, Pugh J. The effect of prior statin use on 30-day mortality for patients hospitalized with community-acquired pneumonia. Respir Res. 2005;6:82. doi: 10.1186/1465-9921-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, Friger M, Zeller L, Danon A. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110:880–885. doi: 10.1161/01.CIR.0000138932.17956.F1. [DOI] [PubMed] [Google Scholar]
- 104.Rosch JW, Boyd AR, Hinojosa E, Pestina T, Hu Y, Persons DA, Orihuela CJ, Tuomanen EI. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J Clin Invest. 2010;120:627–635. doi: 10.1172/JCI39843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Overturf GD. Infections and immunizations of children with sickle cell disease. Adv Pediatr Infect Dis. 1999;14:191–218. [PubMed] [Google Scholar]
- 106.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Rapamycin Strong R. But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J Gerontol A Biol Sci Med Sci. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Methe H, Kim JO, Kofler S, Nabauer M, Weis M. Statins decrease Toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler Thromb Vasc Biol. 2005;25:1439–1445. doi: 10.1161/01.ATV.0000168410.44722.86. [DOI] [PubMed] [Google Scholar]
- 108.Shyamsundar M, McKeown ST, O'Kane CM, Craig TR, Brown V, Thickett DR, Matthay MA, Taggart CC, Backman JT, Elborn JS, McAuley DF. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am J Respir Crit Care Med. 2009;179:1107–1114. doi: 10.1164/rccm.200810-1584OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Novack V, Eisinger M, Frenkel A, Terblanche M, Adhikari NK, Douvdevani A, Amichay D, Almog Y. The effects of statin therapy on inflammatory cytokines in patients with bacterial infections: a randomized double-blind placebo controlled clinical trial. Intensive Care Med. 2009;35:1255–1260. doi: 10.1007/s00134-009-1429-0. [DOI] [PubMed] [Google Scholar]
- 110.Fessler MB, Young SK, Jeyaseelan S, Lieber JG, Arndt PG, Nick JA, Worthen GS. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am J Respir Crit Care Med. 2005;171:606–615. doi: 10.1164/rccm.200406-729OC. [DOI] [PubMed] [Google Scholar]
- 111.Benati D, Ferro M, Savino MT, Ulivieri C, Schiavo E, Nuccitelli A, Pasini FL, Baldari CT. Opposite effects of simvastatin on the bactericidal and inflammatory response of macrophages to opsonized S. aureus. J Leukoc Biol. 2009;87:433–442. doi: 10.1189/jlb.0409273. [DOI] [PubMed] [Google Scholar]
- 112.Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Gallo RL, Monestier M, Wang Y, Glass CK, Nizet V. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. 2010;8:445–454. doi: 10.1016/j.chom.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]