

Abstract
Macroautophagy (often referred to as autophagy) is an evolutionarily conserved intracellular system by which macromolecules and organelles are delivered to lysosomes for degradation and recycling. Autophagy is robustly induced in response to starvation in order to generate nutrients and energy through the lysosomal degradation of cytoplasmic components. Constitutive, basal autophagy serves as a quality control mechanism for the elimination of aggregated proteins and worn-out or damaged organelles, such as mitochondria. Research during the last decade has made it clear that malfunctioning or failure of this system is associated with a vide range of human pathologies and age-related diseases. Our recent data provide strong evidence for the role of autophagy in the pathogenesis of Pompe disease, a lysosomal glycogen storage disease caused by deficiency of acid alpha-glucosidase (GAA). Large pools of autophagic debris in skeletal muscle cells can be seen in both our GAA knockout model and patients with Pompe disease. In this review, we will focus on these recent data, and comment on the not so recent observations pointing to the involvement of autophagy in skeletal muscle damage in Pompe disease.
Keywords: autophagy, muscle, mitochondria, lysosome, glycogen storage
Introduction
The development of enzyme replacement therapy is unquestionably a major scientific and commercial achievement in the history of Pompe disease, a deficiency of the glycogen-degrading lysosomal acid alpha-glucosidase [Van der Ploeg and Reuser, 2008]. The therapy stemmed from an understanding of the pathogenesis of the disease, namely that the accumulation of glycogen within membrane-bound lysosomes eventually leads to damage of skeletal and cardiac muscle, the two major tissues affected by the enzyme deficiency. In theory, the success of therapy would be a validation of the pathogenic concept. In the case of Pompe disease, the verdict is mixed: cardiac muscle responds very well to therapy but skeletal muscle does not. Patients with the most severe, infantile form of the disease survive significantly longer because of the effect of the drug (Myozyme®, Genzyme Corporation, Framingham, MA) on cardiac muscle, but skeletal myopathy, often severe, persists. The poor response of skeletal muscle to therapy led us to question our understanding of the disease mechanisms. Studies in patients with Pompe disease and in the mouse model revealed the role of macroautophagy (often referred to as autophagy) in the pathogenesis of the disease: muscle fibers contain pools of autophagic debris in addition to large glycogen-filled lysosomes [Fukuda et al., 2006b]. Autophagy is a major intracellular catabolic pathway that delivers long-lived proteins and damaged organelles (in particular mitochondria) to lysosomes for degradation and recycling (reviewed in [Weidberg et al., 2011; Yang and Klionsky, 2010a; Yang and Klionsky, 2010b]). The process involves engulfment of a portion of the cytoplasm by double-membrane structures, called autophagosomes, which fuse with lysosomes where the contents of the autophagosomes are broken down (Fig 1). The morphological evidence for abnormal autophagy in Pompe disease was in fact reported long ago [Engel, 1970] but then ignored. Furthermore, a second look at the history of the disease showed that other features of the disorder were noted and then forgotten. In this review we will revisit those neglected clues relevant to the pathogenesis of Pompe disease with an emphasis on autophagy.
Figure 1.
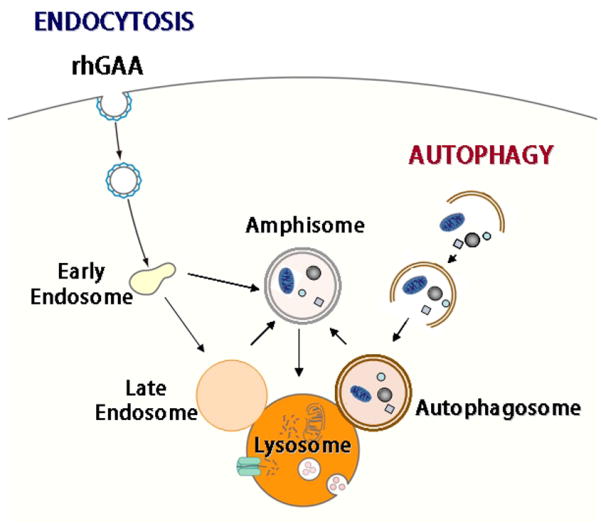
Convergence of endocytic and autophagic pathways
Autophagy: Background
The early studies of autophagy through the 1980s, including the ones in Pompe disease, were based on morphological analyses that allowed researchers to visualize the late stages of the autophagic process, namely, the steps just prior to and following fusion of autophagosomes with lysosomes (Fig 1). Since then, the field has witnessed a dramatic expansion of knowledge concerning the role of autophagy in multiple physiological and pathological conditions, including embryogenesis, immune response, aging, neurodegeneration, cancer, liver and heart diseases, and lysosomal storage diseases. The fundamental role of autophagy is to provide energy and amino acids to maintain cellular function under starvation conditions [Yang and Klionsky, 2010b]. In addition, it became clear that autophagy fulfills housekeeping functions by ridding cells of misfolded proteins, protein aggregates, and worn out organelles such as mitochondria, thus providing physiological renewal for the cells.
The remarkable developments in the field gave researchers the tools for studying the autophagic pathway, from the initiation of autophagosomal formation to the resolution of the autophagosomal content in the lysosome. The range of methods and markers currently available for studying autophagy in different systems is very broad (we refer the reader to a recent publication on the subject) [Klionsky et al., 2007], but for the purpose of this review, we will focus on those that allowed us to evaluate the extent of autophagy and its role in the pathogenesis of skeletal muscle damage in Pompe disease.
The different steps of autophagy - the development of the autophagosomal membrane, the formation of autophagic vesicles, and their fusion with endosomes and lysosomes – are governed by the actions of more than thirty autophagy-related proteins identified to date [Yang and Klionsky, 2010a]. The suppression of autophagy in the whole organism by knocking out critical autophagic genes (Atgs), such as Atg5 or Atg7 is lethal [Komatsu et al., 2005; Kuma et al., 2004]. Therefore, the conventional way of addressing the role of autophagy in a particular tissue is by inactivating one of these genes in a tissue-specific manner. Both Atg5 and Atg7 proteins are involved in the initial steps of autophagosomal formation.
Perhaps the most important discovery that allowed the field to flourish was the identification of a protein, MAP1LC3, commonly referred to as LC3, which can be used as a specific marker of autophagosomes. LC3 exists in two forms – cytosolic LC3-I and membrane-bound LC3-II. The latter can be found on the autophagosomal membrane throughout the whole process of vesicle maturation [Kabeya et al., 2000]. LC3-II can be distinguished from LC3-I by Western blot analysis because these forms migrate differently. Detection of LC3-II became a standard method for evaluating the autophagic process. The functional status of autophagy can be measured by the levels of autophagic substrates normally eliminated through the autophagic pathway. Accumulation of ubiquitinated proteins, for example, is a good indication of autophagic failure [Bjorkoy et al., 2005; Komatsu et al., 2007; Pankiv et al., 2007].
Many studies of autophagy have been conducted in in-vitrosystems, which have allowed researchers to dissect the different steps of the autophagic process and test different pharmacological compounds as inhibitors or activators of autophagy. Unfortunately, Pompe cell lines replicating the autophagic pathology are not yet available, and the development of such lines is a challenge for the future.
Pompe Disease: Historical Perspective
Pompe disease is named after Dutch pathologist J.C. Pompe, who described the syndrome in a seven month-old girl with severe muscle weakness who died of what was thought to be pneumonia. Hypertrophic cardiomyopathy was found on autopsy, and a critical observation was made that glycogen accumulated in tissues throughout the body [Pompe, 1932]. Two German doctors, W. Putschar and G. Bischoff [Bischoff, 1932; Putschar, 1932] independently described the disease in the same year.
The underlying metabolic defect was identified 30 years later by Belgian biochemist H.G. Hers. Not only did he discover the enzyme defective in Pompe patients, but he also made the connection between Pompe disease, lysosomes, and even autophagy [Hers, 1963]. Pompe disease was the first lysosomal storage disorder described, and based on Pompe studies, the concept of inborn lysosomal storage disorders was established.
At the time the field of autophagy was still in its embryonic stage, but the autophagic (literally meaning “self-eating”) function of the lysosomes had already been recognized (reviewed in [Yang and Klionsky, 2010a]). Although Hers did not directly mention autophagy in his publication describing acid alpha-glucosidase, he did link the location of the enzyme and the intra-lysosomal glycogen accumulation when the enzyme was missing to suggest that glycogen traffics from the cytoplasm to lysosomes. Hers' speculation on how this may happen implies the involvement of autophagy. He writes that “the physiological breakdown of tissues occurs by digestion of small and limited areas of the cytoplasm under the action of the hydrolytic enzymes included in the lysosomes.” These words essentially echo the modern day description of macroautophagy.
Clinical heterogeneity of Pompe disease (which initially encompassed only the infantile form) is now taken for granted. In Hers' very first paper on the acid alpha-glucosidase, there is a description of a patient who presented with no cardiomegaly and who survived longer than typical infants with Pompe disease, most of whom die from cardiac failure within the first year. Since then several reports of childhood-onset of Pompe disease have been published, and Engel [Engel, 1970] recognized that acid alpha-glucosidase deficiency can also present “as a syndrome of muscular weakness in adults.” What used to be subdivided into childhood, juvenile, and adult forms of the disease is now as a group referred to as a late-onset form. Unlike in the infantile form of the disease, in which both cardiac and skeletal muscle are affected, in late-onset forms cardiac muscle is usually spared, but a slowly progressive skeletal myopathy eventually leads to premature death from respiratory insufficiency.
In the very first adult case, Engel described autophagic abnormalities in great detail. Electron microscopy of muscle biopsies from adult patients revealed the presence of glycogen in the cytoplasm, in lysosomes, and in autophagic vacuoles. The morphological description of the autophagic vacuoles is remarkably accurate. These vacuoles had “heterogeneous contents. Their border…consisted of double membrane. The vacuoles contained small, dense bodies… membranous fragments, amorphous material, and varying amounts of glycogen…”. Morphological evidence of abnormal autophagy in muscle biopsies from adult patients has been presented in several other reports [Bertagnolio et al., 1978; Fernandez et al., 1999; Lewandowska et al., 2008].
Furthermore, Engel's group reported that autophagic vacuoles in skeletal muscle were associated mainly with childhood and adult cases, and were much less frequent in the infantile form, which appeared to be characterized by intra-lysosomal glycogen accumulation and lakes of glycogen in the cytoplasm [De Bleecker et al., 1993]. This finding, that the autophagic component is prominent in adults but not in infants, in retrospect seems profound and quite unexpected. The subject was revisited only much later in the report by C. Angelini's group [Nascimbeni et al., 2008]. Although autophagic involvement in Pompe disease was noted many years ago, this pathology did not attract the attention of researchers until recently [Raben et al., 2010a].
Autophagy in a Mouse Model
The studies of autophagy in Pompe disease in our lab grew out of experiments in our knockout mouse model (Pompe mice) [Raben et al., 1998] testing the efficacy of the recombinant human acid alpha-glucosidase, the same preparation that was used in clinical trials. Cardiac muscle cleared glycogen very efficiently, but after months on therapy, even with high dosages of the recombinant enzyme, skeletal muscle still contained significant amounts of residual glycogen. Later on, the same proved to be true in the clinic. In mice, oxidative, type I muscle fibers responded to therapy much better than glycolytic, type II muscle fibers despite the significantly higher glycogen burden in type I-rich muscles in the untreated Pompe mice. Electron microscopy showed that the therapy-resistant type II fibers contained large areas of autophagic accumulation, reminiscent of that described by Engel [Engel, 1970] in adult patients with Pompe disease.
The Extent of Autophagy in Skeletal Muscle of Pompe Mice
Electron microscopy clearly established the presence of autophagic accumulation in therapy resistant fibers - classical double membrane autophagosomes with undigested cytoplasmic content can be easily seen in Pompe skeletal muscle (Fig 2, top) - but this method provides a view only of a tiny region of muscle. The extent of autophagic pathology was revealed when we used a novel approach to analyze muscle biopsies in Pompe disease - confocal microscopy of single muscle fibers stained for lysosomal marker LAMP1 and autophagosomal marker LC3. This method allows us to visualize lysosomes and autophagosomes not only in one section, but systematically through the whole depth of the fiber. Unexpectedly, we found that autophagic accumulation was visible in virtually every type II fiber, even in young Pompe mice. In many fibers, the autophagic area often was localized in the core of and spread throughout the length of the fiber, with or without interruptions. The area appeared as an amorphous mass containing clusters of LAMP1- and LC3-positive vesicular structures, sometimes with broken borders, as well as other cellular debris of unknown identity or origin. Thus, in Pompe skeletal muscle, not only were the lysosomes filled with undigested glycogen, but other materials were also backed up outside – unable to reach the recycling place. In the rest of the fiber (that is, outside the core of the fiber) individual or isolated groups of expanded lysosomes with clearly defined borders were seen, the pathology that is expected in Pompe disease (Fig 2, bottom). Autophagosomes in these areas appeared as tiny, dot-like structures.
Figure 2.

Autophagy in skeletal muscle of Pompe mice. Top: electron micrographs of type II-rich muscle (psoas) from a 5 month-old Pompe mouse showing autophagic vacuoles. Bottom: autophagic buildup in the core of a fiber derived from psoas muscle. The fiber is stained for lysosomal marker LAMP (green) and autophagosomal marker LC3 (red). Nuclei are shown in white. Bars: 0.5 microns for EM and 10 microns for the stained fiber.
The autophagic mass increased in size as the animals aged, and in older mice it occupied up to 40% of the volume in some fibers [Raben et al., 2009]. It seemed that the area of autophagic accumulation disrupts muscle architecture much more than the expanded lysosomes in the periphery of the fiber do. From a morphological perspective, one can easily imagine that abnormal autophagy, rather than just lysosomal expansion, eventually leads to skeletal muscle destruction. Skeletal muscle damage and a significant loss of muscle force in Pompe mice [Xu et al., 2010] may also result from the buildup of undigested autophagic substrates. Indeed, the levels of potentially toxic ubiquitinated proteins that can form insoluble aggregates are significantly elevated in Pompe skeletal muscle, suggesting that the recycling process is inefficient [Raben et al., 2008].
Furthermore, we have shown that the autophagic accumulation affects the delivery of the recombinant enzyme - the bulk of the therapeutic enzyme ends up in the autophagic area [Fukuda et al., 2006a]. This finding was not totally unexpected considering the relationship between the autophagic pathway and the endocytic pathway, which delivers extracellular material (including the administered recombinant enzyme) to the lysosome. It has been shown that the autophagic and endocytic pathways converge not only at their common endpoint (the lysosome), but also at other steps along the way. Autophagosomes fuse with late and even early endosomes, resulting in the formation of an intermediate structure called the amphisome [Berg et al., 1998] (Fig 1). So it is as if the therapeutic drug is diverted away from its intended destination, the lysosome, and instead ends up in the autophagic area, which becomes a sink for the recombinant enzyme.
Thus, in Pompe disease, a profoundly disordered intracellular recycling system appears to be an important contributor to the muscle weakness and to the incomplete response to treatment.
Suppression of Autophagy
Elimination of autophagic accumulation by suppressing autophagy looked like a reasonable strategy to improve the effect of ERT. Another reason for suppressing autophagy in Pompe muscle was the assumption that glycogen traffics to the lysosomes by autophagic pathway. This assumption is based on early data showing autophagic degradation of glycogen in skeletal muscle [Schiaffino and Hanzlikova, 1972], liver and heart [Kondomerkos et al., 2005] of the newborn rats (as mentioned above, Hers hinted at such a possibility in his original paper on the discovery of the acid alpha-glucosidase [Hers, 1963]). If true in adult animals, suppression of autophagy may rescue the phenotype in Pompe mice: glycogen will remain in the cytoplasm where it will be degraded by cytoplasmic enzymes, instead of being transported to a compartment where it cannot be broken down (Fig 3).
Figure 3.
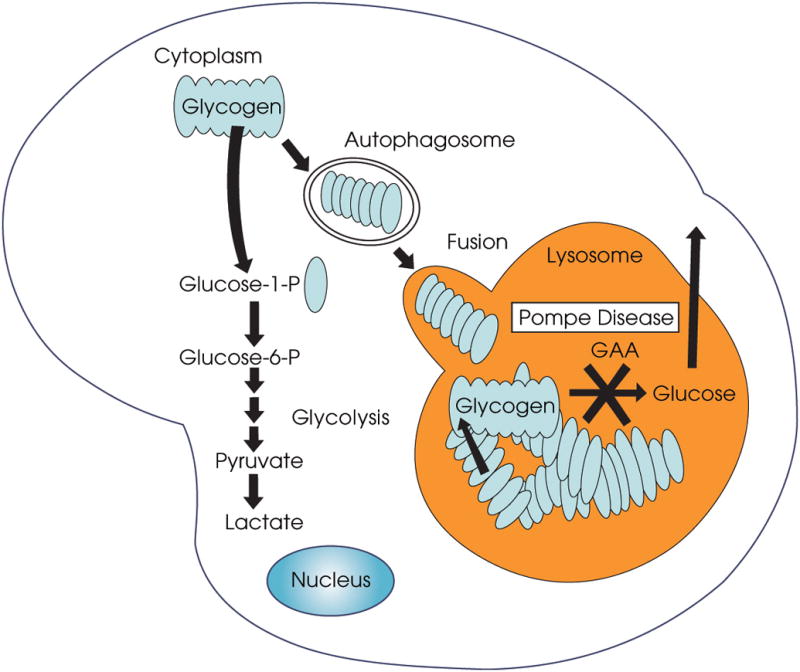
Acid α-glucosidase is responsible for the break-down of glycogen in the lysosomes. When the enzyme is absent or deficient, glycogen accumulates in the lysosomes. It is not clear how glycogen is transported from the cytoplasm to the lysosomes. If this transport involves the delivery of glycogen in the autophagosomes, then suppression of autophagy would reduce the traffic and decrease the amount of lysosomal glycogen. The degradation of the cytoplasmic glycogen would proceed unaffected.
We have generated an autophagy-deficient Pompe mouse model, in which a critical autophagic gene, Atg7, is inactivated specifically in skeletal muscle. As expected, autophagic buildup, which is so prominent in Pompe mice, was not observed in muscle from autophagy-deficient Pompe mice, but glycogen was still present in the lysosomes. However, most lysosomes were smaller than those in Pompe mice, and the level of glycogen accumulation was markedly reduced. In fact, this reduction was more prominent than that in ERT-treated Pompe mice. These data suggested that at least some (but not all) glycogen is delivered to the lysosomes via autophagic pathway. We can only speculate how the remaining glycogen is transported to the lysosome, but another type of autophagy - microautophagy, a process by which cytoplasmic components enter the lysosome through direct invagination of the lysosomal membrane - may play a role [Mijaljica et al., 2011]. Yet another possibility has been recently suggested by P. Roach's group: it was shown that the starch binding domain-containing protein 1 (Stbd1) can bind glycogen and tether it to vesicles which deliver glycogen to the lysosomes by non-classical autophagy (the process is called “non-classical” because the vesicles are not marked by LC3) [Jiang et al., 2010].
Whatever the mechanism, the excess of glycogen, which still remained in skeletal muscle of autophagy-deficient Pompe mice was reduced to near normal levels when the animals were treated with the recombinant enzyme. This outcome observed in both young [Raben et al., 2010b] and older (our unpublished data) mice was never seen in Pompe mice in which autophagy was not tampered with. However, the loss of autophagy in skeletal muscle comes with a price: the accumulation of dysfunctional mitochondria, mild atrophy and age-dependent decrease in muscle strength have been reported in muscle-specific autophagy-deficient wild type mice [Wu et al., 2009; Masiero and Sandri, 2010]. But these changes lead to neither gross phenotypical abnormalities, nor shortened lifespan of the animals. In Pompe disease the benefits clearly outweigh the negatives.
If, as we proposed before, a therapy for a disease is in some sense a test of how well the pathogenesis of the disease is understood, then the success of our therapeutic approach for Pompe disease - a combination of suppression of autophagy and enzyme replacement therapy – would support the idea of the involvement of autophagy in the pathogenesis of Pompe disease.
Autophagy in Pompe Patients
It is well documented that in untreated late-onset patients, muscle pathology is extremely heterogeneous ranging from the unaffected fibers to those completely devoid of contractile machinery. The variability among Pompe patients can be explained by the differences in the levels of residual enzyme activity, but the unevenness of muscle pathology in individual patients remains one of the mysteries in Pompe disease. Confocal microscopy of stained single muscle fibers from a patient's biopsy also shows a great deal of variability, indicating that there is no inherent bias in the selection of fibers. Using this method we have demonstrated that autophagic abnormalities are present in many muscle cells in late-onset patients (both juvenile and adults), thus making the observations in a mouse model relevant to the human study. Furthermore, in many fibers autophagic accumulation is the overwhelming (and in some fibers the only) pathology, because the lysosomes that lie outside the autophagic region appear essentially normal [Raben et al., 2010a; Raben et al., 2007]. Similarly, Lewandowska et al. reported a truly remarkable extent of autophagic accumulation in some fibers in adult onset cases: “the autophagic areas (observed by EM) occupied more than half of the diameter of the fiber, but sometimes the vacuoles filled almost all muscle fibers” [Lewandowska et al., 2008].
The make-up of the autophagic area in patients and the Pompe mice may be different: in addition to clusters of lysosomes and autophagosomes many fibers from patients contain large balloon-like structures (possibly containing lipids), which are not seen in myofibers from mice. These structures can be visualized even without any staining by phase contrast transmitted microscopy (Fig 4). Also, unlike in mice, autophagic accumulation was present in both fast and slow muscle in patients with late-onset disease [Raben et al., 2010a].
Figure 4.

Autophagic area in fibers from an untreated 5-year-old Pompe patient. Top: the fiber was stained for LAMP (red) and LC3 (green). Nuclei are shown in white. Bottom: unstained fixed fibers observed by phase contrast transmitted microscopy.
The role of autophagy in the pathogenesis of infantile Pompe disease is much less obvious than that in late-onset forms (consistent with Engel's early ultrastructural studies [De Bleecker et al., 1993]). Muscle biopsies, in particular from infants, are hard to come by, but we have had access to this rare material through collaboration with Drs. W.L. Hwu and Y.H. Chien in Taiwan where there is a large-scale newborn screening program. This program identifies infantile cases within days (rather than within months by the traditional diagnostic methods) after birth and allows for early initiation of therapy [Chien et al., 2008; Chien et al., 2009]. Unexpectedly, the autophagic component which is so prominent in late-onset cases was insignificant in a group of infants whose biopsies became available for single-fiber analysis. Although the components of the autophagic system are made in excess and occasional enlarged autophagosomes are clearly seen in muscle fibers, the autophagic buildup is absent [Raben et al., 2010a]. Instead, the major characteristic of these fibers is the presence of hugely expanded lysosomes without clear borders, a finding consistent with the hypothesis of lysosomal rupture as a cause of muscle destruction [Griffin, 1984; Thurberg et al., 2006].
The difference between the relative contribution of the lysosomal and autophagic pathologies in untreated infants and adults presents a conundrum in Pompe disease. When the enzyme is completely or nearly completely absent, lysosomal glycogen accumulation occurs prenatally [Hug et al., 1991; Millan et al., 2010; Phupong et al., 2005; Pokorny et al., 1982]. The infants lacking the enzyme are born with already severely damaged muscle fibers filled with giant lysosomes with ruptured membranes and massive glycogen deposits. The lack of autophagic buildup in such infants suggests that the role of autophagy during fetal development is minimal perhaps because of a constant supply of nutrients through the umbilical cord. This hypothesis is consistent with the data in mice showing a low level of autophagy throughout the embryonic period [Kuma et al., 2004]. It has also been shown in a mouse model that autophagy is up-regulated immediately after birth and remains high for several hours before returning to baseline levels within 1-2 days. This massive transitory induction of autophagy in neonates (which is particularly striking in cardiac muscle, but less so in skeletal muscle with the exception of the diaphragm) is probably a response to the nutrient shortage during the sudden cessation of the trans-placental supply [Kuma et al., 2004]. Since many fibers in Pompe infants are already destroyed in the newborn, a surge in autophagy, should it occur in humans, would go unnoticed.
In follow-up biopsies from infantile patients after six months of ERT (the only time point at which the biopsies were available), the lysosomes were smaller in many fibers. Unfortunately for the patients, however, autophagic accumulation resembling that found in skeletal muscle from adults was now present [Raben et al., 2010a]. A long-term study and a larger number of samples are needed to evaluate the fate of this autophagic buildup.
Mitochondrial Abnormalities in Pompe Patients
The history of recognition of the contribution of aberrant mitochondria to the pathophysiology of Pompe disease may turn out to mimic that of abnormal autophagy, noted but ignored for many years. Shortly after identification of the enzyme defect in Pompe disease, Engel and Dale reported the presence of larger than normal mitochondria in skeletal muscle biopsy derived from a patient with adult onset disease. These mitochondria, which were imperfect oval, polygonal or prism shaped, contained dense granular material and paracrystalline inclusions located in intercristae space [Engel and Dale, 1968] (The title of this article from 1968, “Autophagic glycogenosis of late onset with mitochondrial abnormalities: light and electron microscopic observations”could well serve as a title of this review). Decades later, the same observation of paracrystalline inclusions in numerous mitochondria in adult patients was reported [Fernandez et al., 1999; Lewandowska et al., 2008]. In the interim, mitochondrial structural abnormalities were mentioned in the literature but were generally of peripheral rather than central importance. Both histochemical and ultrasturctural studies of biopsied skeletal muscle revealed larger than usual subsarcolemmal mitochondrial aggregates in an adult patient [Hudgson, 1975], and enlarged “pleomorphic” mitochondria with distorted cristae in muscle of an infant [Verity, 1991]. On the other hand, no evidence of mitochondrial proliferation was observed in muscle biopsies from two other infants [Selak et al., 2000]. Dysfunctional mitochondria with swollen cristae have been recently observed in induced pluripotent stem cells (iPSCs) derived from the fibroblasts of two patients with Pompe disease [Huang et al. 2011].
Particularly puzzling as well as intriguing observations have been reported in muscle from two adult patients: accumulation of membrane-enclosed glycogen particles within the mitochondria resulting in interrupted cristae in one [Bertagnolio et al., 1978], and the presence of Hirano bodies in another [Fernandez et al., 1999]. In an infant, electron dense masses reminiscent of those in an adult patient were observed within the cristae of mitochondria, although the nature of these masses was not indentified [Verity, 1991]. Curiously, we too found what appear to be glycogen particles in mitochondria in skeletal muscle of the Pompe mice and autophagy-deficient Pompe mice (Fig 5). How does glycogen gain entry into the mitochondria anyway? The presence of Hirano bodies is an exceptional finding as these entities comprised of actin and actin-associated proteins are usually seen in neuronal tissues of patients suffering from neurodegenerative disorders.
Figure 5.

Some of the mitochondria in muscles of Pompe mice and autophagy-deficient Pompe mice contain glycogen inclusions (white arrowheads). Occasionally, a single mitochondrion may contain several inclusions (not shown). In the example in the top panel, glycogen occupies a limited portion of the mitochondrial space; the mitochondrial cristae (black arrows) have the usual transverse orientation in areas away from the inclusion. In the bottom panel, the inclusion fills a larger portion of the mitochondrion; all visible cristae appear reoriented and abnormal around the inclusion. Glycogen inclusions thus affect the internal structure and, possibly, the function of mitochondria. Bars: 500 nm (top) and 100 nm (bottom).
Mitochondria have also been sighted in autophagic vacuoles [Selak et al., 2000] in muscle biopsies of adult Pompe patients. This is not such a bewildering observation given that anomalous mitochondria are eliminated by the autophagic pathway (a process called mitophagy) (reviewed in [Wang and Klionsky, 2011]). The aberrant mitochondria found in Pompe patients as described above would remain sequestered in autophagic vesicles which are unable to reach the recycling place, the lysosomes. It is still unclear whether mitochondrial abnormalities occur regardless or because of the autophagic dead-end. As mentioned above, suppression of autophagy in skeletal muscle in wild type mice leads to accumulation of enlarged, dysmorphic mitochondria [Wu et al., 2009]. These results suggest the autophagic defect as the culprit, but the alternative hypothesis cannot be ruled out. Our knowledge of the contribution of mitochondria to the pathophysiology of Pompe disease is at the stage previously occupied by autophagy, and awaits further scrutiny.
Acknowledgments
The research was supported by the Intramural research Program of the National Institute of Arthritis and Musculoskeletal and Skin diseases of the National Institutes of Health. Amanda Wong was supported in part by a Cooperative Research and Development Agreement (CRADA) between the NIH and Genzyme Corporation.
Biographies
Author Bio sketches:Nina Raben is a Staff Scientist at the Laboratory of Muscle Stem Cells and Gene Regulation (NIAMS, NIH). Her scientific interests have centered on Pompe disease, in particular, on the role of autophagy in the pathogenesis of the disorder.
Amanda Wong was formerly a postbaccalaureate fellow at the NIAMS, NIH, where she investigated the role of autophagy in murine Pompe disease. She is currently an MD/PhD candidate at the University of Michigan.
Evelyn Ralston is head of the Light Imaging Section in the Office of Science and Technology, NIAMS. Her group focuses on the organization of microtubules and their associated organelles in skeletal muscle.
Rachel Myerowitz was formerly a PI at NIDDK, NIH, where she studied lysosomal storage disorders. She is currently a professor of Biology at St. Mary's College of Maryland and a guest scientist at NIAMS, NIH.
References
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. JBiol Chem. 1998;273:21883–21892. doi: 10.1074/jbc.273.34.21883. [DOI] [PubMed] [Google Scholar]
- Bertagnolio B, Di Donato S, Peluchetti D, Rimoldi M, Storchi G, Cornelio F. Acid maltase deficiency in adults. Clinical, morphological and biochemical study of three patients. Eur Neurol. 1978;17:193–204. doi: 10.1159/000114945. [DOI] [PubMed] [Google Scholar]
- Bischoff G. Zum klinischen Bild der Glykogen-Speicherungs-Krankheit (Glykogenose) Zeitschrift fu Kinderheilkunde. 1932;52:722–725. [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Chen CA, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:e39–e45. doi: 10.1542/peds.2007-2222. [DOI] [PubMed] [Google Scholar]
- Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, Huang AC, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–e1125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- De Bleecker JL, Engel AG, Winkelmann JC. Localization of dystrophin and beta-spectrin in vacuolar myopathies. Am J Pathol. 1993;143:1200–1208. [PMC free article] [PubMed] [Google Scholar]
- Engel AG. Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain. 1970;93:599–616. doi: 10.1093/brain/93.3.599. [DOI] [PubMed] [Google Scholar]
- Engel AG, Dale AJ. Autophagic glycogenosis of late onset with mitochondrial abnormalities: light and electron microscopic observations. Mayo Clin Proc. 1968;43:233–279. [PubMed] [Google Scholar]
- Fernandez R, Fernandez JM, Cervera C, Teijeira S, Teijeiro A, Dominguez C, Navarro C. Adult glycogenosis II with paracrystalline mitochondrial inclusions and Hirano bodies in skeletal muscle. Neuromuscular Disorders: NMD. 1999;9:136–143. doi: 10.1016/s0960-8966(98)00117-5. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in pompe disease. Mol Ther. 2006a;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Roberts A, Ahearn M, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and lysosomes in Pompe disease. Autophagy. 2006b;2:318–320. doi: 10.4161/auto.2984. [DOI] [PubMed] [Google Scholar]
- Griffin JL. Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Arch B Cell Pathol Incl Mol Pathol. 1984;45:23–36. doi: 10.1007/BF02889849. [DOI] [PubMed] [Google Scholar]
- Hers HG. Alpha-glucosidase deficiency in generalize glycogen storage disease (Pompe's disease) Biochem J. 1963;86:11. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HP, Chen PH, Hwu WL, Chuang CY, Chien YH, Stone L, Chien CL, Li LT, Chiang SC, Chen HF, Ho HN, Chen CH, Kuo HC. Human Pompe disease induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human Mol Genet. 2011:1–14. doi: 10.1093/hmg/ddr424. first published online September 15. [DOI] [PubMed] [Google Scholar]
- Hudgson P. Correlations between histochemical and ultrastructural studies of diseased muscle. Riv Istochim Norm Patol. 1975;19:101–110. [PubMed] [Google Scholar]
- Hug G, Chuck G, Chen YT, Kay HH, Bossen EH. Chorionic villus ultrastructure in type II glycogen storage disease (Pompe's disease) N Engl J Med. 1991;324:342–343. doi: 10.1056/NEJM199101313240517. [DOI] [PubMed] [Google Scholar]
- Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli-Roach AA, Wells CD, Skurat AV, Roach PJ. Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem. 2010;285:34960–34971. doi: 10.1074/jbc.M110.150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for Monitoring Autophagy from Yeast to Human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondomerkos DJ, Kalamidas SA, Kotoulas OB, Hann AC. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol Histopathol. 2005;20:689–696. doi: 10.14670/HH-20.689. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lewandowska E, Wierzba-Bobrowicz T, Rola R, Modzelewska J, Stepien T, Lugowska A, Pasennik E, Ryglewicz D. Folia Neuropathologica / Association of Polish Neuropathologists and Medical Research Centre. Vol. 46. Polish Academy of Sciences; 2008. Pathology of skeletal muscle cells in adult-onset glycogenosis type II (Pompe disease): ultrastructural study; pp. 123–133. [PubMed] [Google Scholar]
- Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6:307–309. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–682. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- Millan BS, Teijeira S, Vieitez I, Navarro C. Chorionic villi ultrastructur in the prenatal diagnosis of glycogenosis type II. J Inheri Metab Dis. 2010 doi: 10.1007/s10545-009-9033-6. published online: February 16. [DOI] [PubMed] [Google Scholar]
- Nascimbeni AC, Fanin M, Tasca E, Angelini C. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology. 2008;70:617–626. doi: 10.1212/01.wnl.0000299892.81127.8e. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Phupong V, Shuangshoti S, Sutthiruangwong P, Maneesri S, Nuayboonma P, Shotelersuk V. Prenatal diagnosis of Pompe disease by electron microscopy. Arch Gynecol Obstet. 2005;271:259–261. doi: 10.1007/s00404-004-0620-3. [DOI] [PubMed] [Google Scholar]
- Pokorny KS, Ritch R, Friedman AH, Desnick RJ. Ultrastructure of the eye in fetal type II glycogenosis (Pompe's disease) Invest Ophthalmol Vis Sci. 1982;22:25–31. [PubMed] [Google Scholar]
- Pompe JC. Over idiopatische hypertrophie van het hart. Ned Tijdschr Geneeskd. 1932;76:304. [Google Scholar]
- Putschar M. Uber angeborene Glykogenspeicher-Krankheit des herzens. “Thesaurismosis glycogenica” (v. Gierke) Beitr Pathol Anat Allg Pathol. 1932;90:222. [Google Scholar]
- Raben N, Baum R, Schreiner C, Takikita S, Mizushima N, Ralston E, Plotz P. When more is less: excess and deficiency of autophagy coexist in skeletal muscle in Pompe disease. Autophagy. 2009;5:111–113. doi: 10.4161/auto.5.1.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Jatkar T, Lee A, Lu N, Dwivedi S, Nagaraju K, Plotz P. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther. 2002;6:601–608. [PubMed] [Google Scholar]
- Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, LaMarca M, King C, Ward J, Sauer B, Plotz P. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- Raben N, Ralston E, Chien YH, Baum R, Schreiner C, Hwu WL, Zaal KJ, Plotz P. Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy. Mol Genet Metab. 2010a;108:1383–1388. doi: 10.1016/j.ymgme.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Schreiner C, Baum R, Takikita S, Xu S, Xie T, Myerowitz R, Komatsu M, Van Der Meulen JH, Nagaraju K, Ralston E, Plotz P. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder-murine Pompe disease. Autophagy. 2010b;6:1078–1089. doi: 10.4161/auto.6.8.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Takikita S, Pittis MG, Bembi B, Marie SKN, Roberts A, Page L, Kishnani PS, Schoser BGH, Chien YH, Ralston E, Nagaraju K, Plotz PH. Deconstructing Pompe disease by analyzing single muscle fibers. Autophagy. 2007;3:546–552. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Hanzlikova V. Autophagic degradation of glycogen in skeletal muscles of the newborn rat. J Cell Biol. 1972;52:41–51. doi: 10.1083/jcb.52.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, de Chadarevian JP, Melvin JJ, Grover WD, Salganicoff L, Kaye EM. Mitochondrial activity in Pompe's disease. Pediatr Neurol. 2000a;23:54–57. doi: 10.1016/s0887-8994(00)00145-4. [DOI] [PubMed] [Google Scholar]
- Thurberg BL, Lynch MC, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for pompe disease. Lab Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- Van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- Verity MA. Infantile Pompe's disease, lipid storage, and partial carnitine deficiency. Muscle & Nerve. 1991;14:435–440. doi: 10.1002/mus.880140509. [DOI] [PubMed] [Google Scholar]
- Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, Finkel T. Aging. Vol. 1. Albany.NY: 2009. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy; pp. 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Galperin M, Melvin G, Horowits R, Raben N, Plotz P, Yu LC. Impaired Organization and Function of Myofilaments in Single Muscle Fibers from a Mouse Model of Pompe Disease. J Appl Physiol. 2010;108:1383–1388. doi: 10.1152/japplphysiol.01253.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010a;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010b;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]