

Abstract
A high membrane‐to‐cytoplasm ratio makes axons particularly vulnerable to traumatic injury. Posttraumatic shifts in ionic homeostasis promote spectrin cleavage, disrupt ankyrin linkages and destabilize axolemmal proteins. This study contrasted ankyrin‐G and αII‐spectrin degradation in cortex and corpus callosum following diffuse axonal injury produced by fluid percussion insult. Ankyrin‐G lysis occurred preferentially in white matter, with acute elevation of all fragments and long‐term reduction of a low kD form. Calpain‐generated αII‐spectrin fragments increased in both regions. Caspase‐3 lysis of αII‐spectrin showed a small, acute rise in cortex but was absent in callosum. White matter displayed nodal damage, with horseradish peroxidase permeability into the submyelin space. Ankyrin‐G‐binding protein neurofascin and spectrin‐binding protein ankyrin‐B showed acute alterations in expression. These results support ankyrin‐G vulnerability in white matter following trauma and suggest that ankyrin‐G and αII‐spectrin proteolysis disrupts Node of Ranvier integrity. The time course of such changes were comparable to previously observed functional deficits in callosal fibers.
Keywords: corpus callosum, membrane cytoskeleton, proteolysis, rat, traumatic axonal injury, traumatic brain injury
INTRODUCTION
Extensive clinical and experimental evidence has shown that traumatic axonal injury (TAI) is a consistent pathological feature of head injury affecting widespread areas of the brain. The extent of axonal damage is a major determinant of posttraumatic morbidity and correlates significantly with the degree of functional deficits 1, 19, 63. Laboratory investigations have revealed TAI to be a multiphase pathology, with a rapid primary response, including failures of ionic homeostasis, evolving over hours and days to secondary injury processes, including structural alterations and deleterious biochemical cascades 47, 65, 72, 77.
Prior research has also demonstrated the complex nature of the early events in TAI. Several mechanisms have been implicated in the initial pathogenesis, including perturbations of axolemmal permeability 55, 59, 60, 66, focal cytoskeletal changes 6, 31, 43, 49, 78, alterations in ion channel properties 30, 85 and functional shifts of nodal membrane ion pumps 46, 48. Earlier studies modeling acceleration injury in nonhuman primates also showed extensive pathology at the node/paranode junction, including fragmentation and enlargement of nodal axoplasm (45). However, irrespective of the initial triggering process, the ensuing changes in TAI inevitably include cytoskeleton alterations and breakdown, linked to posttraumatic increases in intracellular calcium 7, 71. Major molecular targets of this pathology include the “membrane skeleton,” comprised largely of a spectrin network located on the cytoplasmic surface of the axolemma. Spectrin is attached to the plasma membrane through interactions involving ankyrin. Results from multiple laboratories have consistently documented the proteolysis of sub‐axolemmal spectrin, mediated by the calpain family of calcium‐dependent neutral proteases 20, 52, 56, 73, 75.
In contrast to the extensive experimental evidence pertaining to calpain‐mediated spectrin proteolysis, injury‐induced changes in ankyrin have not been systematically investigated. However, mounting evidence demonstrates that ankyrin proteins function in roles beyond those of simple linker molecules, as previously regarded. The ankyrins are involved in protein sorting 3, 82 and signal transduction 27, 70. In these roles, ankyrins have a diverse set of binding partners in a variety of tissues, interacting with the cytoplasmic domains of ion channels 42, 86, transporters 37, 50, Na+K+‐ATPase 12, 14, cell adhesion molecules (17) and some classes of receptors 4, 26. Within the mammalian CNS, multiple lines of evidence show that ankyrins stabilize the nodal and paranodal structure of myelinated axons. This role is executed not only through spectrin, but via binding with transmembrane neurofascins (reviewed in 80), as well as directing the clustering of voltage‐gated sodium channels (NaVs) within axonal initial segments and at Nodes of Ranvier 13, 34, 67, 89. Notably, with knockdown of ankyrin‐G expression, Nav channels fail to develop mature clusters at nodes within the dorsal root ganglia (18). Further, when mutant Caspr−/− mice fail to form transverse bands or proper paranodal junctions, the normal distribution of paranodal ankyrin‐B is disrupted (54). In addition, some reports also suggest the expression of distinct ankyrin isoforms within unmyelinated axons 33, 34, 58, which is significant in view of recent evidence that traumatic injury differentially affects subpopulations of axons 15, 68, 78. The need to examine ankyrin response during the pathogenesis of TAI has grown in step with the expanded understanding of the multifunctional nature of these proteins. In view of their complex functional and structural roles, ankyrin molecules may be vulnerable to the mechanical or biochemical perturbations of injury and play a significant role in the pathology of TAI.
The current study examined changes in ankyrin‐G and αII‐spectrin expression following midline fluid percussion brain injury administered to adult rats. Protein levels were assessed in corpus callosum and in cerebral cortex during the postinjury time interval of 3 h–7 days. Analyses were directed at whether ankyrin‐G undergoes a proteolytic fragmentation comparable to that observed for spectrin, and whether injury‐related changes to ankyrin‐G are temporally correlated with those of αII‐spectrin. The study design was also the first to explicitly compare the injury response of these membrane cytoskeletal proteins in white vs. gray matter. Because axons have an intrinsically higher membrane‐to‐cytoplasm ratio than dendritic or parykaryal subcellular compartments, it was reasoned that injury processes targeting membrane‐associated proteins would produce a distinctive proteolytic response in the axon‐rich callosal tissue. Structural correlates of TAI were obtained focusing on pathology at the Node of Ranvier, specifically the effect of injury on the ankyrin‐G‐binding partner neurofascin and on ankyrin‐B, which binds to αII‐spectrin/β‐spectrin heterodimers. An additional goal for the investigation was to search for molecular correlates of conduction deficits observed in corpus callosum axons following traumatic brain injury (TBI) 68, 69, where the function of myelinated and unmyelinated fiber populations exhibited differential degrees of impairment and recovery.
METHODS
The procedures for this study followed all national guidelines for the care and use of experimental animals, and the experimental protocol was approved by the Medical College of Virginia Animal Research Committee. Male Sprague‐Dawley® rats (Harlan Laboratories, Indianapolis, IN, USA) (n = 107) weighing 300–350 g, at the start of the study, were used in these experiments. Animals were housed in individual cages in a temperature‐ (22°C) and humidity‐controlled (50% relative) animal facility on a 12‐h light/dark cycle. Rat chow and water were continually available.
Fluid percussion TBI procedure
Rats were anesthetized with sodium pentobarbital (54 mg/kg, i.p.), and a 4.8‐mm skull craniotomy was prepared over the midline, centered between bregma and lambda. A Leur‐Loc® syringe hub (Becton Dickinson, Bedford, MA, USA) was cemented with cyanoacrylate to the skull surrounding the craniotomy, and dental acrylic was poured around the syringe hub and two small screws placed in the skull for implant rigidity. Bacitracin was applied to the incision, and the animal returned to its home cage.
Twenty‐four hours following implantation of the syringe hub, rats were anesthetized with isoflurane (4% in carrier gas of 70% N2O and 30% O2) and immediately subjected to moderate central fluid percussion injury as described by Dixon et al (16). Briefly, the device consisted of a 60 × 4.5 cm Plexiglas water‐filled cylinder, fitted at one end with a piston mounted on O‐rings, with the opposite end housing a pressure transducer (Entran Devices, Inc., Fairfield, NJ, USA; EPN‐0300A). At the time of injury, the Leur‐Loc® fitting, filled with saline, was attached to the transducer housing. The injury was produced by a metal pendulum that struck the piston, transiently injecting a small volume of saline into the cranial cavity and briefly deforming the brain tissue (20 ms pulse duration). The resulting pressure pulse was recorded extracranially by the transducer and expressed in atmospheres (atm) of pressure. Injury magnitude was controlled by setting the height from which the pendulum was released. Following injury all animals were promptly ventilated with room air until spontaneous breathing resumed. The duration of suppression of the righting reflex was used as an index of traumatic unconsciousness. Sham‐injured controls received the same surgical preparation, anesthesia and connection to the injury device, except that the intracranial pressure pulse was not injected.
Tissue extraction and immunoblot assays
Animals allowed to survive for intervals of 3 h (n = 14), 1 day (n = 14), 3 days (n = 11) or 7 days (n = 8) following TBI, along with their paired sham‐injured cases at 3 h (n = 10), 1 day (n = 9), 3 days (n = 7) and 7 days (n = 6), were decapitated under 4% isoflurane anesthesia. Brains were rapidly removed and blocked into 2‐mm coronal sections. Corpus callosum was dissected bilaterally bounded anteriorly by the ventral hippocampal commissure and posteriorly by the splenium. Cerebral cortex was dissected from the same slices targeting the parieto‐temporal region known to be vulnerable to injury in this model (see Figure 1). Tissue samples for ankyrin‐G and αII‐spectrin Western blots were homogenized in 1X RIPA Lysis Buffer (Millipore, Temecula, CA, USA) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics; Indianapolis, IN, USA), 0.1% SDS and 2 mM EGTA. The homogenates were centrifuged for 20 minutes at 14 000×g at 4°C, supernatants removed, aliquotted and stored at −80°C. Protein concentration of each sample was determined using either Bio‐Rad (Bio‐Rad Laboratories, Hercules, CA, USA) or Pierce (Thermo Scientific, Rockford, IL, USA) protein assay. Aliquots containing either 20–30 ug of protein were mixed with reduced sample buffer and electrophoresed (150 V, 60 minutes) on 3%–8% NuPAGE® Tris‐Acetate gels (Invitrogen, Carlsbad, CA, USA) and subsequently transferred to PVDF membranes (Invitrogen). Post blotted membranes were blocked at room temperature in 5% milk‐TBST (Tris buffered saline containing 0.05% Tween 20) for 1 h before being probed with either mouse monoclonal anti‐ankyrin‐G primary antibody (1:100; Research Diagnostics, Concord, MA, USA) or mouse monoclonal anti‐αII‐spectrin antibody (1:4000; Enzo Life Sciences International, Inc., Plymouth Meeting, PA, USA) in milk‐TBST overnight at 4°C. Blots were then washed with milk‐TBST and incubated for 1 h at room temperature in peroxidase‐conjugated, affinity‐purified goat antimouse IgG secondary antibody (1:20 000; Rockland Laboratories, Gilbertsville, PA, USA) prior to a final washing in TBST. Parallel blots were incubated without primary antibody to confirm signal specificity. Immunoreactive bands were visualized using ECL™ Western Blotting Analysis System (Amersham Biosciences, GE Healthcare, Piscataway, NJ, USA) and membranes exposed to Kodak BioMax® Film (Eastman Kodak Company; Rochester, NY, USA). Digital images were analyzed densitometrically using the Scion Image Analysis System (Scion Corporation, Frederick, MD, USA). Relative optical density was calculated as band area × signal density and injury‐induced effects expressed as percent change from paired sham‐injured cases run on the same blot. After primary antibody binding data were captured, all blots were stripped and re‐probed for β‐actin (mouse monoclonal, Sigma, St. Louis, MO, USA; 1:3000) as a load control. No differences in load between the lanes were detected.
Figure 1.
Location of tissue dissections used for Western blot analyses. Regional boundaries for corpus callosum (CC) and parieto‐temporal cortex (PT) are shown in coronal section (top). Rostrocaudal extent of callosal dissection is shown in midsaggital view (bottom).
A second set of callosal samples was processed for neurofascin and ankyrin‐B Western blot analysis in order to examine the profile of specific nodal and paranodal binding partners of ankyrin‐G and αII‐spectrin. Rats were sacrificed 1 day after TBI for neurofascin (n = 4) or ankyrin‐B (n = 3), along with paired sham‐injured controls (n = 4 for neurofascin; n = 3 for ankyrin‐B) and protein extracts of corpus callosum prepared as described above. Twenty µg of protein was mixed with reduced sample buffer and electrophoresed on either 4%–12% Bis‐Tris Criterion™ XT gels (neurofascin; 200 V, 45 minutes; Bio‐Rad Laboratories) or 3%–8% NuPAGE® Tris‐Acetate gels (ankyrin‐B; 200 V, 45 minutes) and subsequently transferred to PVDF membrane. One membrane was blocked in milk‐TBST and probed overnight at 4°C with mouse monoclonal antibody recognizing an intracellular epitope within neurofascin (L11A141.6, 1:200 dilution) in milk‐TBST, a generous gift from Dr. Matthew Rasband. A second blot was also probed overnight at 4°C with mouse anti‐ankyrin B (1:100, Research Diagnostics, Concord, MA). All membranes were subsequently washed and incubated in peroxidase‐conjugated secondary antibody, either bovine antirabbit (1:20 000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or goat antimouse (1:20 000; Rockland Immunochemicals, Gilbertsville, PA, USA) and immunopositive signal visualized using SuperSignal® West Dura Extended Duration Substrate (Thermo Scientific). Blots were then imaged digitally with the G:Box ChemiHR system for densitometric analysis using GeneSnap software (SynGene, Frederick, MD, USA). After antibody binding data were captured, all blots were stripped and re‐probed for β‐actin as described above. No differences in load between the lanes were detected.
Electron microscopy (EM) of corpus callosum
At 1 day postinjury, rats from both TBI (n = 6) and sham‐injured (n = 4) groups were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and perfused transcardially with 0.9% saline, followed by mixed aldehyde fixative (4% paraformaldehyde and 0.2% glutaraldehyde) in 0.1 M PB, pH 7.2. Brains were removed and postfixed overnight at 4°C. Coronal sections (40 µm) of corpus callosum were collected in 0.1 M PB, placed in 1% osmium (0.1 M phosphate buffer, pH 7.2) and then processed in resin prior to being flat‐embedded on plastic slides. After curing, mid‐dorsal corpus callosum was excised and a series of semithin (0.5 µm) and ultrathin (silver, 600 Å) sections were cut on a Leica EM UC6i ultramicrotome (Leica Microsystems, Wetzlar, Germany). Mid‐dorsal corpus callosum was selected for sampling because the midline fluid percussion insult produces its primary callosal injury in this region and our prior electrophysiological studies revealed differences of axonal conductance properties within mid‐dorsal callosum. The semithin sections were used to generate photographs of the entire block face as a guide for subsequent ultrastructural sampling. Ultrathin sections were collected on membrane‐coated slotted grids and observed on a Jeol JEM‐1230 electron microscope (JEOL USA, Inc., Peabody, MA, USA) equipped with a Gatan UltraScan 4000SP CCD camera (Gatan, Inc., Pleasanton, CA, USA). At 5000 × magnification, a series of 400 µm2 montages were collected at regular intervals across the dorso‐ventral axis of the corpus callosum (for most rats 10 montages were collected, although this ranged from 8 to 12). These regions were examined for profiles cut through Nodes of Ranvier. Node images from injured and sham‐control cases were then compared for qualitative differences in cytoarchitecture.
A second EM experiment was designed to examine the extent of blood brain barrier disruption within the corpus callosum and document the movement of normally excluded molecules into the tissue spaces surrounding callosal axons. At 1 h prior to fluid percussion insult, injured and sham control subjects (n = 3/group) were injected with horseradish peroxidase (HRP type IV; Sigma, St. Louis, MO, USA; 5 mg in 0.1 cc saline, i.c.v.) after the method of Singleton and Povlishock (76). Briefly, a 4.8‐mm craniotomy was made midway between bregma and lambda, after which a blunt 25‐g needle was lowered through the dura into the left lateral ventricle (0.5 mm posterior, 1.4 mm lateral and 4.0 mm depth). Tracer infusion was made over a period of 20–30 minutes and the needle left in place for 10 minutes after infusion prior to removal. Rats were then subjected to moderate central fluid percussion injury and allowed to survive for 1 day postinjury. Animals were then anesthetized, perfused with the EM fixative above and processed for HRP visualization using the cobalt‐glucose oxidase technique of Povlishock et al (64). After visualization, 40 µm sections of the corpus callosum were processed for routine EM observation and thin sections prepared for qualitative analysis of HRP distribution.
Statistical analysis
Changes in Western blot protein expression following injury were evaluated by comparison of relative optical density measures within injured samples to that of paired sham control cases. Results were expressed as percent of control value. The effect of injury on protein levels was evaluated using ANOVA with factors of injury condition (sham vs. TBI) and postinjury survival interval (3 h, 1 day, 3 days and 7 days). Changes in ankyrin‐G, αII‐spectrin, neurofascin or ankyrin‐B following injury were evaluated using simple effects based on planned comparisons (32). Each comparison tested the significance of the mean protein level in sham vs. TBI rats at a single survival interval. A probability of less than 0.05 was considered statistically significant for all experiments. Effect size, estimated using Cohen's d, was also computed for each statistically significant comparison and is reported along with probability levels.
RESULTS
Injury‐induced change in ankyrin‐G expression
Western blot analyses of both corpus callosum and cerebral cortex identified three major immunopositive bands in sham and injured animals. These bands exhibited molecular weights of 220, 212 and 75 kD (Figure 2). Specificity for ankyrin‐G was confirmed using parallel lanes processed without primary antibody and equal gel loading checked by subsequent membrane stripping and re‐probing with an antibody to β‐actin. Given that the commercial antibody used in this study recognizes both intact ankyrin‐G and proteolytic fragments, it was not unexpected that the major signals detected fell in the range of ankyrin‐G breakdown products. Other researchers have similarly observed that the major intact isoforms of ankyrin‐G (480, 270 and 190 kD) are in relatively low abundance within the CNS and sometimes difficult to extract for analysis (5). Indeed, we observed weak to nondetectable signal for these intact forms. Their optical density was well below background and did not fall with the linear range of signal output required for either Scion Image or Syngene Gene Tools software analysis. Therefore, the current study focused on a spatio‐temporal analysis of the three major fragments showing strong ankyrin‐G signal after TBI. Given the expressed molecular weights of these fragments and their different temporal profiles following injury, it is likely that the 220 and 212 kD are derived from either the 480 or 270 kD intact forms, known to be associated with the Nodes of Ranvier in myelinated axons 33, 34, 79, 80. As discussed below, the 75 kD band may represent a proteolytic fragment of the 190 kD isoform of ankyrin‐G, suggested to be abundant in unmyelinated axons (34), although the possibility that the 75 kD band reflects residual fragments of the 270/480 kD forms cannot be eliminated based on the present results.
Figure 2.
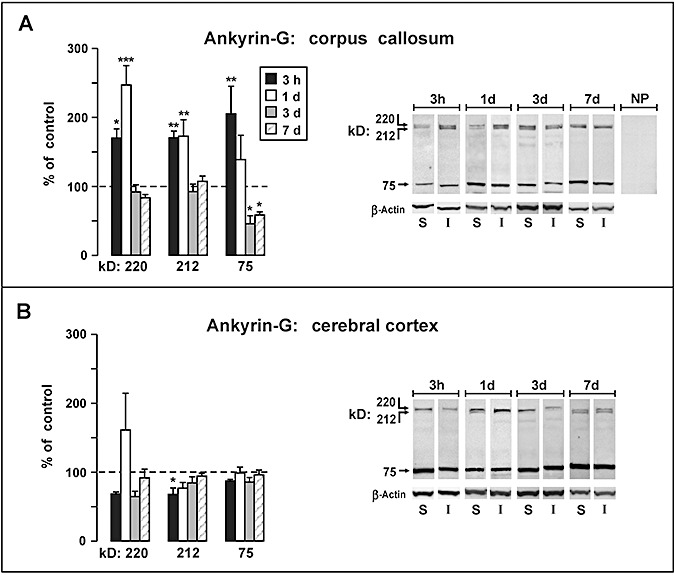
Western blot analysis of ankyrin‐G fragments following moderate fluid percussion injury. Data are plotted as percent change (mean ± SEM) from sham‐injured control rats at survival intervals 3 h, 1 day, 3 days and 7 days. A. In the corpus callosum, a significant surge was observed in 220 and 212 kD ankyrin‐G, which recovered to control levels on 3 days and 7 days. The 75 kD ankyrin‐G product was significantly elevated at 3 h postinjury but decreased to levels significantly below controls at 3 days and 7 days (P < 0.05). B. The injury produced relatively minor changes in levels of ankyrin‐G fragments in the parieto‐temporal cortex, with the singular significant change being a 32% decrease noted for the 212 kD band measured at 3 h postinjury (P < 0.05). *P < 0.05, **P < 0.01, ***P < 0.001, comparing protein levels in injured rats with sham control rats.
Moderate central fluid percussion altered the expression of each of the three major ankyrin‐G bands; however, the predominant changes were observed in the corpus callosum (Figure 2A) with little or no change seen in the cortical samples (Figure 2B). Within the corpus callosum the 220 and 212 kD bands exhibited a similar pattern of significant postinjury increases at early time points (3 h–1 day), followed by a reduction to control levels at later intervals (3–7 days). At 3 h postinjury, an increase of approximately 70% was observed for the 220 kD [P < 0.05; Cohen's d (d) = 2.76] and 212 kD (P < 0.01; d = 3.53) bands. However, at 1 day injury‐induced change in the 220 kD band climbed to a significant 147% increase over control levels (P < 0.001; d = 2.67), while the 212 kD band remained significantly elevated at approximately the same range measured at 3 h (72% increase over controls; P < 0.01; d = 1.66). The postinjury changes in the 75 kD ankyrin‐G band contrasted sharply with the pattern observed for the 220/212 kD doublet bands. Specifically, levels of the 75 kD protein showed a significant 105% increase at 3 h (P < 0.05; d = 1.47), but the band density decreased steadily to 39% at 1 day and continued to fall to levels significantly below controls at 3 and 7 days. At 3 days the 75 kD band density was 54% (P < 0.05; d = −1.82) below that of the sham controls, and at 7 days was 41% (P < 0.05; d = −2.99) below control levels.
Analyses of ankyrin‐G within cortical tissue revealed a dramatically different profile of posttraumatic alterations than those observed in the corpus callosum. The major pattern for all bands (220, 212, 75 kD) was to remain at, or slightly below, control levels. The magnitudes of these injury effects were small, however, attaining statistical significance only for a 32% decrease noted for the 212 kD band measured at 3 h postinjury (P < 0.05; d = −2.00). There was one possible exception to the general pattern of postinjury suppression in cortical ankyrin‐G levels, which was a 61% increase in the mean density of the 220 kD band seen at 1 day, although this narrowly missed significance (P = 0.063).
Injury‐induced change in αII‐spectrin expression
Analysis of Western blots showed antibody recognition of five major bands, a result consistent with prior descriptions of banding patterns for this antibody 20, 61, 62, 81. The five bands migrated at 280, 180, 150, 145 and 120 kD, and densitometric analyses revealed injury‐induced increases in some bands.
Within the corpus callosum, levels of the 280 kD band, representing the intact spectrin molecule, were suppressed during the initial postinjury period and showed a significant 28% decrease at 1 day (P < 0.01; d = −1.70) (Figure 3A). However, callosal levels of intact 280 kD αII‐spectrin recovered and were not significantly different from control values at 3 and 7 days. The corpus callosum exhibited a marked upsurge in levels of the calpain‐derived 145 kD fragment of αII‐spectrin. This increase reached significance at 1 day, with a 4.2‐fold elevation over controls (P < 0.001; d = 1.43), and peaked at 3 days when the 145 kD fragment was 566% of control levels (P < 0.001; d = 3.14). By 7 days, levels of the 145 kD fragment receded to a level 2.5‐fold above the control value, but this was no longer significant. The 150 kD fragment, which may represent a mix of fragments from calpain and caspase‐3 activity 2, 84, showed modest elevations in the corpus callosum, with significant (P < 0.05) increases of 76% (d = 0.92) and 94% (d = 1.78) at 1 and 3 days, respectively. In contrast to the substantial increases seen in the 145/150 kD calpain‐associated fragments, the 120 kD caspase‐3‐derived breakdown product showed no significant change in response to TBI and remained within 20% of control levels.
Figure 3.
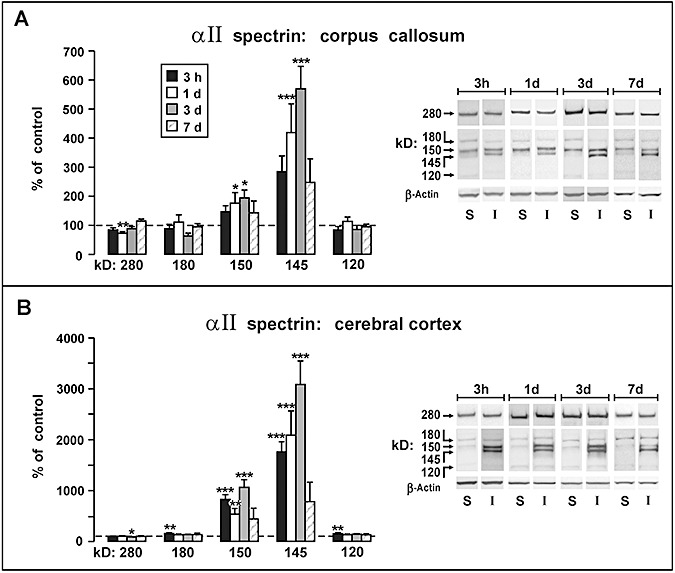
Western blot analysis of αII‐spectrin following moderate fluid percussion injury. Data are plotted as percent change (mean ± SEM) from sham‐injured control rats at survival intervals 3 h, 1 day, 3 days, and 7 days. A. Analysis of αII‐spectrin and fragments in the corpus callosum indicated an injury response primarily mediated by calpain proteolysis, with significant increases at 1 day and 3 days in the 145/150 kD products. No significant injury related changes were found for the 120 kD fragment associated with caspase‐3 activity. B. Analysis of αII‐spectrin and fragments in the parieto‐temporal cortex revealed massive increases in the 145/150 kD fragments, exceeding control levels by approximately 30‐fold (P < 0.001) at 3 days postinjury. Comparatively minor postinjury increases were seen for the 180 kD and 120 kD bands at the 3 h measurement, both showing approximately 50% increases above controls (P < 0.01). *P < 0.05, **P < 0.01, ***P < 0.001, comparing protein levels in injured rats with sham control rats.
Analyses of cortical tissue extracts revealed a similar pattern of injury‐induced change, with sharp escalations in the 145 kD band and more modest increases in the 150 kD band (Figure 3B). However, the magnitudes of the percent change relative to control cases for these bands were 5–10‐fold higher than the corresponding injury‐induced changes observed in the callosum. At the earliest postinjury measurement (3 h) significant increases were seen in the 145 kD band (1757% of controls; P < 0.001; d = 3.94) and in the 150 kD band (826% of controls; P < 0.001; d = 3.66). The largest magnitude response measured in the study was a 3086% increase above controls observed in the 145 kD band at 3 days postinjury (P < 0.001; d = 3.32). The remaining bands quantified in cortical tissue (280, 180, 120 kD) exhibited posttraumatic alterations comparable in magnitude to those same bands in the corpus callosum. Postinjury decreases in intact cortical αII‐spectrin (280 kD) were small and reached significance only at 3 days with a 14% decrease from control levels (P < 0.05; d = −1.84). The 180 and 120 kD bands were transiently elevated at the 3 h measurement, both showing approximately 50% increases above controls (P < 0.01; d = 1.61).
The magnitude of effect of changes in protein level, as assessed using the Cohen's d statistic, suggested these injury‐induced alterations are robust. In the case of ankyrin‐G, the absolute value of d ranged from 1.47 to 3.53, and for αII‐spectrin from 0.92 to 3.94. These values are well above the approximate guideline suggested by (9), that values above 0.80 correspond to “high” magnitudes of effect.
Altered node/paranode structure correlated with changes in cytoskeletal binding partner expression
Ultrastructural analysis of injured corpus callosum showed diffuse axonal pathology, with compaction of axonal cytoskeleton (43), and damage to Nodes of Ranvier, often appearing as cytoplasmic extravasations, or nodal blebs, previously described in an optic nerve model of axonal injury by Maxwell and colleagues (44). In control callosal samples the axolemma was continuous across the node, with profiles of intact paranodal loops and normal nodal interface with glial processes (Figure 4A). By contrast, injured axons showed evidence of disrupted submembrane cytoarchitecture and axolemmal disorganization at the node, often presenting with expansion of axoplasm into the extracellular space (Figure 4B, C). Such profiles are consistent with the development of secondary axotomy, where cytoskeletal networks are disrupted over time, culminating in axonal disconnection. In some cases paranodal loops exhibited darkened, floccular cytoplasm (Figure 4D), while other injured axons displayed frank paranodal membrane degeneration (Figure 4E). When the nodal/paranodal adhesion protein neurofascin was assayed at 1 day postinjury by Western blot (Figure 5A), the primary neuronal and glial isoforms (186, 155 kD) showed no change in expression. However, a third band at 120 kD, most likely a lytic fragment of neurofascin, was increased relative to sham controls (60.4 ± 18.7%; P < 0.02; d = 2.40). In a second blot experiment, we found reduction (29.5 ± 3.69%; P < 0.001; d = 3.46) of a 230‐kD band representing the adult isoform of ankyrin‐B at 1 day postinjury (Figure 5B). These results show that the injury‐induced lysis of ankyrin‐G and αII‐spectrin is concomitant with altered expression of their white matter‐binding partners, neurofascin and ankyrin‐B. These differences are spatially and temporally correlated with structural pathology seen at the Node of Ranvier.
Figure 4.
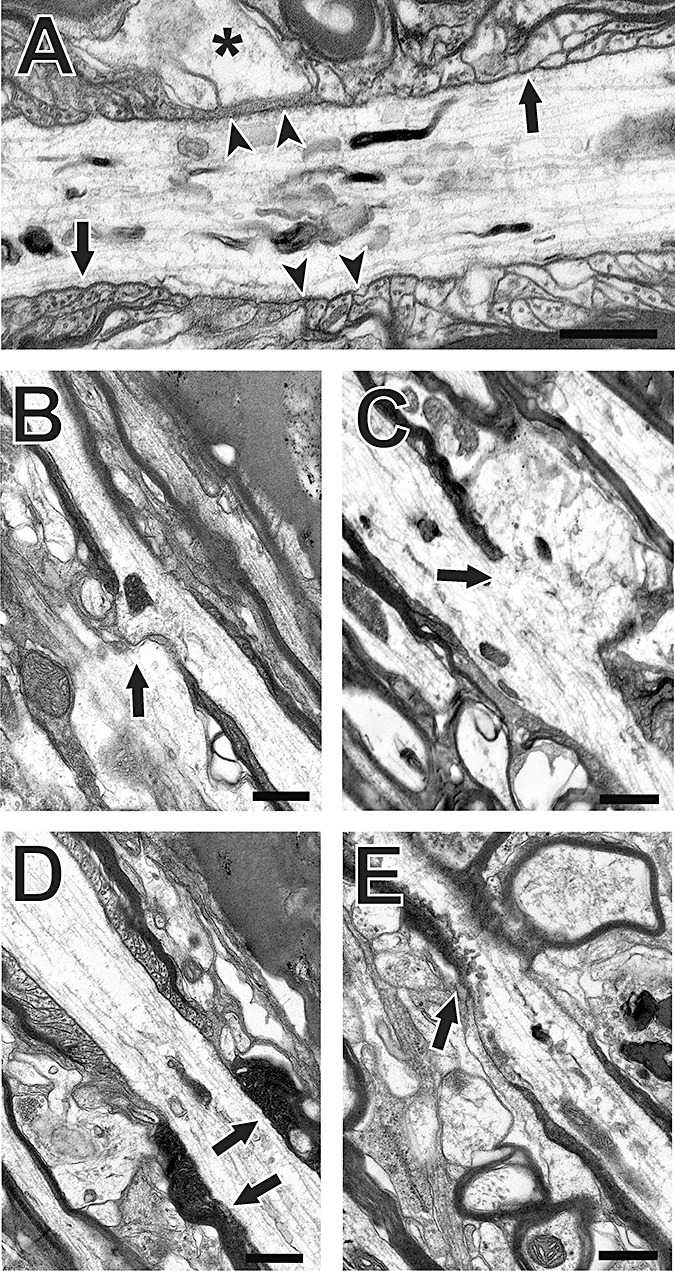
Effect of moderate fluid percussion injury on Node of Ranvier ultrastructure: (A) sham‐injured control; (B–E) injured cases. Sham controls show typical nodal cytoarchitecture, with intact axolemmal membranes (arrowheads in A), normal profiles of adjacent paranodal loops (arrows in A) and axonal/glial interfaces (asterisk in A). Following injury, the axolemma is disrupted, with sites of extruded axonal cytoplasm visible (arrows in B,C), in some cases showing shifts of axial cytoskeleton into these regions. Further, paranodal loops of injured cases may appear abnormally electron dense (arrows in D), or show clear membrane disorganization (arrows in E). Bars = 0.5 µm.
Figure 5.
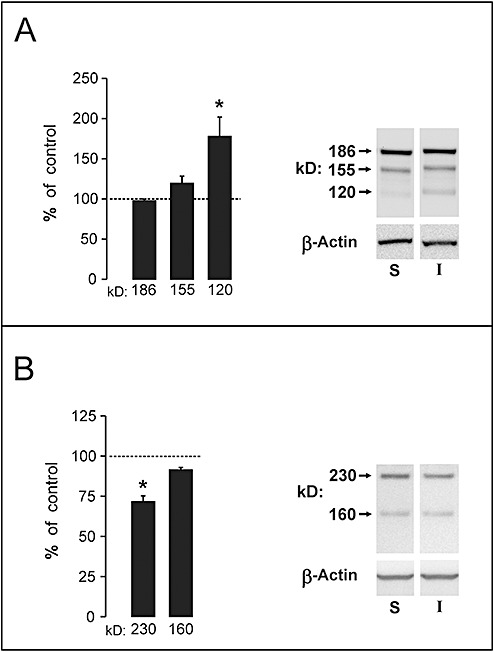
Neurofascin and ankyrin‐B expression in corpus callosum at 1 day postinjury. A. Results show a 60% elevation of signal within a novel 120 kD form of neurofascin, likely a proteolytic fragment. The prominent 186 and 155 kD forms of the protein failed to change at the 1 day time interval. B. A 230 kD isoform of ankyrin‐B, paranodal ligand for αII‐spectrin, was reduced by 30% at 1 day after injury. Representative gel images for each antibody and load control of β‐actin are shown at right. *P < 0.05.
Injury‐induced compromise of node/paranode integrity and movement of HRP into submyelin space
Examination of blood brain barrier disruption in corpus callosum at 1 day postinjury showed that HRP, normally retained within brain vasculature, had passed through the vessel walls and entered into the extracellular space around myelinated and unmyelinated fibers. Movement of HRP into the submyelin space adjacent to damaged axons was also evident. A similar pattern of barrier vulnerability has been reported for brainstem white matter tracts subjected to impact acceleration injury (60). At low magnification, we observed the HRP tracer confined within callosal vasculature of controls but clearly extruded into the parenchyma of injured animals (Figure 6A, B). When unstained thin sections were examined ultrastructurally, HRP was extensively distributed within the spaces surrounding myelinated axons and outlining bundles of small unmyelinated fibers (Figure 6C). This view also revealed a thin band of tracer between myelin sheath and the axolemma of some fibers. In counterstained thin sections, details of cytoarchitecture confirm that HRP reaches the extracellular interface between myelinated and unmyelinated axons, as well as spaces surrounding glial processes (Figure 6D). At injured nodes, HRP appears to fill the space around paranodal loops and extends along the initial juxtaparanodal axon/glial interface (Figure 6E). From these observations, damaged nodes exhibiting reduced cytoskeletal integrity and membrane adhesion may permit axonal exposure to molecules normally excluded from the internodal region.
Figure 6.
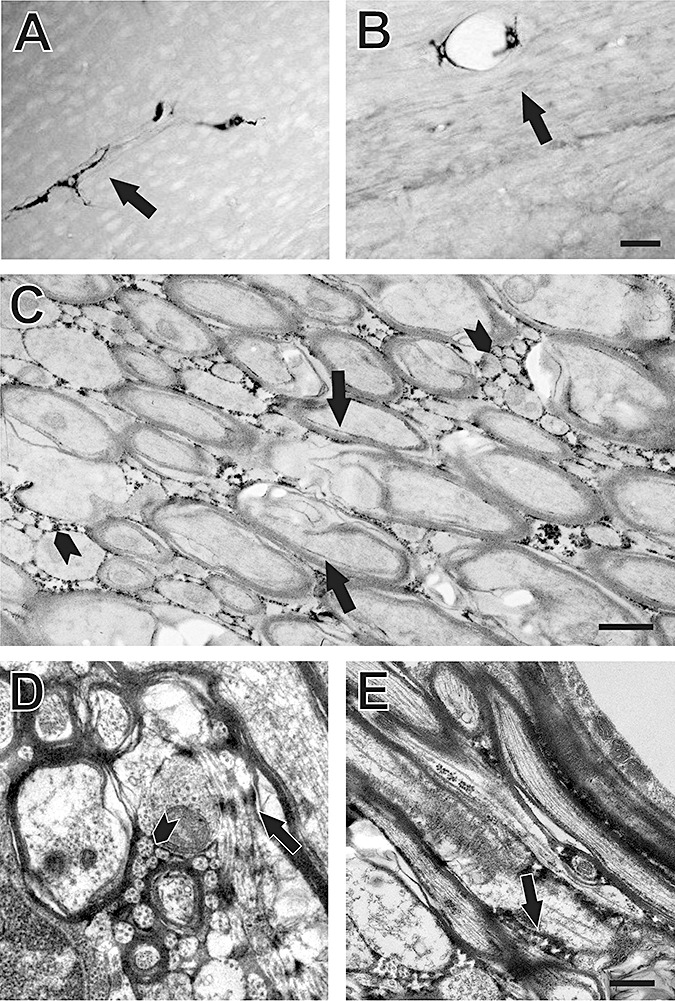
Effects of moderate fluid percussion injury on white matter blood brain barrier disruption and nodal integrity. A. Low magnification view of corpus callosum from control case infused with HRP 1 h before sham injury. Intact blood brain barrier retards extravasation of HRP into neuropil, with tracer restricted to the callosal vasculature (arrow). B. Corpus callosum from animal administered HRP 1 h prior to moderate central fluid percussion injury and sacrificed 24 h postinjury. Movement of HRP from vascular bed into space around axons is evident throughout the white matter (arrow). C. Ultrastructure of non‐counterstained injured case showing HRP diffusion into extracellular space around small unmyelinated fibers (arrowheads) and within the submyelin space between axolemma and inner myelin sheath (arrows). D,E. Higher magnification of counterstained ultrastructure from an injured case. In D HRP localization is seen around axon bundles (arrowhead) and along the surface of glial membranes (arrows) and infiltrating paranodal/submyelin space at arrow in E. Bar = 30 µm in A,B; 10 µm in C; 0.5 µm in D, E.
DISCUSSION
The present study examined TBI‐induced changes in ankyrin‐G and αII‐spectrin fragments over 3 h–7 days following moderate central fluid percussion injury. Although others have documented widespread posttraumatic spectrin proteolysis, the present study is the first to investigate a role for ankyrin‐G in the pathology of brain trauma and the first to specifically compare injury‐induced changes in both cytoskeletal binding proteins. We show divergent injury‐induced changes in expression of these molecules dependent upon brain region, with ankyrin‐G alterations primarily a white matter phenomenon, while spectrin breakdown is disproportionately localized to gray matter.
Postinjury alterations in ankyrin‐G fragments
Injury‐induced changes in ankyrin‐G fragments were strikingly different from those produced in αII‐spectrin, being more rapid, smaller in percent postinjury change and confined almost exclusively to the white matter samples of the study. Significant postinjury increases in the 220/212 kD forms were noted at 3 h and 1 day, in the callosum, and the 75 kD product was significantly elevated only at 3 h. Unlike the 220/212 kD bands, the 75 kD fragment was significantly reduced below control levels at 3 and 7 days. In the cerebral cortex, the singular significant finding was a transient decrease in the 212 kD fragment noted at 3 h. This supports a predominantly white‐matter locus for the ankyrin‐G vulnerability in central fluid percussion TBI.
The comparison of injury‐induced ankyrin proteolysis in the callosum with that in the cortex is important for at least two reasons. First, relative to other subcellular compartments, axons have a high membrane‐to‐cytoplasm ratio, which may constitute an elevated risk factor for membrane‐targeting pathological processes. Secondly, multiple lines of evidence point to increased calpain proteolytic activity following brain injury 7, 52, 61, 71, 73, and calpain isozymes are unequally distributed in white vs. gray matter. Calpain I (or µ‐calpain) is preferentially located in neuronal perikarya and dendrites and less abundant in axons (57), whereas calpain II (or m‐calpain) is more prevalent in axons (23). In this regard, regional differences in calpain activation kinetics may determine, in part, susceptibility to elevated intracellular calcium. The present observation of early postinjury elevation of callosal ankyrin‐G fragments, followed by a 3–7‐day reduction, is consistent with an injury‐induced shift in the normal functional role of axonal ankyrin following TBI. In contrast to the extensive characterization of calpain‐mediated spectrin breakdown after TBI, calpain proteolysis of brain ankyrin has been described only after ischemic injury (24). However, cleavage of ankyrins by calpain is a critical pathogenic event in non‐neural organ systems, and consistently observed after myocardial ischemia 29, 87, 88. Notably, calpains are capable of proteolyzing a diverse set of proteins, including enzymes such as Ca2+‐ATPase, and cytoskeletal proteins such as α‐actinin, talin, tubulin, spectrin and ankyrin (10), which may be enriched in white matter. In view of this generalized distribution and varied substrates of calpain, it is likely that ankyrin fragments observed in the present study are generated, at least in part, by calpain. While a specific role for calpain‐mediated ankyrin proteolysis has not been established, the regulatory domain of ankyrin is known to be highly acidic and sensitive to proteolysis 38, 39. This domain modifies the affinities of the spectrin‐binding and membrane‐binding domains to their target proteins (21). Such flexibility in binding affinity may underlie distinct roles for different ankyrins during normal neuronal function. It is notable that ankyrin‐B promotes neurite initiation possibly by coupling actin flow to extracellular substrates via L1‐CAM (53), although it is not known if proteolysis is involved.
Postinjury alterations in αII‐spectrin fragments
Analysis of αII‐spectrin fragments revealed significant changes in the cerebral cortex at the earliest time point sampled, 3 h postinjury, which reached a maximum at 3 days. These posttraumatic alterations were almost entirely confined to the 145 and 150 kD forms, which are the signature fragments of a calpain‐mediated breakdown process (25). There was no comparable increase in the 120 kD fragment, known to signify caspase‐3‐mediated apoptosis 51, 83, with the single exception being a modest 20% increase detected at 3 h after injury. Postinjury changes in callosal αII‐spectrin fragments, measured presently, were similar to those observed in the cortex, although attenuated in magnitude and not reaching statistical significance until 1 day. The detection of αII‐spectrin proteolysis in white matter at 1 day after injury is consistent with prior morphological evidence for increased axonal damage using antibodies to amyloid precursor protein (69) or the G*SAHEVQ cleavage site of αII‐spectrin (40).
The postinjury changes in spectrin observed in this study were generally consistent with the time course of spectrin proteolysis and cytoskeletal degeneration reported in prior studies. Notably, calpain‐mediated spectrin breakdown products were reported to occur as early as 90 minutes after lateral (71) or 3 h after midline (40) fluid percussion TBI, becoming prominent at 1–3 days postinjury, and persisting up to 7 days. Moreover, the contrasting pattern of calpain‐ and caspase‐3‐related processing of αII‐spectrin, observed in our study, was similar to prior findings using traumatic (61) and ischemic (62) injury models. These studies consistently noted massive cortical increases in 145‐kD calpain‐mediated spectrin breakdown product (up to 30‐fold) without injury‐related changes in 120 kD caspase‐3‐associated spectrin fragments. Time‐dependent changes in spectrin fragments, observed in this study, agreed well with prior descriptions of posttraumatic degeneration. In this regard, fluoro‐Jade staining of degenerating neurons increased at 1 day after rat lateral fluid percussion injury and persisted through 7 days postinjury (22). The close spatial correspondence previously noted between the appearance of spectrin breakdown products and subsequent loss of Nissl stain (71) suggests cell populations exhibiting calpain‐mediated spectrin proteolysis eventually undergo significant cell death. Similarly, following impact acceleration injury in the mouse, both calpain and caspase‐3 proteolysis of αII‐spectrin was reported (36), showing a peak fragment formation at 3 days postinjury as observed in the present study. It should be noted, however, that we also observed measurable proteolytic fragments in uninjured tissue, suggesting that some level of cytoskeletal proteolysis is operative under normal physiological processes, such as neurite extension, synaptogenesis and long‐term potentiation (reviewed in [11]). There are multiple lines of evidence to suggest this is true in the case of spectrin. In postmortem human brain, calpain‐induced spectrin breakdown products can be present without a direct correlation to either postmortem interval or clinical pathology (28).
Nodal injury and the generation of neurofascin and ankyrin‐B fragments
Ankyrin‐G and αII‐spectrin are integral to the stable linkage between a variety of membrane proteins and the cytoskeletal framework of the neuron. The present results show that, following moderate central percussive injury, there is significant degradation of ankyrin‐G and αII‐spectrin, with prominent acute ankyrin‐G lysis in white matter. Several structural correlates of this breakdown are evident at the Node of Ranvier, where axolemma is disrupted and paranodal loop sealing of the myelinated internode fails. Specific binding partners of ankyrin‐G and αII‐spectrin which are critical to node integrity also change following injury. Taken together, these results suggest that injury‐induced proteolysis of ankyrin‐G and αII‐spectrin exacerbates nodal dysfunction after TBI.
Evidence for structural pathology at the Node of Ranvier was initially described by Maxwell and colleagues following rodent optic nerve injury (44) and nonhuman primate TBI (45). Here, we also observed key features of axolemmal disruption at the node and paranodal loop disorganization. While animal studies of percussive TBI often show axonal damage with a signature of αII‐spectrin fragmentation 7, 40, little emphasis has been placed on the role of ankyrins or the extent of cytoskeletal proteolysis at the node/paranode interface. The present evidence for nodal/paranodal breakdown, along with altered neurofascin and ankyrin‐B immunobinding, are consistent with extensive callosal ankyrin‐G and αII‐spectrin lysis after TBI. Together, these results suggest at least two additional mechanisms for the evolution of axonal pathology after injury. At the node, injury‐induced proteolysis of ankyrin‐G would reduce binding to the adhesion protein neurofascin 186, destabilizing the axolemma, contributing to loss of membrane integrity. Indeed, we found evidence for an injury‐induced rise in a lower kD form of neurofascin, suggesting limited proteolysis of the intact 186 or 155 proteins after TBI. Further, this shift in neurofascin expression occurred 1 day after injury, the same survival interval showing significant disruption of node cytoarchitecture. At present, the source of the 120 kD neurofascin is unclear. A 125 kD neurofascin peptide, shed from the extracellular domain, has been described in cultured oligodendrocytes, proposed to mediate spatio‐temporal parameters of myelination (41). However, it is unlikely that our 120 kD band represents this peptide as its extracellular sequence would not be recognized by the intracellular c terminus‐targeted antibody used in our study. Alternatively, it remains possible that the 120 kD peptide is a unique neurofascin isoform induced by either neurons or glia after injury. While more specific assessment of ankyrin‐G‐/neurofascin‐binding properties will be required to resolve the role of the 120 kD neurofascin in TBI, it is clear that node destabilization is spatially and temporally correlated with ankyrin‐G proteolysis and altered neurofascin protein expression.
The current findings also suggest a second mechanism of TAI focused within the adjacent paranode. Here we provide structural evidence that paranode junctions are compromised after TBI and show HRP permeability at the myelin/axolemma interface. In this case, injury‐induced proteolysis may disrupt the ankyrin‐B/αII‐spectrin/β‐spectrin complex, which causes dissociation of transmembrane adhesion proteins and detachment of oligodendrocyte paranodal loops from the axolemma. As a result, extracellular molecules could have free access to the internode submyelin space. Collectively, the present evidence points to reduced binding between ankyrin‐B and the αII‐spectrin/β‐spectrin heterodimer complex as a mechanism of paranode pathology. We show that the predicted lower 220–230 kD ankyrin‐B isoform (8) is predominant in adult callosum and that protein expression of this isoform is reduced nearly 30% at 1 day following TBI, a time when the callosal ultrastructure shows damaged paranodal loops and loose nodal myelin. At this same interval, we detected the first significant elevation in the 145 kD calpain fragment of αII‐spectrin. A similar rapid reduction in ankyrin‐B protein expression was also reported in a rodent ischemia‐reperfusion model (24), where a 212 kD ankyrin peptide was reduced approximately 20% at 90 minutes postinjury. Selective loss of ankyrin‐B was also documented in vitro using cerebellar neurons (74) and NB‐1 cells (35), after each was subjected to neurotoxic methylmercury insult. In these studies, the early stages of neuronal degeneration were associated with dose‐dependent reduction of the 440 kD ankyrin‐B splice variant, including attenuated mRNA expression in the NB‐1model. While such studies often attribute reduced ankyrin‐B protein expression to calpain proteolysis, the pathways responsible for TBI‐induced reductions in ankyrin‐B remain to be determined. It is possible that reduced ankyrin‐B in our trauma model could also result from attenuation of mRNA transcription or translation rather than proteolysis.
Potential relationship of ankyrin‐G fragments and suppression of axonal conductance after injury
It is useful to compare the postinjury changes in white matter ankyrin‐G fragments to prior findings regarding posttraumatic alterations in axonal conduction properties. Our laboratory previously demonstrated that compound action potentials (CAPs), evoked through corpus callosum axons, exhibited decreased amplitudes following fluid percussion injury 68, 69. A key aspect of these results was that CAPs generated in myelinated axons showed a postinjury amplitude decrement which subsequently recovered, while the CAPs evoked through unmyelinated axons remained suppressed in amplitude for at least a week postinjury. As part of our effort to understand this differential deficit and recovery pattern, the present investigation examined changes in ankyrin‐G, a protein likely to be critical to axonal electrophysiology because it secures ion channels and transporters essential for bioelectric function, and spatially organizes diverse proteins integral to the axolemma. Although the present Western blot findings do not permit an unequivocal identification of which full‐length ankyrin isoforms generated the observed fragments, the pattern of postinjury protein changes suggests specific hypotheses to further explore molecular correlates of pathology in evoked CAPs. One question is whether there is a relationship between time‐dependent changes in protein fragments and the pattern of postinjury CAP changes observed previously. In Figure 7 the mean amplitudes of the unmyelinated and myelinated components of the callosal CAP are plotted along with the present measurements of ankyrin‐G fragments, with all data normalized to uninjured control values. For this comparison, the results for the 220 and 212 kD ankyrin‐G bands were averaged together, because they migrated in the gel as a doublet, and exhibited similar profiles of postinjury change. Several features of the summary plots in Figure 7 suggest a possible association of the ankyrin fragments with postinjury CAP alterations. Injury effects reach a peak, for both protein levels and evoked CAPs, at the earlier (3 h–1 day) time points. However, at 3 and 7 days the myelinated CAP component and the 220/212 kD ankyrin level exhibit a return to control levels. A dissimilar pattern was seen for the unmyelinated CAP signal and levels of the 75 kD ankyrin, both of which remained significantly below control levels.
Figure 7.
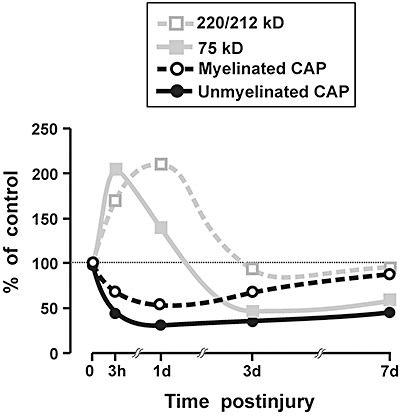
Comparison of present study results with previous measurements of compound action potential amplitudes recorded from the corpus callosum. All data are normalized as percent of sham‐injured control levels. Data for the 220 and 212 kD ankyrin‐G fragments are pooled together. Several features of this graphical comparison suggest an association of the 220/212 kD fragments with myelinated axons, and the 75 kD product with unmyelinated axons (see text). CAP data are adapted from Reeves et al (68).
One hypothesis, suggested in the protein/CAP plots of Figure 7, is that the upsurge and resolution of ankyrin 220/212 represents breakdown and clearance of higher isoforms of ankyrin, specifically the 270‐ and 480‐kD isoforms of ankyrin‐G which are highly localized to myelinated axons. In contrast, time‐dependent changes in levels of the 75‐kD form may represent primarily a breakdown of the 190‐kD ankyrin‐G, which has been associated with unmyelinated axons (reviewed in 70), although the possibility that some of the 75‐kD surge reflects residual fragments of the 270/480 kD forms cannot be eliminated based on existing data. The processes leading to the rapid postinjury increase at 3 h in the 75 kD fragment may be distinct from the reductions below control levels observed at 3 and 7 days. Specifically, the 3‐ and 7‐day decreases indicate some constitutive presence for 75 kD ankyrin‐G, and the loss of this protein may contribute to the persistent suppression of conduction in unmyelinated axons.
The major finding of this study was the documentation that diffuse brain injury differentially alters αII‐spectrin and ankyrin‐G protein expression as a function of brain region. Effects on ankyrin‐G expression were more pronounced in the corpus callosum, differing in both direction and degree from those in the cerebral cortex. White matter ankyrin‐G fragments showed a general acute elevation, while at least one form of callosal ankyrin‐G remained reduced up to a week postinjury. Injury effects on αII‐spectrin were similar between the white and gray matter samples but 5–10‐fold larger in the cortex. Callosal shifts in ankyrin‐G and αII‐spectrin at 1 day postinjury were correlated with altered neurofascin and ankyrin‐B expression. Corpus callosum ultrastructure linked these protein profiles to specific membrane disruption at Nodes of Ranvier of damaged axons. Comparing injury‐induced changes in the membrane skeletal proteins with prior recordings of evoked callosal CAPs also suggested that shifts in ankyrin‐G expression were correlated with deficits in myelinated and unmyelinated CAP signals.
ACKNOWLEDGMENTS
The authors wish to thank Lesley Harris, Raiford Black, Nancy Lee and Judy Williamson for their excellent technical assistance in this study. We also acknowledge Dr. Jiepei Zhu for his important contribution to the HRP permeability experiments. These studies were supported by funding from NIH grants NS057758 and NS056247 and from Virginia Commonwealth Neurotrauma Initiative 07‐302F. Microscopy was performed at VCU, Department of Anatomy and Neurobiology Microscopy Facility which is supported, in part, with funding from NIH‐NINDS Center Core Grant NS047463.
REFERENCES
- 1. Adams JH, Graham DI, Murray LS, Scott G (1982) Diffuse axonal injury due to nonmissle head injury: an analysis of 45 cases. Ann Neurol 12:557–563. [DOI] [PubMed] [Google Scholar]
- 2. Aikman J, O'Steen B, Silver X, Torres R, Boslaugh S, Blackband S et al (2006) Alpha‐II‐spectrin after controlled cortial impact in the immature rat brain. Dev Neurosci 28:457–465. [DOI] [PubMed] [Google Scholar]
- 3. Bennett V, Chen L (2001) Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol 13:61–67. [DOI] [PubMed] [Google Scholar]
- 4. Bourguignon LY, Jin H (1995) Identification of the ankyrin‐binding domain of the mouse T‐lymphoma cell inositol 1,4,5‐trisphosphate (IP3) receptor and its role in the regulation of IP3‐mediated internal Ca2+ release. J Biol Chem 270:7257–7260. [DOI] [PubMed] [Google Scholar]
- 5. Bouzidi M, Tricaud N, Giraud P, Kordeli E, Caillol G, Deleuze C et al (2002) Interaction of the Nav1.2a subunit of the voltage‐dependent sodium channel with nodal ankyrin‐G: in vitro mapping of the interacting domains and association in synaptosomes. J Biol Chem 32:28996–29004. [DOI] [PubMed] [Google Scholar]
- 6. Bramlett HM, Kraydieh S, Green EJ, Dietrich WD (1997) Temporal and regional patterns of axonal damage following traumatic brain injury: a betaamyloid precursor protein immunocytochemical study in rats. J Neuropathol Exp Neurol 56:1132–1141. [DOI] [PubMed] [Google Scholar]
- 7. Büki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain‐mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58:365–375. [DOI] [PubMed] [Google Scholar]
- 8. Chan W, Kordeli E, Bennett V (1993) 440‐kD ankyrinB: structure of the major developmentally regulated domain and selective localization in unmyelinated axons. J Cell Biol 123:1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen J (1988) Statistical Power Analysis for the Behavioral Sciences, 2nd edn. LEA: Hillsdale, NJ. [Google Scholar]
- 10. Croall DE, Demartino GN (1991) Calcium‐activated neutral protease (Calpain) system: structure, function and regulation. Physiol Rev 71:813–847. [DOI] [PubMed] [Google Scholar]
- 11. Czogalla A, Sikorski AF (2005) Spectrin and calpain: a “target” and a “sniper” in the pathology of neuronal cells. Cell Mol Life Sci 62:1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis JQ, Bennett V (1990) The anion exchanger and Na+K(+)‐ATPase interact with distinct sites on ankyrin in in vitro assays. J Biol Chem 265:17252–17256. [PubMed] [Google Scholar]
- 13. Davis JQ, Lambert S, Bennett V (1996) Molecular composition of the Node of Ranvier: identification of ankyrin‐binding cell adhesion molecules neurofascin (mucin+/third FNIII domain‐) and NrCAM at nodal axon segments. J Cell Biol 135:1355–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devarajan P, Scaramuzzino DA, Morrow JS (1994) Ankyrin binds to two distinct cytoplasmic domains of Na,K‐ATPase alpha subunit. Proc Natl Acad Sci U S A 91:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DiLeonardi AM, Huh HW, Raghupathi R (2009) Impaired axonal transport and neurofilament compaction occur in separate populations of injured axons following diffuse brain injury in the immature rat. Brain Res 1263:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A et al (1987) A fluid percussion model of experimental brain injury in the rat. J Neurosurg 67:110–119. [DOI] [PubMed] [Google Scholar]
- 17. Dubreuil RR, MacVicar G, Dissanayake S, Liu C, Homer D, Hortsch M (1996) Neuroglian‐mediated cell adhesion induces assembly of the membrane skeleton at cell contact sites. J Cell Biol 133:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL (2007) Nodes of Ranvier and axon initial segments are ankyrinG‐dependent domain that assemble by distinct mechanisms. J Cell Biol 177:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graham DI, Adams JH, Gennarelli TA (1988) Mechanisms of nonpenetrating head injury. Prog Clin Biol Res 234:159–168. [PubMed] [Google Scholar]
- 20. Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW (2005) Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma 22:252–265. [DOI] [PubMed] [Google Scholar]
- 21. Hall TG, Bennett V (1987) Regulatory domains of erythrocyte ankyrin. J Biol Chem 262:10537–10545. [PubMed] [Google Scholar]
- 22. Hallam TM, Floyd CL, Folkerts MM, Lee LL, Gong QZ, Lyeth BG et al (2004) Comparison of behavioral deficits and acute neuronal degeneration in the rat lateral fluid percussion and weight‐drop brain injury models. J Neurotrauma 21:521–539. [DOI] [PubMed] [Google Scholar]
- 23. Hamakubo T, Kannagi R, Murachi T, Matus A (1986) Distribution of calpains I and II in rat brain. J Neurol Sci 6:3103–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harada K, Fukuda S, Kunimoto M, Yoshida K (1997) Distribution of ankyrin isoforms and their proteolysis after ischemi and reperfusion in rat brain. J Neurochem 69:371–376. [DOI] [PubMed] [Google Scholar]
- 25. Harris AS, Morrow JS (1988) Proteolytic processing of human brain alpha spectrin (fodrin): identification of a hypersensitive site. J Neurosci 8:2640–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayashi T, Su TP (2001) Regulating ankyrin dynamics: roles of sigma‐1 receptors. Proc Natl Acad Sci U S A 98:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hryniewicz‐Jankowska A, Czogalla A, Bok E, Sikorski AF (2002) Ankyrins, multifunctional proteins involved in many cellular pathways. Folia Histochem Cytobiol 40:239–249. [PubMed] [Google Scholar]
- 28. Huh GY, Glantz SB, Je S, Morrow JS, Kim JH (2001) Calpain proteolysis of aII‐spectrin in the normal adult human brain. Neurosci Lett 316:41–44. [DOI] [PubMed] [Google Scholar]
- 29. Inserte J, Garcia‐Dorado D, Hernando V, Soler‐Soler J (2005) Calpain‐mediated impairment of Na+/K+–ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ Res 97:465–473. [DOI] [PubMed] [Google Scholar]
- 30. Iwata A, Stys PK, Wolf JA, Chen X‐H, Taylor AG, Meaney DF, Smith DH (2004) Traumatic axonal injury induces proteolytic cleavage of the voltage‐gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci 24:4605‐ 5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jafari SS, Maxwell WL, Neilson M, Graham DI (1997) Axonal cytoskeletal changes after non‐disruptive axonal injury. J Neurocytol 26:207–221. [DOI] [PubMed] [Google Scholar]
- 32. Keppel G (1991) Design and Analysis: A Researcher's Handbook, 3rd edn. Prentice Hall: Englewood Cliffs, NJ. [Google Scholar]
- 33. Kordeli E, Bennett V (1991) Distinct ankyrin isoforms at neuron cell bodies and nodes of Ranvier resolved using erythrocyte ankyrin‐deficient mice. J Cell Biol 114:1243–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural‐specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359. [DOI] [PubMed] [Google Scholar]
- 35. Kunimoto M, Suzuki T (1995) Selective down‐regulation of 440 kDa ankyrinB associated with neurite retraction. Neuroreport 6:2545–2548. [DOI] [PubMed] [Google Scholar]
- 36. Kupina NC, Detloff MR, Bobrowski WF, Snyder BJ, Hall ED (2003) Cytoskeletal protein degredation and neurodegeneration evolves differently in males and females following experimental head injury. Exp Neurol 180:55–72. [DOI] [PubMed] [Google Scholar]
- 37. Li ZP, Burke EP, Frank JS, Bennett V, Philipson KD (1993) The cardiac Na+‐Ca2+ exchanger binds to the cytoskeletal protein ankyrin. J Biol Chem 268:11489–11491. [PubMed] [Google Scholar]
- 38. Lambert S, Yu H, Prchal JT, Lawler J, Ruff P, Speicher D et al (1990) cDNA sequence for human erythrocyte ankyrin. Proc Natl Acad Sci USA 87:1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lux SE, John KM, Bennett V (1990) Analysis of cDNA for human erythrocyte ankyrin indicates a repeated structure with homology to tissue‐differentiation and cell‐cycle control proteins. Nature 344:36–42. [DOI] [PubMed] [Google Scholar]
- 40. McGinn MJ, Kelly BJ, Akinyi L, Oli MW, Liu MC, Hayes RL et al (2009) Biochemical, structural and biomarker evidence for calpain‐mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J Neuropathol Exp Neurol 68:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maier O, Van Der Heide T, Johnson R, De Vries H, Baron W, Hoekstra D (2006) The function of neurofascin 155 in oligodendrocytes is regulated by metalloprotease‐mediated cleavage and ectodomain shedding. Exp Cell Res 312:500–511. [DOI] [PubMed] [Google Scholar]
- 42. Malhotra JD, Koopmann MC, Kazen‐Gillespie KA, Fettman N, Hortsch M, Isom LL (2002) Structural requirements for interaction of sodium channel beta‐1 subunits with ankyrin. J Biol Chem 277:26681–26688. [DOI] [PubMed] [Google Scholar]
- 43. Marmarou CR, Walker S, Davis CL, Povlishock JT (2005) Quantitative analysis of the relationship between intra‐axonal neurofilament compaction and impaired axonal transport following diffuse traumatic brain injury. J Neurotrauma 22:1066–1080. [DOI] [PubMed] [Google Scholar]
- 44. Maxwell WL, Irvine A, Graham DI, Adams JH, Gennarelli TA, Tipperman R, Sturatis M (1991) Focal axonal injury: the early axonal response to stretch. J Neurocytol 20:157–164. [DOI] [PubMed] [Google Scholar]
- 45. Maxwell WL, Watt C, Graham DI, Gennarelli TA (1993) Ultrastructural evidence of axonal shearing as a result of lateral acceleration of the head in non‐human primates. Acta Neuropathol 86:136–144. [DOI] [PubMed] [Google Scholar]
- 46. Maxwell WL, McCreath BJ, Graham DI, Gennarelli TA (1995) Cytochemical evidence for redistribution of membrane pump calcium‐ATPase and ecto‐Ca‐ATPase activity, and calcium influx in myelinated nerve fibers of the optic nerve after stretch injury. J Neurocytol 24:925–942. [DOI] [PubMed] [Google Scholar]
- 47. Maxwell WL, Povlishock JT, Graham DI (1997) A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma 14:419–440. [DOI] [PubMed] [Google Scholar]
- 48. Maxwell WL, Kosanlavit R, McCreath BJ, Reid O, Graham DI (1999) Freeze‐fracture and cytochemical evidence for structural and functional alteration in the axolemma and myelin sheath of adult guinea pig optic nerve fibers after stretch injury. J Neurotrauma 16:273–284. [DOI] [PubMed] [Google Scholar]
- 49. Maxwell WL, Domleo A, McColl G, Jafari SS, Graham DL (2003) Post‐acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma 20:151–168. [DOI] [PubMed] [Google Scholar]
- 50. Michaely P, Bennett V (1995) Mechanism for binding site diversity on ankyrin. Comparison of binding sites on ankyrin for neurofascin and the Cl‐/HCO3‐ anion exchanger. J Biol Chem 270:31298–31302. [DOI] [PubMed] [Google Scholar]
- 51. Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H et al (1996) Nonerythroid‐spectrin breakdown by calpain and interleukin 1 ‐converting‐enzyme‐like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J 319:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Newcomb JK, Kampfl A, Posmantur RM, Zhao X, Pike BR, Liu S‐J et al (1997) Immunohistochemical study of calpain‐mediated breakdown products to a‐spectrin following controlled cortical impact injury in the rat. J Neurotrauma 14:369–383. [DOI] [PubMed] [Google Scholar]
- 53. Nishimura K, Yoshihara F, Tojima T, Ooashi N, Yoon W, Mikoshiba K et al (2003) L1‐dependent neuritogenesis involves ankyrinB that mediates L1‐CAM coupling with retrograde actin flow. J Cell Biol 163:1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ogawa Y, Schafer DP, Horresh I, Bar V, Hales K, Yang Y et al (2006) Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci 26:5230–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Okonkwo DO, Pettus EH, Moroi J, Povlishock JT (1998) Alteration of the neurofilament sidearm and its relation to neurofilament compaction occurring with traumatic axonal injury. Brain Res 784:1–6. [DOI] [PubMed] [Google Scholar]
- 56. Park E, Liu E, Shek M, Park A, Baker AJ (2007) Heavy neurofilament accumulation and alpha‐spectrin degradation accompany cerebellar white matter functional deficits following forebrain fluid percussion injury. Exp Neurol 204:49–57. [DOI] [PubMed] [Google Scholar]
- 57. Perlmutter LS, Gall C, Baudry M, Lynch G (1990) Distribution of calcium‐activated protease calpain in the rat brain. J Comp Neurol 296:269–275. [DOI] [PubMed] [Google Scholar]
- 58. Peters LL, John KM, Lu FM, Eicher EM, Higgins A, Yialamas M et al (1995) Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J Cell Biol 130:313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pettus EH, Christman CW, Giebel ML, Povlishock JT (1994) Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma 11:507–522. [DOI] [PubMed] [Google Scholar]
- 60. Pettus EH, Povlishock JT (1996) Characterization of a distinct set of intraaxonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res 722:1–11. [DOI] [PubMed] [Google Scholar]
- 61. Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KKW, Hayes RL (1998) Regional calpain and caspase‐3 proteolysis of α‐spectrin after traumatic brain injury. Neuroreport 9:2437–2442. [DOI] [PubMed] [Google Scholar]
- 62. Pike BR, Flint J, Dave JR, May Lu X‐C, Wang KK, Tortella FC, Hayes RL (2003) Accumulation of calpain and caspase‐3 proteolytic fragments of brain‐derived aII‐spectrin in cerebral spinal fluid after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 24:98–106. [DOI] [PubMed] [Google Scholar]
- 63. Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2:1–12. [PubMed] [Google Scholar]
- 64. Povlishock JT, Becker DP, Sullivan HG, Miller JD (1978) Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res 153:223–239. [DOI] [PubMed] [Google Scholar]
- 65. Povlishock JT, Christman CW (1995) The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma 12:555–564. [DOI] [PubMed] [Google Scholar]
- 66. Povlishock JT, Pettus EH (1996) Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochir Suppl (Wien) 66:81–86. [DOI] [PubMed] [Google Scholar]
- 67. Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P (1999) Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19:7516–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reeves TM, Phillips LL, Povlishock JT (2005) Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol 196:126–139. [DOI] [PubMed] [Google Scholar]
- 69. Reeves TM, Phillips LL, Povlishock JT (2007) Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res 1154:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rubtsov AM, Lopina OD (2000) Ankyrins. FEBS Lett 482:1–5. [DOI] [PubMed] [Google Scholar]
- 71. Saatman KE, Bozyczko‐Coyne D, Marcy V, Siman R, McIntosh TK (1996) Prolonged calpain‐mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55:850–860. [DOI] [PubMed] [Google Scholar]
- 72. Saatman KE, Graham DI, McIntosh TK (1998) The neuronal cytoskeleton is at risk after mild and moderate brain injury. J Neurotrauma 15:1047–1058. [DOI] [PubMed] [Google Scholar]
- 73. Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF (2003) Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab 23:34–42. [DOI] [PubMed] [Google Scholar]
- 74. Sakaue M, Takanaga H, Adachi T, Hara S, Kunimoto M (2003) Selective disappearane of an axonal protein, 440‐kDa ankyrinB, associated with neuronal degeneration induced by methylmercury. J Neurosci Res 73:831–839. [DOI] [PubMed] [Google Scholar]
- 75. Serbest G, Burkhardt MF, Siman R, Raghupathi R, Saatman KE (2007) Temporal profiles of cytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res 32:2006–2014. [DOI] [PubMed] [Google Scholar]
- 76. Singleton RH, Povlishock JT (2004) Identification and characterization of heterogenous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci 24:3543–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE et al (1999) Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol 58:982–992. [DOI] [PubMed] [Google Scholar]
- 78. Stone JR, Singleton RH, Povlishock JT (2001) Intra‐axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol 172:320–331. [DOI] [PubMed] [Google Scholar]
- 79. Srinirasan Y, Elmer L, Davis J, Bennett V, Angelides K (1988) Ankyrin and spectrin associate with voltage‐dependent sodium channels in brain. Nature 333:177–180. [DOI] [PubMed] [Google Scholar]
- 80. Susuki K, Rasband MN (2008) Spectrin and ankyrin‐based cytoskeletons at polarized domains in myelinated axons. Exp Biol Med 233:394–400. [DOI] [PubMed] [Google Scholar]
- 81. Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED (2006) Relationship of calpain‐mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol 201:253–265. [DOI] [PubMed] [Google Scholar]
- 82. Tuvia S, Buhusi M, Davis L, Reedy M, Bennett V (1999) Ankyrin‐B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J Cell Biol 147:995–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV et al (1998) Simultaneous degradation of alphaII‐ and betaII‐spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem 273:22490–22497. [DOI] [PubMed] [Google Scholar]
- 84. Wang KK (2000) Calpain and caspase: can you tell the difference. Trends Neurosci 23:20–26. [DOI] [PubMed] [Google Scholar]
- 85. Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin‐sensitive sodium channels. J Neurosci 21:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wood SJ, Slater CR (1998) beta‐Spectrin is colocalized with both voltagegated sodium channels and ankyrin G at the adult rat neuromuscular junction. J Cell Biol 140:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yoshida K (2000) Myocardial ischemia‐reperfusion injury and proteolyisis of fodrin, ankyrin, and calpastatin. Methods Mol Biol 144:267–275. [DOI] [PubMed] [Google Scholar]
- 88. Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T et al (1995) Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ Res 77:603–610. [DOI] [PubMed] [Google Scholar]
- 89. Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998) AnkyrinG is required for clustering of voltage‐gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]