

Abstract
This paper provides the first review of the memory-enhancing and neuroprotective metabolic mechanisms of action of methylene blue in vivo. These mechanisms have important implications as a new neurobiological approach to improve normal memory and to treat memory impairment and neurodegeneration associated with mitochondrial dysfunction. Methylene blue’s action is unique because its neurobiological effects are not determined by regular drug-receptor interactions or drug-response paradigms. Methylene blue shows a hormetic dose-response, with opposite effects at low and high doses. At low doses, methylene blue is an electron cycler in the mitochondrial electron transport chain, with unparalleled antioxidant and cell respiration-enhancing properties that affect the function of the nervous system in a versatile manner. A major role of the respiratory enzyme cytochrome oxidase on the memory-enhancing effects of methylene blue is supported by available data. The memory-enhancing effects have been associated with improvement of memory consolidation in a network-specific and use-dependent fashion. In addition, low doses of methylene blue have also been used for neuroprotection against mitochondrial dysfunction in humans and experimental models of disease. The unique auto-oxidizing property of methylene blue and its pleiotropic effects on a number of tissue oxidases explain its potent neuroprotective effects at low doses. The evidence reviewed supports a mechanistic role of low-dose methylene blue as a promising and safe intervention for improving memory and for the treatment of acute and chronic conditions characterized by increased oxidative stress, neurodegeneration and memory impairment.
Keywords: methylene blue, methylthioninium chloride, memory, neuroprotection, cytochrome oxidase, mitochondria
1. Introduction
In 1886, Paul Ehrlich injected methylthioninium chloride, also known as methylene blue (MB) into rats for the first time. He used the name “magic bullet” to describe MB’s selective uptake by respiring nervous tissue (Wainwright and Crossley, 2002). MB was then just one of many new synthetic chemicals used as dyes in the textile industry. But the early work of Ehrlich with MB fueled a scientific revolution that included the use of MB in numerous and novel experimental applications. For over more than 120 years, researchers have found in MB an unparalleled bioactive versatility. The uses of MB have ranged from redox indicator to antimalarial agent, and from supravital stain to photosensitizer and cancer chemotherapy agent (Peter et al., 2000; Wainwright and Crossley, 2002). For example, Santiago Ramon y Cajal used the “Ehrlich reaction” in live rodents to stain neurons supravitally to demonstrate that his discovery of synaptic spines was not an artifact of the Golgi stain. In the early twentieth century, psychiatrists were already using MB as an experimental treatment for schizophrenia (Allexsaht, 1938). After more than a century, Ehrlich’s key observation of the high affinity of MB for nervous tissue has been rediscovered in investigations revealing significant effects of MB as a memory-enhancing and neuroprotective agent. Specifically, MB has been reintroduced as a potential pharmacotherapy in mild cognitive impairment (MCI), early Alzheimer’s disease (AD), Parkinson’s disease, Leber’s optic neuropathy and other neurodegenerative disorders bearing in common fundamental deficits in mitochondrial function. MB not only has great affinity for nervous tissue, it has also been recognized as one of the most potent chain-breaking antioxidants (Ohlow and Moosmann, 2011). Recent evidence supports that MB effectively improves memory in healthy animals and humans. These enhancing effects have been shown in a variety of experimental learning and memory paradigms ranging from habituation to spatial memory. In addition, MB has been used in the therapy of mental disorders and cardiac arrest-associated brain damage (Naylor et al., 1986; Kelner et al., 1988b; Deutsch et al., 1997; Pelgrims et al., 2000; Wainwright and Crossley, 2002; Clifton and Leikin, 2003; Miclescu et al., 2006, 2007; Sharma et al., 2011). The data suggest that the mechanism for these effects is based on MB’s redox cycling properties and its effects on the energy metabolism machinery in mitochondria. This is all in addition to its well-established role as an FDA-grandfathered antidote for the treatment of methemoglobinemia, a condition characterized by elevated blood levels of an oxidized form of hemoglobin with decreased ability to release oxygen to tissues (Wright et al., 1999). Thus, more than one hundred years later, studies with MB seem to support a new revolution with promising horizons for the field of neurotherapeutics. What follows is the summary of evidence implicating MB as a nootropic and neuroprotective agent. An initial discussion of the chemical properties and the proposed neurochemical mechanisms underlying MB’s effects is also provided, with special emphasis on its remarkable antioxidant and metabolic-enhancing properties in mitochondria (Rojas and Gonzalez-Lima, 2010).
The goal of this review is to summarize, for the first time, the evidence supporting the memory-enhancing and metabolic neuroprotective effects of MB at the neurochemical, network and behavioral levels. This evidence has important implications for the development of treatments against memory impairment and neurodegeneration associated with mitochondrial dysfunction. The rationale for the present work is that no expert has properly reviewed these findings in a detailed and integrated manner that explains how the mechanism of action of MB is related to both facilitation of memory and mitochondrial neuroprotection. In addition, there is a need for an accurate historical review that gives the proper attribution to the various groups that discovered the effects of MB relevant to memory enhancement and neuroprotection, because recent papers have not properly credited the sources and ideas in this field.
The review by Oz et al. (2011) described general physicochemical features and actions of MB on multiple cellular and molecular targets in the nervous system, including the cGMP pathway. But it is misleading to generally refer to MB effects without mentioning dose because MB has a hormetic dose-response with opposite effects at high and low doses (Bruchey and Gonzalez-Lima, 2008). For example, high intravenous doses of MB cause methemoglobinemia, whereas low-dose MB is the treatment of choice for methemoglobinemia (Wright et al., 1999). Oz et al. (2009) also reviewed the influence of MB on β-amyloid deposits and neurofibrillary tangles. The MB effects reviewed were based on relatively large concentrations of MB that impair memory in vivo, whereas the data and mechanisms presented below describe how low-dose MB is beneficial to memory processing. In turn, Atamna (2009) described a β-amyloid mechanism of toxicity based on disruption of heme metabolism and cytochrome oxidase depletion, and proposed this pathway as a target to delay the onset and progression of AD. In contrast, the current review distinguishes itself from the existing literature because it addresses the evidence of in vivo memory-enhancing effects of MB in the normal brain as well as the in vivo neuroprotective effects against mitochondrial failure independently of β-amyloid toxicity. This review also highlights the existence of a common mechanism of action for both memory improvement and neuroprotection based on the dual antioxidant and metabolic enhancing effects of low-dose MB, focusing on the central role of the mitochondrial enzyme cytochrome oxidase in neuronal physiology and homeostasis. This common mechanism is discussed for the first time in the context of the pharmacological concept of hormesis. Finally, this review offers a novel mechanistic perspective by summarizing data on two important topics in the neurobiology of memory that previous reviews have failed to address: the neural network effects of MB and crucial observations from behavioral studies on the effects of MB on different types and phases of memory. Acknowledgment of these data is expected to provide important mechanistic insights for neurotherapeutic applications of MB.
2. Mechanisms of action of MB relevant for memory enhancement and neuroprotection
2.1. The unique biochemical and pharmacokinetic properties of MB
MB is a synthetic cationic tri-heterocyclic redox compound that contains a central aromatic thiazine ring system. This structure allows it to carry a delocalized positive charge at neutral pH, which explains its high reduction potential similar to that of oxygen (Wainwright and Crossley, 2002). The delocalized positive charge in the MB molecule is favored by the presence of an imine group that also confers radical stability (Moosmann et al., 2001). The combination of a thiazine ring system conferring a high reduction potential and an imine group facilitating antioxidant function are two structural features in the MB molecule that allow MB to retain its redox activity despite being reduced. In other words, unlike most conventional short-lived radical traps, MB has the potential to autoxidize, which means that its reduction-oxidation capacity allows electron cycling, without MB gaining any permanent stoichiometric or net reduction (Figure 1). This redox cycling activity has been shown to be contingent upon its concentration, the redox state of the medium and the presence of molecular oxygen (Buchholz et al., 2008).
Figure 1. Chemical structure and redox balance of methylene blue.
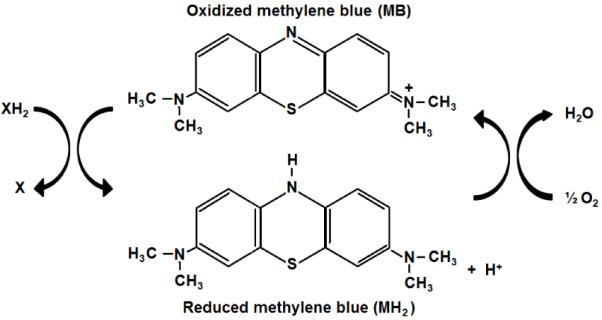
The tri-heterocyclic thiazide ring of MB allows the presence of a delocalized positive charge that confers a high reduction potential. At the same time, the presence of imine groups (C = N − R) confers high antioxidant activity to the MB molecule. In its oxidized form, methylene blue (MB) accepts electrons from an electron donor (XH2). In its reduced form, leucomethylene blue (MH2) is colorless, acts as an electron donor, and it can transfer electrons to oxygen to form water. In vivo and at low concentrations methylene blue and leucomethylene blue are at equilibrium, so that they form a reversible reduction-oxidation system. The auto-oxidizing capacity of MB provides a mechanism for electron transfer to oxygen, which accounts for its antioxidant and metabolic-enhancing properties, as well as its hormetic dose-response effects at the biochemical, physiological and behavioral levels.
Its aromatic nature confers MB a high lipophilicity. MB has a remarkably high permeability through biomembranes, which is unparalleled by redox compounds also displaying neuroprotective properties in experimental conditions including creatine, α-lipoic acid, nicotinamide and coenzyme Q (Rainer et al., 2000; Teichert et al., 2003; Artuch et al., 2004; Lensman et al., 2006). Due to its redox nature, MB has a notable affinity for a wide variety of tissue oxidases, including those localized to mitochondria (Salaris et al., 1991; Visarius et al., 1997). Such affinity for tissue oxidases in mitochondria is evidenced by higher MB concentrations in subcellular isolates with the highest concentrations of mitochondrial membranes (Gabrielli et al., 2004). MB easily crosses the blood-brain barrier and accumulates in nervous tissue after intravenous or oral administration (Peter et al., 2000; O’Leary et al., 2010). Once inside the cell, MB easily concentrates in the mitochondrial matrix in a manner stimulated by the mitochondrial proton potential (Gabrielli et al., 2004). Depending on the medium redox state and pH, MB can in turn display one of its most remarkable effects: the transfer electrons to oxygen or alternate electron acceptors. Such electron transfer resembles the activity of native mitochondrial electron carriers. This observation that MB acts as an electron shuttle in mitochondria is not new and it is a well-known fact in the biochemistry literature (Lindahl and Oberg, 1961; Scott and Hunter, 1966; Visarius et al., 1997). Such high bioavailability to mitochondria and autoxidizable potential are unique properties among pharmacologic agents with bioactive effects in nervous tissue.
MB is also a sui generis drug because its physiological, pharmacological and clinical effects are not determined by regular drug-receptor interactions or best explained by classical drug-response pharmacological paradigms. MB may have as many receptors as oxidoreductases are available, and may potentially exert a wide range of pleiotropic effects. MB also displays distinctive hormetic pharmacological effects. Hormesis is a dose-response with opposite effects at low and high doses that is recognized as a general biological phenomenon with major relevance for pharmacological responses (Calabrese et al., 2007). Hormetic effects have been described for numerous agents of diverse structures and mechanisms of action including antibiotics (Calabrese et al., 2010a), chemotherapeutic agents (Nascarella et al., 2009), antioxidants (Calabrese et al., 2010b), steroids (Lupien et al., 2005), radiation (Vaiserman, 2010) and low-level light therapy (Rojas and Gonzalez-Lima, 2011). Thus, it is likely that hormetic responses are not contingent upon a particular chemical structure, but have instead multiple pharmacokinetic and pharmacodynamic determinants. The most common form of hormesis follows the widely recognized inverted U-shaped relationship (Calabrese and Baldwin, 1997) (Figure 2). The hormetic response to MB consists of an increase in the effect at a low dose, followed by a decrease in the same effect with an intermediate dose, until the effect is equal to a control-type effect. With doses increasing beyond the hormetic zone, the effect decreases even further, until it is below the control effect. Hormetic effects of MB at the neurochemical and behavioral levels have been recently described (Bruchey and Gonzalez-Lima, 2008).
Figure 2. Inverted U-shaped curve typical of hormesis.
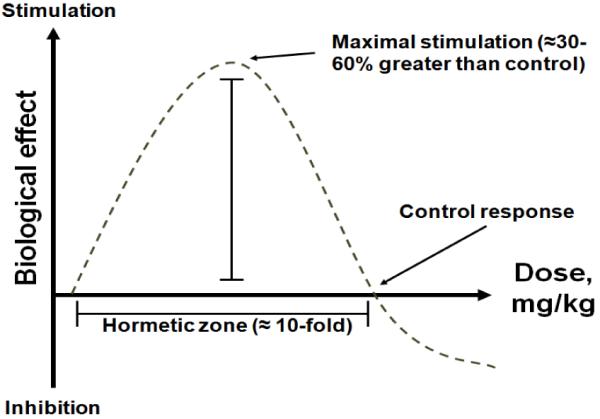
Increasing doses induce stimulatory or beneficial effects. Maximal stimulation is seen at intermediate doses and corresponds to 30-60% increases compared to control, as opposed to several fold-increases typical of linear-non-threshold dose-response curves. As the dose increases, the biological response becomes less stimulatory and can be no different than control. With even higher doses, inhibitory or toxic effects are observed. This hormetic dose-response is also called the β curve. Behavioral and neurochemical hormetic effects of methylene blue have been described in vivo (Bruchey and Gonzalez-Lima, 2008).
MB hormesis can be explained by the pharmacokinetics of MB in mitochondria, which is determined by the mitochondrial membrane potential and the relative local concentration of MB. Higher membrane potentials induce higher MB accumulation (i.e. binding to mitochondrial proteins) which in turn promotes higher MB aggregation (i.e. dimerization of MB molecules). However, MB aggregation is also affected by the proportion of MB molecules to binding sites, with less aggregation at very high, and very low binding site concentrations. Production of radicals has been shown to increase in the presence of MB monomers and be minimal in the presence of MB dimers (Gabrielli et al., 2004). Thus, in the presence of an optimal mitochondrial membrane potential, low MB concentrations favor dimerization and reduction, whereas high concentrations promote oxidation and reaction with endogenous electron donors such as nicotine adenine dinucleotide (NADH) and nicotine adenine dinucleotide phosphate (NADPH). Therefore, it is expected that low MB doses or concentrations will be, in general, more effective than large ones at facilitating physiological effects within mitochondria. In fact, at high local concentrations, MB can potentially “steal” electrons away from the electron transport chain complexes, disrupting the redox balance and acting as a pro-oxidant (Vutskits et al., 2008). Consistent with this, cell cultures exposed to high (micromolar range) but not to low (nanomolar range) concentrations of MB induce high levels of oxidants, and show a compensatory up regulation of antioxidant enzymes with decreased heme expression and iron uptake by 50% (Atamna et al., 2008). Several detrimental effects of MB on neural structure or function have been reported in vivo, including humans. These effects have been associated with administration of large doses of the compound, contact with connective tissue or concomitant use of psychotropic drugs (Arieff and Pyzik, 1960; Poppers et al., 1970; Blass and Fung, 1976; Martindale and Stedeford, 2003; Sweet and Standiford, 2007; Vutskits et al., 2008).
It is also important to note that adverse effects of MB are not explained solely on the basis of hormesis or oxidative damage, but also on that of its chemical purity. Even pharmaceutical (USP) grade MB contains impurities, such as arsenic, aluminum, cadmium, mercury and lead. At low doses, the presence of contaminants is not of great concern, but at higher doses non-specific effects due to accumulation of various toxic and bioactive substances are possible. Industrial-grade and chemical-grade MB sold as a dye or stain can consist of more than 8% or 11% of various contaminants (NTP, 2008, Sigma Chemical Co, St. Louis, MO) and should not be administered to humans or animals. For example, commercial chemical suppliers routinely warn that their non-USP MB products are of a chemical grade not suitable for use in living applications. Nevertheless, some recent studies have used these impure chemical-grade MB products with living cells and animals, obtaining potentially misleading dose-response and toxic effects (Atamna et al., 2008; Auerbach et al., 2010).
2.2. Metabolic-enhancing and antioxidant effects of MB
Early experiments in biochemistry using artificial electron donors including MB, noted that MB increases oxygen consumption of living cells at the expense of carbohydrate metabolism. This effect was attributed to its autoxidizable capacity that allows it to play a catalyst role in “some oxidative process” (Guzman-Barron and Hoffmann, 1930). Subsequent investigations of cell respiration revealed that MB enhances oxidative metabolism by interacting with components of the mitochondrial respiratory chain (Scott and Hunter, 1966). These components are protein complexes embedded in the inner mitochondrial membrane (i.e., complex I-IV, coenzyme Q, cytochrome c) that are adapted to shuttle electrons to adjacent proteins and finally to oxygen, releasing energy in a tightly regulated fashion. This MB action supports the pumping of protons into the mitochondrial intermembrane space against a concentration gradient, whose electromotive energy is later used in the synthesis of adenosine triphosphate (ATP). Normally, the electron transport chain complexes receive electrons from reduced co-substrates such as NADH and flavin adenine dinucleotide (FADH2), but MB has been regularly used as an artificial electron donor in experimental conditions and has shown metabolic enhancing properties. It is well established that reduced MB can donate electrons to coenzyme Q and possibly to cytochrome c (Figure 3), thus increasing cytochrome oxidase (complex IV) activity and oxygen consumption (Scott and Hunter, 1966). In fact, MB is able to stimulate glucose metabolism in anoxic conditions (Lee and Urban, 2002), and increase NADH oxidation by mitochondria (Gabrielli et al., 2004). Additional evidence supports that MB also influences biochemical mechanisms dependent on mitochondrial respiration such as lipid β-oxidation (Visarius et al., 1999), glycolysis, ATP synthesis, extracellular matrix production (Lee and Urban, 2002) and Na+/K+ ATPase activity (Furian et al., 2007). As mentioned before, MB can be reduced or oxidized inside mitochondria, where most of the intracellular reactive oxygen species are produced. At low concentrations, MB can enter a reversible redox cycle and interact with oxygen to form water, which would decrease the superoxide radicals produced during the process of oxidative phosphorylation. MB can also trap leaking electrons produced by mitochondrial inhibitors and preserve the metabolic rate by bypassing blocked points of electron flow, thus improving mitochondrial respiration (Lindahl and Oberg, 1961; Scott and Hunter, 1966; Zhang et al., 2006) (Figure 3).
Figure 3. Mitochondrial mechanism of action of methylene blue in memory enhancement and neuroprotection.
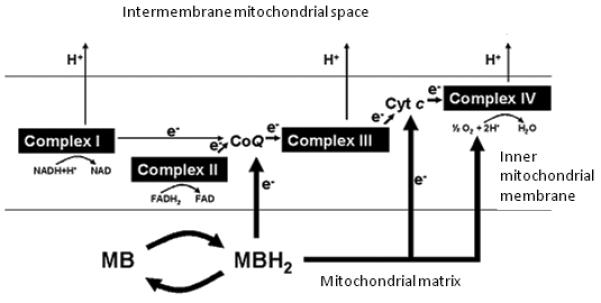
Normally, electron donors (NADH and FADH2) reduce mitochondrial complex I or II. Electrons (e−) are subsequently transferred to ubiquinone (CoQ), complex III, cytochrome c (Cyt c) and complex IV. As this electron transfer occurs in a tightly regulated fashion, the energy released from each redox reaction is used to pump protons (H+) into the intermembrane space to generate an electrochemical gradient that is used to activate the enzyme ATP synthase. Methylene blue (MB) is a synthetic chemical compound and does not occur endogenously. However, in optimal conditions MB can emulate the activity of endogenous electron carriers within the inner mitochondrial membrane. MB is an auto-oxidizable compound that becomes readily available to mitochondria, where it can be reduced to leucomethylene blue (MBH2). In physiological conditions characterized by high energy demands, MB enters a reversible redox cycle, increasing cytochrome oxidase (complex IV) activity, interacting with oxygen to form water and thus facilitating cell respiration. During the oxidizing conditions prevailing during excessive energy demands or mitochondrial failure, low concentrations of MB exert antioxidant and electron shuttling actions that support the respiratory chain function. At high concentrations, methylene blue can take electrons away from the electron transport chain complexes, thereby impairing their activity.
The effects of MB are not limited to its interactions with mitochondrial complexes. To a lesser degree, MB is hypothesized to interact with a number of less abundant alternate tissue oxidases such as xanthine oxidase and monoamine oxidase (Kelner et al., 1988a, b). Based on the high prevalence of redox reactions in biological systems, it is not surprising that MB has been found to have a wide range of pleiotropic effects. Some of them may be dose-dependently related to neurotransmission, including inhibition of monoamine oxidase activity (Gillman, 2008), inhibition of cholinesterase activity (Pfaffendorf et al., 1997), bolstering of cortico-limbic serotonin and I-norepinephrine levels (Ramsay et al., 2007), and modification of the cGMP pathway (Harvey et al., 2010). But it is unclear whether in vitro effects of MB on neurotransmission may play a significant role in its in vivo systemic effects. For example, intravenous MB can release serotonin from blood platelets that can result in increased circulating serotonin unrelated to the in vitro action of MB on monoamine oxidase activity. Further description of interactions of MB with neurotransmitter systems have been reviewed recently (Oz et al., 2009; Oz et al., 2011).
An enzyme of particular interest with which MB interacts is nitric oxide synthase (NOS). Inhibition of NOS mediates the systemic cardiovascular effects of MB that have granted its clinical use in septic shock, a life-threatening condition that happens when an infection lowers blood pressure. In septic shock patients, MB increases systemic vascular resistance (Schneider et al., 1992), improves tissue oxygenation and oxygen consumption and left ventricular filling and function, which induce a rise in cardiac ejection volume (Daemen-Gubbels et al., 1995). Through these effects, MB indirectly affects mean arterial, mean pulmonary artery and pulmonary artery occlusion pressures, and systemic lactate levels (Andresen et al., 1998; Juffermans et al., 2010). Patients with septic shock that receive MB show decreased plasma concentrations of nitrates and nitrites and have decreased requirements for vasopressors such as norepinephrine, epinephrine and dopamine. Interestingly, the hemodynamic dose-response effects of MB occur in a hormetic manner, with a tendency of an intermediate dose (3 mg/kg) to improve systemic and pulmonary vascular resistance and oxygen uptake, compared to less improvement with a higher (7 mg/kg) or a lower dose (1 mg/kg) (Juffermans et al., 2010). MB inhibits NOS perhaps oxidizing the ferrous ion in its heme group or directly inactivating its product nitric oxide (NO) (Klatt et al., 1992; Mayer et al., 1993; Volke et al., 1999; Cohen et al., 2000). Regardless of the mechanism, the final theoretical consequence of inhibition of the NO pathway is a decrease in oxidative stress derived from the formation of nitrogen reactive species, and modulation of not only hemodynamics, but also neuroendocrine function in which NO is implicated. The NO molecule itself resembles MB in several aspects. It is a small, uncharged molecule that can pass through cell membranes by simple diffusion. Although it has a very short half-life of less than 30 sec it has the ability to regulate the electron transport chain involved in mitochondrial respiration. NO can act as a substrate, inhibitor and effector of cytochrome oxidase, controlling its function independently of the redox state of the rest of the respiratory chain components (Cleeter et al., 1994; Cooper, 2002).
A valuable property of MB inherent to its metabolic enhancing effects is its antioxidant potential. Under the right conditions (i.e. low pH, high redox potential, low local concentrations), MB is expected to scavenge electrons produced in oxidation-reduction reactions in all cell compartments, thus decreasing the generation of oxygen reactive species (Salaris et al., 1991). It is predicted that MB will decrease oxidative damage in pro-oxidant conditions, including physiological mitochondrial function. Consistent with this predictions, MB has been shown to prevent nerve cell death induced by oxidative stress (Moosmann et al., 2001), inhibit rotenone-induced lipid peroxidation (Zhang et al., 2006) and methylmalonic acidemia-induced protein carbonylation in nervous tissue (Furian et al., 2007). MB also reduces oxidative stress-induced AD-like tau and β-amyloid aggregation in vitro (Wischik et al., 1996; Taniguchi et al., 2005; Necula et al., 2007) and decreases the oxidative damage induced by ischemic-reperfusion injury to brain in vivo (Miclescu et al., 2006, 2007). In summary, MB is a potent redox agent, with autoxidizable properties and high bioavailability to mitochondria. The metabolic enhancing properties and potent antioxidant effects have been documented in a wide variety of biological systems. Recent research has determined the potential of MB to support memory enhancement and neuroprotection based on these peculiar properties. These data are briefly summarized in the following sections.
3. MB as a memory-improving drug
Neurons are highly specialized cells that have a major dependence on sustained aerobic energy production. They rely on mitochondrial aerobic metabolism in order to carry out functions ranging from basic processes for cell survival to those favored by their complex structure and function, such as memory. It has been demonstrated that impaired mitochondrial oxidative metabolism is associated with memory deficits and neurodegeneration similar to those observed in AD and related disorders (Bennett et al., 1992; Gonzalez-Lima et al., 1997; Gonzalez-Lima et al., 1998a; Liang et al., 2008). Therefore, interventions aimed at improving mitochondrial metabolism are hypothesized to benefit the function of both the diseased and normal brain. MB seems to be an ideal drug to achieve this goal. The studies implicating MB as an effective memory enhancer are summarized in Table 1. These data allow establishing some principles that may be useful at maximizing the potential clinical benefits of MB as a memory-improving drug.
Table 1.
Summary of memory-enhancing effects of methylene blue (MB) in various experimental memory tasks.
| Behavioral paradigm |
MB dose (mg/kg) | Results | References |
|---|---|---|---|
| Inhibitory avoidance |
Single i.p. 0.05, 0.5,1,5, 50 | Enhancement of avoidance memory at 1 mg/kg dose. Retention deficits at highest (50 mg/kg) dose. |
Martinez, Jr. et al., 1978 |
| Holeboard spatial search appetitive task |
1, i.p. repeated over 5 days | Improved spatial memory retention in normal rats. Restoration of spatial memory retention impaired by an inhibitor of cytochrome oxidase. |
Callaway et al., 2002; 2004 Riha et al., 2011 |
| Conditioned fear extinction |
4, i.p. repeated over 5 days | Improved retention of extinction memory in normal rats. Decreased fear renewal in congenitally helpless rats. |
Gonzalez-Lima and Bruchey, 2004; Wrubel et al., 2006 |
| Object recognition |
Single i.p. 1,4,10 | Improved object recognition at 4 mg/kg dose. | Riha et al., 2005 |
| Open field habituation |
Single i.p. 1,4,10 | Improved long-term habituation at 4 mg/kg dose. |
Riha et al., 2005 |
| Discrimination learning |
1, i.p. repeated over 5 days | Improved reward/no-reward discrimination learning. |
Wrubel et al., 2007 |
| Morris Water Maze |
Single i.p. 0.15-4 | Reversed scopolamine-induced spatial learning impairment in a dose-dependent manner. |
Deiana et al., 2009 |
| Oral 9-30/day estimate over 12-16 weeks |
Improved spatial memory in transgenic mouse model of AD and in transgenic mouse model of human tauopathy. |
Medina et al., 2010; O’Leary et al., 2010 |
i.p. = intraperitoneal
3.1. Low-dose MB improves memory by enhancing consolidation in both aversive and appetitive tasks
Memory processing involves different time-dependent phases such as encoding, consolidation and retrieval, which appear to be mediated by different neural mechanisms. The first study of MB as a memory-modulating agent was reported by Martinez Jr. et al. (1978). This leading group closely analyzed the temporal relationship between MB administration and memory enhancement in rats and mice, and concluded that MB acts on the consolidation phase. The study used an inhibitory avoidance test to measure memory retention 24 h following a learning session. The results showed that 1 mg/kg MB injected after training enhanced retention of the inhibitory avoidance response. Remarkably, retention was not affected by the same low dose given 15 min before training, 6 h after training or 15 min before testing. This crucial observation led to the conclusion that the memory-enhancing effects of MB were based on its effects during the consolidation phase of memory processing, as opposed to encoding or retrieval phases. These effects were thought to be mediated by the interaction of MB with hemoglobin. A low MB dose was thought to oxidize hemoglobin, increasing the oxygen carrying capacity of red blood cells and inducing a counterintuitive retrograde enhancement of memory. But not only timing of MB administration for memory improvement purposes was highly relevant. A large MB dose of 50 mg/kg induced a memory retention deficit when given 30 min before training, while doses lower than 1 mg/kg had no effects on memory. Similarly, a high MB dose was hypothesized to reduce hemoglobin into methemoglobin, thus inducing hypoxia and decreased glucose metabolism. Although these mechanisms were not formally tested, the data supported the notion that the consolidation phase of memory processing was highly suitable to enhancement through a redox mechanism. Also, it evidenced a negative dose-response effect of MB on memory consolidation that was contingent upon the time of administration, which supports hormetic pharmacokinetics.
The memory-enhancing effects of MB were not further investigated for over twenty years. The next MB memory studies were done by Callaway et al. (2002; 2004) in our lab. We confirmed the memory-enhancing effects of 1 mg/kg USP MB given post-training using a spatial task for food search (Figure 4). In that study, adult male Sprague-Dawley rats were treated with intraperitoneal MB 1 mg/kg daily after each of five training sessions using a baited holeboard maze to examine spatial memory retention 24 h after the end of training. Post-training MB enhanced memory retention (percent visits to training baited holes compared to all visits) in probe trials. These results were not biased by olfactory cues, since MB-treated subjects showed a higher percentage of visits to training baited holes when the baiting pattern was altered. Also, there were no group differences in motor activity and no overall differences in performance in day 1-5 training sessions that could account for the results. These studies were conducted with both normal rats and with rats having memory impairment caused by chronic cytochrome oxidase inhibition, a metabolic model of AD (Bennett et al., 1992; Bennett and Rose, 1998). Together these studies with low-dose MB given during the memory consolidation phase were the first to show MB’s enhancement of memory retention in both aversive (Martinez Jr. et al., 1978) and appetitive tasks in normal animals (Callaway et al., 2004), and to show restoration of memory in a model of AD (Callaway et al., 2002). Therefore, the reported beneficial effects of MB on memory are selective for the consolidation phase, but they do not appear to be task-specific since thus far all the kinds of memory tested have been enhanced by low-dose MB (Table 1).
Figure 4. Memory enhancement with methylene blue.

Methylene blue (MB) improved memory retention examined in a holeboard spatial memory task, in which rats used spatial cues to learn the location of sweetened cereal placed in different holes (Callaway et al., 2004). A) MB USP 1 mg/kg was given daily after learning sessions using a consistent baiting pattern (pattern 1). Solid circles represent baited holes. Memory retention was subsequently tested in an unbaited probe trial. After the unbaited trial, the baiting pattern was reversed (reversal), and the animals were tested again after re-learning the task. B) The graph shows mean (± standard error bar) of memory performance (% visits to correct holes) in groups of vehicle control and MB-treated subjects. The MB group had twice as many correct responses as the control group for the first baiting pattern (pattern 1), the second baiting pattern (Reversal), and the averaged total (Overall). *p = 0.037, **p = 0.000014, ***p = 0.00017
3.2. The effects of MB on memory follow a hormetic dose-response curve
Further analysis of the dose-response effects of MB on memory were evaluated in a study by Riha et al. (2005). This study also analyzed the effect of MB on non-specific variables that could account for improved memory scores such as effects on general activity levels or anxiety. Different doses of USP MB were administered to Long-Evans rats that were evaluated 24 h after MB administration in two general activity tests and two memory tests. The general activity measures included wheel running and feeding, whereas the memory tests consisted of open field habituation and an object recognition test. MB doses used in this study ranged from 1 to 100 mg/kg, administered intraperitoneally as single doses after training. Rats injected with MB 100 mg/kg displayed adverse reactions to the drug and one subject died following this high-dose MB administration. Similarly, subjects injected with 50 mg/kg displayed decreased wheel running activity and decreased daily food consumption. As predicted by its hormetic pharmacology (Bruchey and Gonzalez-Lima, 2008), these studies confirmed that MB doses of 50 mg/kg or higher have adverse effects on general locomotor behavior and well-being of experimental subjects, and should be avoided. In contrast, MB at low (1-4 mg/kg) and intermediate (10 mg/kg) doses did not result in any non-specific behavioral effects. Remarkably the 4 mg/kg dose was the most reliable at enhancing memory after single administration, significantly improving both long-term behavioral habituation and object memory recognition (Riha et al., 2005). An equivalent MB dose has been given to humans chronically without side effects (Naylor et al., 1986).
3.3. The memory-improving effects of MB are mediated by enhancement of neuronal oxidative metabolic capacity
The redox interaction between MB and hemoglobin is well-known. MB is able to non-enzymatically reduced or oxidize the iron atom in hemoglobin’s heme group (Smith and Thron, 1972). This interaction is the basis for the clinical use of MB to treat methemoglobinemia. As described above, Martinez Jr. et al. (1978) hypothesized that low-dose MB could facilitate memory by oxidizing methemoglobin and thereby increasing the concentration of hemoglobin with oxygen-carrying capacity. Nevertheless, in normal animals the reduction of methemoglobin can be, at best, from two to zero percent in blood (Juffermans et al., 2010). This would represent only a small improvement in the oxygen content in blood, especially because oxygen reaches a plateau in the hemoglobin dissociation curve at hemoglobin saturations of 90 percent and above. In fact, MB has been demonstrated to improve hemodynamic parameters including mean cardiac index, mean arterial blood pressure and oxygen consumption, without major effects on blood oxygen partial pressure or methemoglobin concentration (Juffermans et al., 2010). However, a memory enhancing effect could be easily explained by the fact that MB has a direct interaction not only with the oxygen carrying system, but also with the tissues consuming oxygen. Neurons have an abundant content of cytochrome oxidase, a heme/copper-containing metaloenzyme with allosteric properties and great affinity for oxygen. For purposes of aerobic metabolism, cytochrome oxidase is a non-circulating version of hemoglobin. Cytochrome oxidase is the terminal enzyme of the mitochondrial respiratory chain. It finalizes the electron transport by using electrons and molecular oxygen to produce water. It is a highly conserved molecule and a key enzyme for aerobic life (Michel et al., 1998). In contrast to the fixed content of hemoglobin in each red blood cell, cytochrome oxidase activity in the brain varies widely in response to energetic demands (Wong-Riley, 1989). Cytochrome oxidase displays an exquisite molecular plasticity, changing its levels of expression in response to neuronal activation and constitutes the best known intraneuronal marker of oxidative metabolic capacity (Gonzalez-Lima and Cada, 1998; Wong-Riley et al., 1998; Wong-Riley et al., 2000). Based on this, a direct interaction of MB with cytochrome oxidase is believed to underlie its memory-enhancing effects, at least in the normal brain. As discussed below, this mechanistic hypothesis is supported by a large body of experimental data across different levels of analysis including neurochemical, systems and behavioral (Callaway et al., 2002; Callaway et al., 2004; Gonzalez-Lima and Bruchey, 2004; Riha et al., 2005; Wrubel et al., 2007a; 2007b; Riha et al., 2011). Such data permit to establish that even if memory enhancement by MB is not directly mediated by increases in cytochrome oxidase, the changes in this mitochondrial complex at least constitute a reliable marker of memory effects (Gonzalez-Lima, 1992; Poremba et al., 1997, 1998).
Increases in brain cytochrome oxidase and oxygen consumption in relation to memory enhancement have been indeed demonstrated after administration of MB. For example, Callaway et al. (2004) was the first to report significant increases in brain cytochrome oxidase activity after MB administration. When a single dose of 1 mg/kg MB was injected in vivo to rats, a 30% increase in their brain cytochrome oxidase activity was detected at 24h, but not one or two hours following the MB injection. Similarly, when MB was applied to rat brain homogenates in vitro, 500 nM MB increased cytochrome oxidase activity by 25%. This nanomolar concentration in vitro was estimated to correspond to the 1 mg/kg dose administered to the rats in the spatial memory experiment (Callaway et al., 2004). In addition, the hormetic pharmacologic effect of MB was confirmed when a 5 μM MB concentration did not change cytochrome oxidase activity in vitro, and an even higher MB concentration of 10 μM significantly decreased cytochrome oxidase activity. The low MB dose (1 mg/kg) which improved both brain cytochrome oxidase activity and spatial memory retention in this study is the same dose which improved memory retention of the inhibitory avoidance response in the aforementioned Martinez Jr. et al. (1978) study. These results were replicated in another study that analyzed the effects of daily MB administration on spatial discrimination memory in a baited holeboard maze (Wrubel et al., 2007b). Following three days of discrimination training, adult male Long-Evans rats treated daily with post-training MB (1 mg/kg, intraperitoneally) reliably discriminated between rewarded and non-rewarded trials, while saline-treated subjects did not. Cytochrome oxidase activity in the brains from rats treated with three daily 1 mg/kg MB injections was measured 24 h following the last injection using spectrophotometry. It was found that three repeated low-dose MB injections resulted in about 70% enhancement of cytochrome oxidase activity, as compared to saline-injected control. Such substantial MB-induced increases in cytochrome oxidase in normal healthy animals are best explained by initial increases in cytochrome oxidase catalytic activity, which translates in enzyme induction and up-regulation of the holloenzyme subunit pool (Wong-Riley et al., 1998).
Increases in nuclear respiratory factors and cytochrome oxidase expression occur upon persistent neural activation, and are thought to regulate brain oxygen consumption to support intracellular processes with high-energy demand involved in memory consolidation, such as neurotransmission and synaptogenesis (Govindaiah et al., 2000; Dhar et al., 2009b, a; Dhar and Wong-Riley, 2009) (Figure 5). Riha et al. (2005) showed that low concentrations (5 and 10 μM) of leucomethylene blue (reduced form of MB) applied to rat brain homogenates increased brain oxygen consumption in vitro. The same group was also the first to report that low-dose MB in vivo was associated with significant increases in brain oxygen consumption 24 h after administration, using doses that improved behavioral habituation and object recognition memory. Thus, low-dose MB in vivo is believed to display potent and consistent memory enhancing effects by targeting the respiratory process of oxygen-dependent brain energy formation. This proposed mechanism of action of MB based on boosting of mitochondrial respiration and aerobic metabolic capacity in the brain constitute an optimal fit between the biochemical properties of MB and the current understanding of the neurobiology of memory.
Figure 5. Activity-dependent effects of methylene blue.

A) MB acts on pre-synaptic and postsynaptic mitochondria (Mit) and will preferentially accumulate in neurons within activated networks with high energy demands and active mitochondria. This generalized but at the same time activity-dependent enhancing effect is explained by the existence of elevated proton gradients in highly active mitochondria, which drive the location of MB within the cell. B) MB enhances the activity of the electron transport chain and modulates nitric oxide synthase (NOS). NOS is normally activated by glutamatergic receptors (AMPAR and NMDAR) to produce nitric oxide, which acts as a second messenger and, among other actions, regulates cytochrome oxidase activity, thus exerting a strong influence on respiration. Metabolically active mitochondria communicate with the nuclear and mitochondrial genome (red arrows) to up-regulate all cytochrome oxidase (COX) and some nitric oxide synthase (Nos1), NMDA receptor (Grin 1 and Grin 2b) and AMPA receptor (Gria2) subunit genes, all under the influence of the nuclear respiratory transcription factor NRF-1. C) Thus, gene expression orchestrated by NRF-1 allows for co-localization of proteins critical for energy metabolism and synaptic transmission that aid in synaptic strengthening and improved memory function.
3.4. The enhancing effects of MB on the brain are potentially global but show activational specificity
Studies of the effects of MB on the metabolic capacity of the brain at the network level provide further evidence that the memory-enhancing effects of MB are mediated by increases in cytochrome oxidase. In a fear extinction paradigm (Gonzalez-Lima and Bruchey, 2004), male Long-Evans rats were trained to associate a tone conditioned stimulus with an aversive foot shock unconditioned stimulus. Subsequently, subjects underwent extinction training, in which unreinforced tones were presented repeatedly until subjects displayed very low conditioned responses to the tones. Subjects were then injected with saline or MB 4 mg/kg intraperitoneally and tested in tone-alone probe trials spaced 24 hours apart over the next five days. MB was administered following each probe trial, targeting the consolidation phase of fear extinction. As predicted, MB-treated animals showed better extinction memory retention than the saline-treated animals during the fourth and the fifth probe trial, as well as overall better retention of extinction memory. Those rats with higher retention of extinction also showed a relative increase in cytochrome oxidase activity only in prefrontal cortical regions thought to contribute to extinction memory processing. Such effect was less evident in other brain regions with weaker contributions to this type of memory (Barrett et al., 2003; Quirk et al., 2006). The selected regional metabolic increases were also correlated with the behavioral index of conditioned responding used to assess retention of extinction. This suggested that MB administered after the extinction training enhanced retention of extinction memory through a region-specific increase in brain cytochrome oxidase activity.
Riha et al. (2011) tested the regional memory-enhancing properties of systemic, low-dose MB (4 mg/kg) in rats that received an infusion of the cytochrome oxidase inhibitor sodium azide directly into the posterior cingulate cortex (PCC). The PCC displays the earliest signs of energy hypometabolism in patients with amnestic MCI who later develop AD (Minoshima et al., 1997; Ishiwata et al., 2006). Using metabolic mapping based on cytochrome oxidase histochemistry and network-level computational modeling of brain function, it was demonstrated that MB’s metabolic-enhancing effects are contingent upon the neural context, and that its memory-improving effects are paralleled by network adaptations after injury. Specifically, sodium azide infusions induced PCC hypometabolism and impaired visuospatial memory in a holeboard food search task through disruption of the cingulo-thalamo-hippocampal effective connectivity. Sodium azide also decreased the PCC functional networks and the normal functional redundancy found within the thalamus. In contrast, MB prevented the memory impairment and this effect was associated with preserved cingulo-thalamo-hippocampal effective connectivity. In addition, the modeling strategy used in this study showed that MB had no effects on regions remotely involved in the spatial memory task, while engaged regions showed remarkable sub-network alterations in functional connectivity (Figure 6).
Figure 6. The network effects of MB relevant to visuospatial memory.

Patients with mild cognitive impairment show early impairments in visuospatial memory. A) The diagram shows key structures of the circuit of Papez, a neuroanatomical network involved in the processing of visuospatial memory. Increases in metabolic activity in both the thalamus and hippocampus induce increased metabolic activity in the posterior cingulate cortex (solid arrows). The posterior cingulate cortex is a region considered as a major “hub” in the neural network for visuospatial function, receiving and sending numerous projections. B) Hypometabolism in the posterior cingulate cortex (cross) induces a network disruption in which high metabolic activity in the thalamus and hippocampus induces a further decrease (dashed arrow) in activation of the posterior cingulate. This disruption is behaviorally evident as a subtle visuospatial memory retrieval deficit. C) Methylene blue concentrates in areas with high metabolic activity and facilitates energy metabolism. By doing this, MB facilitates the adaptive changes in connectivity within the thalamus and hippocampus, which are represented by the patchwork pattern shown in these regions. Such changes consist of tendencies of subregions to act as either functional clusters or independent functional nodes. This internal subregional reconfiguration allows for the recruitment of additional brain regions that can “make up” for the faulty connectivity and mediate an appropriate behavioral output. These network effects possibly occur in addition to the direct local neuroprotective effects exerted by MB in those regions originally affected by hypometabolism (Riha et al., 2011).
These data reveal that the effects of MB are not mediated by a global enhancement of brain metabolism but occur in a use-dependent manner. The data are in agreement with the notion that the pattern of MB distribution is determined by the electrochemical gradients created within the intracellular compartments, particularly in mitochondria during the electron transport. Thus, brain regions with the highest metabolic demands during memory consolidation in a particular task will show the largest increases in cytochrome oxidase activity, and those will be the regions that will benefit the most from the metabolic-enhancing effects of MB (Gonzalez-Lima and Bruchey, 2004). This specificity of MB implies that, even when it has a potential homogeneous distribution and bioavailability in the brain, it will preferentially accumulate in the regions with a higher metabolic demand, which are those engaged in a particular computational task. These exciting findings have prompted attempts to use MB as a therapeutic intervention in a number of animal models of neuropsychiatric diseases including post-traumatic stress disorder (PTSD), amnestic MCI and AD. The memory-enhancing effects of MB are also currently being tested in clinical trials for improvement of cognitive ability in AD and extinction learning for PTSD (NIH Clinical Trials NCT00684944 and NCT01188694). For example, MB’s potential to enhance consolidation and retention of extinction memory has been tested as an adjunct for psychotherapy, as a novel way to improve exposure therapy outcome for anxiety disorders. We have administered MB to phobic subjects following exposure therapy, and found that post-training MB administration decreases fear renewal one month following exposure (unpublished data). Furthermore, MB-treated subjects remembered spatial cues presented in the treatment context better than the placebo-treated subjects, suggesting that MB’s memory-enhancing effects are not limited to one type of memory, but can affect consolidation of any memories that are being processed during the time that MB is present as a redox agent in brain mitochondria.
3.5. Acute and chronic MB facilitates memory in conditions associated with abnormal cytochrome oxidase activity
If MB exerts its metabolic-enhancing effects by interacting with cytochrome oxidase, it should promote rescue of function in conditions of abnormal cytochrome oxidase activity. To test this hypothesis, MB was used in an animal model of vulnerability to PTSD and depression, the congenitally helpless rat (Wrubel et al., 2007a). Congenitally helpless rats have dramatic fear extinction deficits, due to alterations in regional brain metabolic capacity which resemble neurobiological abnormalities detected in PTSD patients (Shumake and Gonzalez-Lima, 2003). When 4 mg/kg MB was given systemically following each extinction trial over the period of five days, it significantly improved memory retention of extinction in the acquisition context in congenitally helpless subjects. While MB did not improve extinction learning in extinction context (possibly due to a floor effect on conditioned responding in the extinction context), it did improve the generalization of that learning to a different context. Since in rats MB has a half-life of 5-6.5 h (Peter et al., 2000), it is unlikely that the facilitation of the extinction memory observed 24 h after the last MB injection reflects a continued action of the drug. Rather, the memory retention effects of low dose MB in this study can be explained by an enhanced oxidative energy metabolism occurring during a critical time in memory consolidation.
The beneficial effect of chronic MB on memory has also been shown using chronic cytochrome oxidase inhibition. Chronic inhibition of cytochrome oxidase activity with systemic sodium azide in behaving rats has been used to model the memory deficits and cytochrome oxidase inhibition found in AD (Bennett et al., 1992; Bennett and Rose, 1998; Gonzalez-Lima and Cada, 1998; Gonzalez-Lima et al., 1998a; 1998b; Berndt et al., 2001). In a more direct test of the ability of MB to prevent deficits in cytochrome oxidase, Callaway et al. (2002), administered MB in animals chronically treated with the specific cytochrome oxidase inhibitor sodium azide. Sodium azide has been shown to decrease oxygen consumption in brain tissue, increase oxidative stress and induce neurodegeneration (Riha et al., 2008). Male Sprague-Dawley rats were implanted with osmotic pumps providing a subcutaneous infusion of sodium azide at 0.9 mg/kg/h for 28 days. Following the osmotic pump implantation, subjects were trained five days in the baited holeboard maze designed to test spatial memory, followed by an unbaited probe trial, another five days of training and another probe trial. Some of the subjects also received post-training 1 mg/kg MB intraperitoneally, while control subjects had sham surgery and received saline injections. As anticipated, chronic sodium azide administration produced deficits in learning performance during training trials, as well as deficits in spatial memory retention during probe trials. Repeated administration of MB after each training session, during the memory consolidation period, improved memory retention in probe trials, without improving training-trial performance. These remarkable memory-enhancing effects of chronic MB were later replicated in an acute model of amnestic MCI induced by posterior cingulate cortex hypometabolism as described above (Riha et al., 2011). Together, these data support that both chronic and acute administration of MB are able to improve memory in normal conditions and in those associated with abnormal mitochondrial function. At the same time, the data suggest that the mitochondrial metabolic machinery plays an important role in the processes of memory formation and may constitute a major mechanistic target for the memory-enhancing effects of MB.
3.6. MB improves spatial memory in animal models of cholinergic depletion and tau and amyloid aggregation
Scopolamine treatment is frequently used in neuroscience research to induce memory dysfunction because scopolamine antagonizes acetylcholine, which is the most abundant neurotransmitter in the nervous system. Deiana et al. (2009) treated NMRI mice (a strain particularly sensitive to scopolamine treatment) with 0.5 mg/kg scopolamine and tested their spatial reference memory in a water maze task. Mice that received intraperitoneal injections of scopolamine along with intraperitoneal MB 4 mg/kg showed improved learning curves compared to mice treated with scopolamine alone. MB-treated subjects also performed significantly better in a spatial memory probe trial after 4 days of training. In this study, MB was injected 5 min after scopolamine and 30 min before the learning trials with the intention of acting as a reversing agent on a pre-existent condition of cholinergic disruption. MB was as effective as rivastigmine, a cholinesterase inhibitor, at preventing the scopolamine-induced spatial learning and memory deficits. These data demonstrated that in conditions of acute cholinergic disruption, MB has an enhancing effect during learning. The authors suggested that their data were in agreement with observed in vitro cholinesterase inhibition by MB (Pfaffendorf et al., 1997) and increased brain cytochrome oxidase activity produced by in vivo MB (Callaway et al., 2004), but the effects of MB on brain cholinesterase and energy metabolism were not tested in this study.
Two additional studies have tested the memory-enhancing effects of MB in animal models of human neurodegeneration associated with protein aggregation. O’Leary et al. (2010) used a transgenic mouse model of human tauopathy, progressive supranuclear palsy and fronto-temporal dementia with parkinsonism linked to chromosome 17 to test the memory-enhancing effects of MB. They injected 1 mM MB or saline to the right hippocampi of 7-monthold rTg4510 mice for 1 month using mini-osmotic pumps. Morris water maze testing during the final week of treatment showed that MB-treated mice had significant improvements in learning the location of the escape platform. They also showed significantly improved memory retention of the platform location, based on target quadrant discrimination, number of platform crossings and search strategy. This group also confirmed that oral MB showed concentrations in the brain 100 times higher than plasma at 4 hr after administration. MB was given chronically ad libitum in drinking water at an estimated dose of 9.3 mg/kg per day for 12 weeks. Transgenic mice treated with MB showed improved spatial memory and motor learning compared to non-treated transgenic mice. Remarkably, MB concentrations in the brain showed a high positive correlation with spatial memory in the treated group. MB-treated subjects also showed increased cell counts in the hippocampus, cortex and striatum, compared with non-treated subjects. Tau deposition in the brain was unaffected, but MB decreased the levels of soluble Tau in the hippocampus.
Medina et al. (2011) used a transgenic mouse model of AD, the 3xTg-AD mice, and also reported that chronic MB oral administration significantly improved spatial learning and memory in these mice that show memory impairment. Unfortunately the MB dose used in this study was unclear; and dosing is critical because MB’s hormetic dose-response in normal rodents is behaviorally effective within 1-4 mg/kg and becomes ineffective by 10 mg/kg (Bruchey and Gonzalez-Lima, 2008). A standard rodent diet was supplemented with 25 mg of MB per each 100 g of food daily for 16 weeks. Assuming a mouse of 25 g eats 3 g of food daily; this would supply 30 mg/kg of MB, but no dose estimate was given by the authors. If the transgenic mice consumed about 30 mg/kg of MB, this would be a fairly large dose for normal mice, but since mice were group housed it was not possible to know how much MB they consumed daily. Furthermore, the body weight of the mice was only reported as a percentage of initial unreported weight, making it impossible to accurately estimate a dose of MB per mg body weight. Nevertheless, MB dietary supplementation improved spatial reference memory in the Morris water maze in the 3xTg-AD model. Mice were given four training trials per day for 5 days. During the probe trials, transgenic mice treated with MB reached the platform faster, spent more time in the target quadrant and crossed the hidden platform more than the transgenic mice fed the control diet. In contrast to what was shown by O’Leary et al. (2010), this MB treatment had no effect on the steady-state levels of phosphorylated tau or somatodendritic tau localization. Also, cytochrome oxidase activity showed a small increase that was not reliably significant in the MB-treated transgenic mice. This may be due to the presumably higher MB dose used or to the commercial kit used to measure enzymatic activity, which has very low sensitivity and reliability as compared to optimized methods (Hevner et al., 1993; Gonzalez-Lima and Cada, 1998). The apparently high daily dose of MB used in this study was associated with decreases in soluble Aβ40 and Aβ42 fragments, as well as increases in trypsin and chemotrypsin-like activity of the proteosomal system. These two studies demonstrated a potential beneficial effect of MB in memory before protein aggregation could be detectable by histopathology. Both studies also provided evidence that MB decreased soluble levels of tau or amyloid. These results are of interest because they are in agreement with evidence showing that mitochondrial dysfunction, including changes in cytochrome oxidase expression precede protein aggregation pathology in models of amyloid deposition (Valla et al., 2010; Poirier et al., 2011).
An argument could be made for MB improving memory through its effects on protein aggregation. Nevertheless, compelling data support that this cannot be the sole mechanism through which MB improves memory. First, as discussed above, there is an extensive body of evidence describing memory enhancement effects of MB in normal subjects (Table 1). MB has been effective in models of memory dysfunction and neurodegeneration associated with mitochondrial dysfunction in which tau or amyloid pathology plays no role. If the effect of MB on protein aggregation is acknowledged, it should be considered in the light of in vitro studies showing that tau-like fragments impair cytochrome oxidase activity (Atlante et al., 2008) and that amyloid beta decreases cytochrome oxidase activity and energy production both in vitro (Rhein et al., 2009) and in vivo (Du et al., 2010). Interestingly, the higher doses and concentrations of MB needed to prevent protein aggregation in the transgenic mice studies by Medina et al. and O’Leary et al. may be different from low-dose MB’s memory-enhancement associated with increased cytochrome oxidase activity in normal animals. One way to reconcile these findings would be to test whether MB mainly targets the redox stress induced by the presence of protein aggregates in the animal models of protein aggregation. Such effect could indeed be relevant in a state where protein aggregates are assumed to play a major role in the pathophysiology of memory impairment. However, if protein aggregates only represent a physiological response to oxidative stress and metabolic impairment, the effects of high-dose MB on protein aggregation could be separate from low-dose MB’s effects on memory improvement. The relevance of the effects of MB on memory in relation to its effects on protein aggregation and the physiologic mechanisms for energy metabolism in memory remain to be determined.
4. MB as a neuroprotective agent
Some of the studies testing the memory-enhancing effects of MB discussed above have provided interesting data supporting that MB also has the potential to specifically prevent structural and functional neural damage. In the past few years, a number of additional studies have pioneered the use of MB for preventing neurodegeneration induced by metabolic insults. As discussed below, available data support the potential of MB as a neuroprotective agent in the setting of mitochondrial dysfunction and hypoxia.
4.1. Neuroprotection against ifosfamide-induced encephalopathy
Kupfer et al. (1994) reported two cases in which MB was successfully used to prevent the metabolic encephalopathy associated with the chemotherapeutic agent ifosfamide in humans. Ifosfamide is an alkylating agent used alone or in combination for the treatment of solid tumors. About ten percent of patients receiving ifosfamide develop a clinical picture of somnolence, stupor and coma over several (3-7) days that can be fatal (Pelgrims et al., 2000). This encephalopathy is believed to be caused by phase II ifosfamide metabolites, which have been demonstrated to be mitochondrial toxins. One ifosfamide metabolite is a potent flavoprotein inhibitor, which leads to impaired oxidation of NADH to NAD. A decrease in NAD, which is a cofactor of several aldehyde dehydrogenases, leads to accumulation of toxic aldehyde-related compounds (Kupfer et al., 1996). After the original report, several series have confirmed that either intravenous or oral MB administered in doses of 50 mg/day × 6 after the onset of the encephalopathy or even at 50 mg/day × 4 for prophylaxis of ifosfamide-induced encephalopathy, reduces the duration of encephalopathy to less than 1 day (Pelgrims et al., 2000; Dufour et al., 2006). It is possible that MB substitutes for flavoprotein deficiency and accelerates NADH oxidation, which forms during ifosfamide metabolism. These reports and case series provided evidence that systemic MB administration can be effectively used to prevent and treat acute brain functional changes caused by mitochondrial metabolic insults in humans.
4.2. Neuroprotection against experimental optic neuropathy induced by mitochondrial dysfunction
Using a novel in vivo mouse model of retinal optic neuropathy designed for efficient toxicological screening (Rojas and Gonzalez-Lima, 2010), Zhang et al. (2006) demonstrated that MB effectively prevents the morphologic retinal damage induced by a single intravitreal injection of rotenone. Rotenone is a natural inhibitor of mitochondrial complex I, regarded as a potential environmental neurotoxin that produces neurotoxicity by inducing oxidative stress and energy depletion. The neurotoxic effects of rotenone have been implicated in the pathogenesis of neurodegenerative disorders such as Parkinson’s disease and Leber’s hereditary optic neuropathy (Larsson et al., 1991; Betarbet et al., 2000). The neuroprotective effects of MB in the retina were observed in a dose-response manner. At 24 h, an intravitreal dose of 70 μg/kg prevented the 40% decrease in ganglion cell layer (GCL) thickness induced by rotenone, as well as the 30% decrease in GCL cell density. An MB intravitreal dose of 7 μg/kg was similarly effective at preventing retinal damage, whereas a dose of 0.7 μg/kg had no effect. Using a similar optic neuropathy model, Rojas et al. (2009a) analyzed the long-term effects of MB against rotenone-induced neurodegeneration in the rat retina. In addition to structural damage, we also evaluated retinal function by analyzing changes in both visually-guided behavior and energy metabolism in the central visual pathway. At 14 days, intravitreal 70 μg/kg MB prevented the approximate fifty percent reductions in retinal layer thickness and GCL cell density induced by rotenone. Behaviorally, MB preserved visual function at levels comparable to pre-treatment and vehicle-treated animals. Specifically, MB prevented the 44% decrease in the successful completion of a visually-guided task and the 3.8-fold increase in the log illuminance sensitivity threshold produced by rotenone. In addition, it was shown that the morphologic and functional effects of MB were associated with an improvement of retinal energy metabolism. Whereas rotenone-treated subjects showed a 32.6% decrease in the mean retinal cytochrome oxidase activity compared to control, MB-cotreated subjects showed a 17.2% increase in retinal cytochrome oxidase activity. These studies validated the relevant principle that MB can effectively be used to prevent the structural and functional deficits caused by inhibition of an electron transfer chain complex implicated in the pathogenesis of neurodegenerative diseases.
4.3. Neuroprotection against cardiac arrest-induced brain damage
A series of elegant studies with low-dose MB have demonstrated its potential value in vivo to prevent the neural damage induced by hypoxia-reperfusion injury. Miclescu et al. (2006) used an experimental pig model of extended circulatory arrest to study the effects of systemic MB on myocardial and neural damage. MB was added to a hypertonic saline-dextran solution as an adjunct treatment. This treatment was administered after 1 min of cardiac arrest while cardiopulmonary resuscitation was given, and during the initial reperfusion period after cardiac arrest. MB was given at a loading rate of 7.5 mg/kg/h and then maintained at 2.25 mg/kg/h for up to 50 min for a total dose of 3 mg/kg. MB improved the 4 h arrest survival and improved hemodynamic variables after the first minute of restoration of spontaneous circulation. Additionally, MB was shown to prevent neural damage, as demonstrated by a decrease in the plasma level of the astroglial marker of hypoxic brain injury (protein S-100β), compared to control groups. MB-treated subjects also showed decreased levels of systemic 8-iso-PGF2α, a marker of oxidative injury, suggesting that the neuroprotective effects of MB may be related to an in vivo antioxidant effect. In a follow-up study, Miclescu et al. (2007) used the same animal model to show that MB decreases reperfusion injury after a more prolonged period of 12 minutes of cardiac arrest including 8 min of cardiopulmonary resuscitation. An additional study demonstrated that MB treatment ameliorates blood-brain barrier disruption, brain edema, myelin damage and albumin leakage following cardiac arrest in the same porcine animal model (Sharma et al., 2011). Martijn and Wiklund (2010) have also examined the genomic responses to reperfusion injury after MB treatment in the porcine brain. They found that low-dose MB treatment induced expression of critical neuroprotective genes, such as CX43, THBD and CART. Furthermore, low-dose MB treatment reversed the shutdown of translation induced by endoplasmatic reticulum stress that was observed in the brains of animals treated with saline only. MB treatment also stimulated several anti-apoptotic genes and activated many brain repair/regeneration genes. MB’s regulation of the expression of soluble guanylate cyclase was also confirmed, and MB was found to restore the functional cellular trafficking through regulation of proteins which mediate transport between endoplasmatic reticulum, Golgi, cytoskeleton and vesicles.
4.4. Neuroprotection against striatal neurodegeneration
Rojas et al. (2009b) used the neurotoxin rotenone to induce large anatomical lesions of the rat striatum which resembled “metabolic strokes” observed in patients with inborn-errors of metabolism. Administration of MB greatly reduced the size of these lesions, and prevented the decrease in cytochrome oxidase activity and the perilesional increase in oxidative stress associated with rotenone infusion in the striatum (Figure 7). Furthermore, MB prevented the functional decoupling of basal ganglia-thalamocortical motor circuits induced by rotenone, and also prevented the lesion-induced indirect effects of rotenone on cytochrome oxidase activity in motor regions such as the substantia nigra and the pedunculopontine nucleus. Functionally, MB partially prevented the behavioral sensorimotor asymmetries elicited by rotenone. These findings provided additional evidence of protective effects of MB against neurotoxic damage in the brain parenchyma.
Figure 7. Neuroprotection with methylene blue.
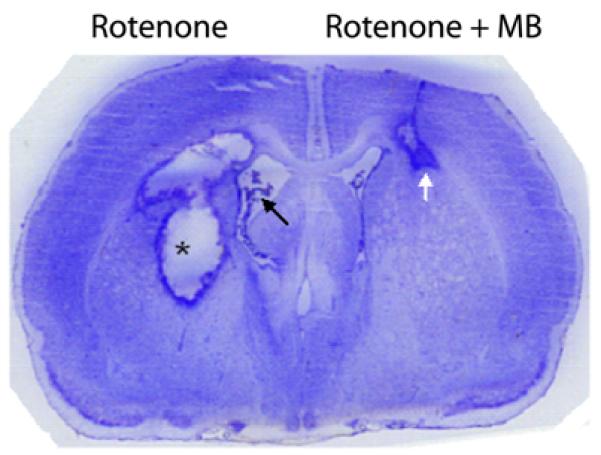
MB prevented the neurodegeneration induced by the neurotoxin rotenone in the brain (Rojas et al., 2009b). The figure is a Nissl-stained coronal section of the forebrain of a rat receiving bilateral intrastriatal infusions of rotenone alone (left hemisphere) and rotenone plus MB (right hemisphere). The rotenone-treated hemisphere showed a large area of neurodegeneration, characterized by a cavity of liquefactive necrosis surrounded by a rim of reactive gliosis (asterisk). This lesion was accompanied by an enlarged lateral ventricle (black arrow). In contrast, the MB co-treated hemisphere showed significantly less damage, with a comparatively smaller lesion limited to the corpus callosum (white arrow). Similar dramatic neuroprotective results were found in other experiments with rats and mice that used bilateral or unilateral infusions of rotenone as compared to rotenone plus MB in the eye (Zhang et al., 2006; Rojas et al., 2009a) or in the brain (Rojas et al., 2009b).
An additional study has demonstrated that MB prevents the acute biochemical and electrophysiological effects of a neurotoxic metabolite in vivo. Methylmalonate (MMA) is an endogenous metabolite elevated in patients with methylmalonic acidemia, a condition caused by a deficiency in the enzyme methylmalonil CoA-mutase and by defects in the metabolism of cobalamin (Morrow et al., 1975). MMA is a mitochondrial toxin that produces a decrease in oxygen consumption, energy depletion and acidosis, with a mitochondrial inhibitory effect that appears to be associated with impairment of complex II activity and disruption of the Krebs cycle (Brusque et al., 2002; Okun et al., 2002; Furian et al., 2007). Methylmalonic acidemia can produce a clinical picture characterized by failure to thrive, lethargy, hypotonia, hypoglycemia and myopathy (Henriquez et al., 1994). Most importantly, methylmalonic acidemia produces neural impairment characterized by seizures and signs of acute striatal damage including choreoathetosis, dystonia, dysarthria and dysphagia (Heidenreich et al., 1988). Furian et al. (2007) modeled the neurotoxicity-related seizure activity of methylmalonic acidemia by giving intrastriatal injections of MMA and tested the neuroprotective effects of MB against this mitochondrial neurotoxin. Intrastriatal pre-treatment with MB increased seizure latency and decreased seizure duration after MMA administration. MB treatment also prevented the electroencephalographic alterations induced by MMA. A total amount of 560 ng MB was also effective at preventing the 35% MMA-induced increase in striatal protein carbonyl and nitrite/nitrate content. Similarly, the MMA induced a nearly 50% decrease in Na+/K+ ATPase activity that was reversed by the co-administration of MB. All these biochemical effects were not observed with MB doses one and two log units lower.
Whether the beneficial use of MB can be generalized to other contexts of neurodegeneration should be further investigated. Surprisingly, the studies reviewed above demonstrating the dramatic neuroprotective effects of MB are not commonly cited or acknowledged in the literature on neuroprotection. For example, a recent paper by Wen et al. (2011) reporting neuroprotective effects of MB in animal models of rotenone-induced Parkinsonism and cerebral ischemia did not cite any of the previous MB studies showing neuroprotection against rotenone-induced neurodegeneration and ischemia/reperfusion damage. Moreover, the mechanism of action proposed by Wen et al. (2011) is the same mitochondrial mechanism reviewed above that has been proposed in previous MB studies of neuroprotection (Zhang et al., 2006; Bruchey and Gonzalez-Lima, 2008; Rojas et al., 2009a; 2009b; Rojas and Gonzalez-Lima, 2010).
5. Conclusion
Together, these studies provide conclusive evidence that low doses of pharmaceutical grade (USP) MB are effective for improving different forms of memory and for preventing various neurochemical, structural, and functional deficits derived from mitochondrial inhibition and oxidative stress. Ongoing experiments in the authors’ laboratory will determine the effectiveness of MB as a neuroprotective agent in various other animal models of neurodegeneration and amnesia, and its effectiveness as a nootropic agent in human volunteers. These and similar studies should provide further testing of the potential clinical use of MB to improve memory and prevent neurodegenerative disorders. It is proposed that MB or related compounds with similar neurometabolic mechanisms of action could be used to enhance memory and prevent aging-related and disease-related memory loss, especially in the treatment of acute or chronic conditions associated with mitochondrial dysfunction in humans.
Highlights.
Methylene blue has unique antioxidant and metabolic-enhancing hormetic properties
Low-dose methylene blue improves memory retention by enhancing consolidation
Memory-improving effects of methylene blue are mediated by cytochrome oxidase
Methylene blue local brain effects on mitochondria function are activity-dependent
Low-dose methylene blue prevents neurodegeneration induced by metabolic insults
Acknowledgements
Supported in part by NIH grants MH65728 and MH076847 to FGL.
Abbreviations
- AD
Alzheimer’s disease
- ATP
Adenosine triphosphate
- FADH2
Flavin adenine dinucleotide
- GCL
Ganglion cell layer
- MB
Methylene blue
- MCI
Mild cognitive impairment
- MMA
Methylmalonate
- NADH
Nicotine adenine dinucleotide
- NADPH
Nicotine adenine dinucleotide phosphate
- NOS
Nitric oxide synthase
- NO
Nitric oxide
- PCC
Posterior cingulate cortex
- PTSD
Post-traumatic stress disorder
- USP
United States Pharmacopeia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allexsaht WJ. The use of methylene blue in the treatment of catatonic dementia praecox patients. Psychiatric Quarterly. 1938;12:245–252. [Google Scholar]
- Andresen M, Dougnac A, Diaz O, Hernandez G, Castillo L, Bugedo G, et al. Use of methylene blue in patients with refractory septic shock: impact on hemodynamics and gas exchange. J. Crit. Care. 1998;13:164–168. doi: 10.1016/s0883-9441(98)90001-6. [DOI] [PubMed] [Google Scholar]
- Arieff AJ, Pyzik SW. Quadriplegia after intrathecal injection of methylene blue. J. Am. Med. Assoc. 1960;173:794–796. doi: 10.1001/jama.1960.73020250011008d. [DOI] [PubMed] [Google Scholar]
- Artuch R, Aracil A, Mas A, Monros E, Vilaseca MA, Pineda M. Cerebrospinal fluid concentrations of idebenone in Friedreich ataxia patients. Neuropediatrics. 2004;35:95–98. doi: 10.1055/s-2004-815830. [DOI] [PubMed] [Google Scholar]
- Atamna H. Amino acids variations in amyloid-beta peptides, mitochondrial dysfunction, and new therapies for Alzheimer’s disease. J Bioenerg Biomembr. 2009;41:457–464. doi: 10.1007/s10863-009-9246-2. [DOI] [PubMed] [Google Scholar]
- Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, et al. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta. 2008;1777:1289–1300. doi: 10.1016/j.bbabio.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Auerbach SS, Bristol DW, Peckham JC, Travlos GS, Hebert CD, Chhabra RS. Toxicity and carcinogenicity studies of methylene blue trihydrate in F344N rats and B6C3F1 mice. Food Chem. Toxicol. 2010;48:169–177. doi: 10.1016/j.fct.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett D, Shumake J, Jones D, Gonzalez-Lima F. Metabolic mapping of mouse brain activity after extinction of a conditioned emotional response. J. Neurosci. 2003;23:5740–5749. doi: 10.1523/JNEUROSCI.23-13-05740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MC, Diamond DM, Stryker SL, Parks JK, Parker WD., Jr. Cytochrome oxidase inhibition: a novel animal model of Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 1992;5:93–101. doi: 10.1177/002383099200500206. [DOI] [PubMed] [Google Scholar]
- Bennett MC, Rose GM. Behavioral, electrophysiological and biochemical consequences of chronic cytochrome oxidase inhibition in rats. In: F G-L, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. Plenum Press; New York: 1998. pp. 217–232. [Google Scholar]
- Berndt JD, Callaway NL, Gonzalez-Lima F. Effects of chronic sodium azide on brain and muscle cytochrome oxidase activity: a potential model to investigate environmental contributions to neurodegenerative diseases. J. Toxicol. Environ. Health A. 2001;63:67–77. doi: 10.1080/152873901750128380. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Blass N, Fung D. Dyed but not dead--methylene blue overdose. Anesthesiology. 1976;45:458–459. doi: 10.1097/00000542-197610000-00020. [DOI] [PubMed] [Google Scholar]
- Bruchey AK, Gonzalez-Lima F. Behavioral, physiological and biochemical hormetic responses to the autoxidizable dye methylene blue. Am. J. Pharm. & Toxicol. 2008;3:72–79. doi: 10.3844/ajptsp.2008.72.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusque AM, Borba Rosa R, Schuck PF, Dalcin KB, Ribeiro CA, Silva CG, et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002;40:593–601. doi: 10.1016/s0197-0186(01)00130-9. [DOI] [PubMed] [Google Scholar]
- Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, et al. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob. Agents Chemother. 2008;52:183–191. doi: 10.1128/AAC.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, Bachmann KA, Bailer AJ, Bolger PM, Borak J, Cai L, et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007;222:122–128. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Hoffmann GR, Stanek EJ, Nascarella MA. Hormesis in high-throughput screening of antibacterial compounds in E coli. Hum. Exp. Toxicol. 2010a;29:667–677. doi: 10.1177/0960327109358917. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Mattson MP, Calabrese V. Resveratrol commonly displays hormesis: occurrence and biomedical significance. Hum. Exp. Toxicol. 2010b;29:980–1015. doi: 10.1177/0960327110383625. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol. Biochem. Behav. 2004;77:175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Wrubel KM, McCollum D, Gonzalez-Lima F. Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neurosci. Lett. 2002;332:83–86. doi: 10.1016/s0304-3940(02)00827-3. [DOI] [PubMed] [Google Scholar]
- Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Clifton J, 2nd, Leikin JB. Methylene blue. Am. J. Ther. 2003;10:289–291. doi: 10.1097/00045391-200307000-00009. [DOI] [PubMed] [Google Scholar]
- Cohen N, Robinson D, Ben-Ezzer J, Hemo Y, Hasharoni A, Wolmann Y, et al. Reduced NO accumulation in arthrotic cartilage by exposure to methylene blue. Acta Orthop. Scand. 2000;71:630–636. doi: 10.1080/000164700317362299. [DOI] [PubMed] [Google Scholar]
- Cooper CE. Nitric oxide and cytochrome oxidase: substrate, inhibitor or effector? Trends Biochem. Sci. 2002;27:33–39. doi: 10.1016/s0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- Daemen-Gubbels CR, Groeneveld PH, Groeneveld AB, van Kamp GJ, Bronsveld W, Thijs LG. Methylene blue increases myocardial function in septic shock. Crit. Care Med. 1995;23:1363–1370. doi: 10.1097/00003246-199508000-00009. [DOI] [PubMed] [Google Scholar]
- Deiana S, Harrington CR, Wischik CM, Riedel G. Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine. Psychopharmacology (Berl) 2009;202:53–65. doi: 10.1007/s00213-008-1394-2. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Schwartz BL, Fay-McCarthy M, Rosenberg PB, Fearing K. Methylene blue adjuvant therapy of schizophrenia. Clin. Neuropharmacol. 1997;20:357–363. doi: 10.1097/00002826-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Liang HL, Wong-Riley MT. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: tight coupling of glutamatergic transmission and energy metabolism in neurons. J. Neurochem. 2009a;108:1595–1606. doi: 10.1111/j.1471-4159.2009.05929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Liang HL, Wong-Riley MT. Transcriptional coupling of synaptic transmission and energy metabolism: role of nuclear respiratory factor 1 in co-regulating neuronal nitric oxide synthase and cytochrome c oxidase genes in neurons. Biochim. Biophys. Acta. 2009b;1793:1604–1613. doi: 10.1016/j.bbamcr.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Wong-Riley MT. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 2009;29:483–492. doi: 10.1523/JNEUROSCI.3704-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour C, Grill J, Sabouraud P, Behar C, Munzer M, Motte J, et al. Arch. Pediatr. 2006;13:140–145. doi: 10.1016/j.arcped.2005.10.021. Ifosfamide induced encephalopathy: 15 observations. [DOI] [PubMed] [Google Scholar]
- Furian AF, Fighera MR, Oliveira MS, Ferreira AP, Fiorenza NG, de Carvalho Myskiw J, et al. Methylene blue prevents methylmalonate-induced seizures and oxidative damage in rat striatum. Neurochem. Int. 2007;50:164–171. doi: 10.1016/j.neuint.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Gabrielli D, Belisle E, Severino D, Kowaltowski AJ, Baptista MS. Binding, aggregation and photochemical properties of methylene blue in mitochondrial suspensions. Photochem. Photobiol. 2004;79:227–232. doi: 10.1562/be-03-27.1. [DOI] [PubMed] [Google Scholar]
- Gillman PK. Methylene blue is a potent monoamine oxidase inhibitor. Can. J. Anaesth. 2008;55:311–312. doi: 10.1007/BF03017212. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lima F. Brain imaging of auditory learning functions in rats: Studies with fluorodeoxyglucose autoradiography and cytochrome oxidase histochemistry. In: Gonzalez-Lima F, Finkenstädt T, Scheich H, editors. Advances in Metabolic Mapping Techniques for Brain Imaging of Behavioral and Learning Functions. Kluwer Academic Publishers; Dordrecht/Boston/London: 1992. pp. 39–109. [Google Scholar]
- Gonzalez-Lima F, Bruchey AK. Extinction memory improvement by the metabolic enhancer methylene blue. Learn. Mem. 2004;11:633–640. doi: 10.1101/lm.82404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Cada A. Quantitative histochemistry of cytochrome oxidase activity: Theory, methods, and regional vulnerability. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. Plenum press; New York: 1998. [Google Scholar]
- Gonzalez-Lima F, Valla J, Cada A. Brain cytochrome oxidase activity and how it relates to the pathophysiology of memory and Alzheimer’s disease. In: Ozben T, editor. Free Radicals, Oxidative Stress and Antioxidants: Pathological and Physiological Significance. Plenum Press; New York: 1998a. pp. 205–227. [Google Scholar]
- Gonzalez-Lima F, Valla J, Lorandby L. Cytochrome oxidase inhibition in Alzheimer’s disease. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. New York; Plenum Press: 1998b. pp. 171–202. [Google Scholar]
- Gonzalez-Lima F, Valla J, Matos-Collazo S. Quantitative cytochemistry of cytochrome oxidase and cellular morphometry of the human inferior colliculus in control and Alzheimer’s patients. Brain Res. 1997;752:117–126. doi: 10.1016/s0006-8993(96)01464-3. [DOI] [PubMed] [Google Scholar]
- Govindaiah, Shankaranarayana Rao BS, Ramamohan Y, Singh YK, Dhingra NK, Raju TR. Cytochrome oxidase activity in rat retinal ganglion cells during postnatal development. Brain. Res. Dev. Brain Res. 2000;124:117–120. doi: 10.1016/s0165-3806(00)00092-4. [DOI] [PubMed] [Google Scholar]
- Guzman-Barron ES, Hoffmann LA. The catalytic effect of dyes on the oxygen consumption of living cells. J. Gen. Physiol. 1930;13:483–494. doi: 10.1085/jgp.13.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey BH, Duvenhage I, Viljoen F, Scheepers N, Malan SF, Wegener G, et al. Role of monoamine oxidase, nitric oxide synthase and regional brain monoamines in the antidepressant-like effects of methylene blue and selected structural analogues. Biochem. Pharmacol. 2010;80:1580–1591. doi: 10.1016/j.bcp.2010.07.037. [DOI] [PubMed] [Google Scholar]
- Heidenreich R, Natowicz M, Hainline BE, Berman P, Kelley RI, Hillman RE, et al. Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. J. Pediatr. 1988;113:1022–1027. doi: 10.1016/s0022-3476(88)80574-2. [DOI] [PubMed] [Google Scholar]
- Henriquez H, el Din A, Ozand PT, Subramanyam SB, al Gain SI. Emergency presentations of patients with methylmalonic acidemia, propionic acidemia and branched chain amino acidemia (MSUD) Brain Dev. 1994;16(Suppl):86–93. doi: 10.1016/0387-7604(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Liu S, Wong-Riley MT. An optimized method for determining cytochrome oxidase activity in brain tissue homogenates. J. Neurosci. Methods. 1993;50:309–319. doi: 10.1016/0165-0270(93)90038-s. [DOI] [PubMed] [Google Scholar]
- Ishiwata A, Sakayori O, Minoshima S, Mizumura S, Kitamura S, Katayama Y. Preclinical evidence of Alzheimer changes in progressive mild cognitive impairment: a qualitative and quantitative SPECT study. Acta Neurol. Scand. 2006;114:91–96. doi: 10.1111/j.1600-0404.2006.00661.x. [DOI] [PubMed] [Google Scholar]
- Juffermans NP, Vervloet MG, Daemen-Gubbels CR, Binnekade JM, de Jong M, Groeneveld AB. A dose-finding study of methylene blue to inhibit nitric oxide actions in the hemodynamics of human septic shock. Nitric Oxide. 2010;22:275–280. doi: 10.1016/j.niox.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Kelner MJ, Bagnell R, Hale B, Alexander NM. Methylene blue competes with paraquat for reduction by flavo-enzymes resulting in decreased superoxide production in the presence of heme proteins. Arch. Biochem. Biophys. 1988a;262:422–426. doi: 10.1016/0003-9861(88)90393-1. [DOI] [PubMed] [Google Scholar]
- Kelner MJ, Bagnell R, Hale B, Alexander NM. Potential of methylene blue to block oxygen radical generation in reperfusion injury. Basic Life Sci. 1988b;49:895–898. doi: 10.1007/978-1-4684-5568-7_146. [DOI] [PubMed] [Google Scholar]
- Klatt P, Heinzel B, John M, Kastner M, Bohme E, Mayer B. Ca2+/calmodulin-dependent cytochrome c reductase activity of brain nitric oxide synthase. J. Biol. Chem. 1992;267:11374–11378. [PubMed] [Google Scholar]
- Kupfer A, Aeschlimann C, Cerny T. Methylene blue and the neurotoxic mechanisms of ifosfamide encephalopathy. Eur. J. Clin. Pharmacol. 1996;50:249–252. doi: 10.1007/s002280050102. [DOI] [PubMed] [Google Scholar]
- Kupfer A, Aeschlimann C, Wermuth B, Cerny T. Prophylaxis and reversal of ifosfamide encephalopathy with methylene-blue. Lancet. 1994;343:763–764. doi: 10.1016/s0140-6736(94)91839-2. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Andersen O, Holme E, Oldfors A, Wahlstrom J. Leber’s hereditary optic neuropathy and complex I deficiency in muscle. Ann. Neurol. 1991;30:701–708. doi: 10.1002/ana.410300511. [DOI] [PubMed] [Google Scholar]
- Lee RB, Urban JP. Functional replacement of oxygen by other oxidants in articular cartilage. Arthritis Rheum. 2002;46:3190–3200. doi: 10.1002/art.10686. [DOI] [PubMed] [Google Scholar]
- Lensman M, Korzhevskii DE, Mourovets VO, Kostkin VB, Izvarina N, Perasso L, et al. Intracerebroventricular administration of creatine protects against damage by global cerebral ischemia in rat. Brain Res. 2006;1114:187–194. doi: 10.1016/j.brainres.2006.06.103. [DOI] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl PE, Oberg KE. The effect of rotenone on respiration and its point of attack. Exp. Cell Res. 1961;23:228–237. doi: 10.1016/0014-4827(61)90033-7. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, Buss C, Schramek TE, Maheu F, Pruessner J. Hormetic influence of glucocorticoids on human memory. Nonlinearity Biol. Toxicol. Med. 2005;3:23–56. doi: 10.2201/nonlin.003.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martijn C, Wiklund L. Effect of methylene blue on the genomic response to reperfusion injury induced by cardiac arrest and cardiopulmonary resuscitation in porcine brain. BMC Med. Genomics. 2010;3:27. doi: 10.1186/1755-8794-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale SJ, Stedeford JC. Neurological sequelae following methylene blue injection for parathyroidectomy. Anaesthesia. 2003;58:1041–1042. doi: 10.1046/j.1365-2044.2003.03415_23.x. [DOI] [PubMed] [Google Scholar]
- Martinez J, Jr., Jensen RA, Vasquez B, McGuiness T, McGaugh JL. Methylene blue alters retention of inhibitory avoidance responses. Physiological Psychology. 1978;6:387–390. [Google Scholar]
- Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem. Pharmacol. 1993;45:367–374. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miclescu A, Basu S, Wiklund L. Methylene blue added to a hypertonic-hyperoncotic solution increases short-term survival in experimental cardiac arrest. Crit. Care Med. 2006;34:2806–2813. doi: 10.1097/01.CCM.0000242517.23324.27. [DOI] [PubMed] [Google Scholar]
- Miclescu A, Basu S, Wiklund L. Cardio-cerebral and metabolic effects of methylene blue in hypertonic sodium lactate during experimental cardiopulmonary resuscitation. Resuscitation. 2007;75:88–97. doi: 10.1016/j.resuscitation.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Michel H, Behr J, Harrenga A, Kannt A. Cytochrome c oxidase: structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 1998;27:329–356. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Skutella T, Beyer K, Behl C. Protective activity of aromatic amines and imines against oxidative nerve cell death. Biol. Chem. 2001;382:1601–1612. doi: 10.1515/BC.2001.195. [DOI] [PubMed] [Google Scholar]
- Morrow G, 3rd, Mahoney MJ, Mathews C, Lebowitz J. Studies of methylmalonyl coenzyme A carbonylmutase activity in methylmalonic acidemia. I. Correlation of clinical, hepatic, and fibroblast data. Pediatr. Res. 1975;9:641–644. doi: 10.1203/00006450-197508000-00006. [DOI] [PubMed] [Google Scholar]
- Nascarella MA, Stanek EJ, 3rd, Hoffmann GR, Calabrese EJ. Quantification of hormesis in anticancer-agent dose-responses. Dose Response. 2009;7:160–171. doi: 10.2203/dose-response.08-025.Nascarella. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor GJ, Martin B, Hopwood SE, Watson Y. A two-year double-blind crossover trial of the prophylactic effect of methylene blue in manic-depressive psychosis. Biol. Psychiatry. 1986;21:915–920. doi: 10.1016/0006-3223(86)90265-9. [DOI] [PubMed] [Google Scholar]
- Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, et al. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- O’Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol. Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlow MJ, Moosmann B. Phenothiazine: the seven lives of pharmacology’s first lead structure. Drug Discov. Today. 2011;16:119–131. doi: 10.1016/j.drudis.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Okun JG, Horster F, Farkas LM, Feyh P, Hinz A, Sauer S, et al. Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and the tricarboxylic acid cycle, and synergistically acting excitotoxicity. J. Biol. Chem. 2002;277:14674–14680. doi: 10.1074/jbc.M200997200. [DOI] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of Methylene Blue in the nervous system. Med Res Rev. 2011;31:93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Petroianu GA. Methylene blue and Alzheimer’s disease. Biochem Pharmacol. 2009;78:927–932. doi: 10.1016/j.bcp.2009.04.034. [DOI] [PubMed] [Google Scholar]
- Pelgrims J, De Vos F, Van den Brande J, Schrijvers D, Prove A, Vermorken JB. Methylene blue in the treatment and prevention of ifosfamide-induced encephalopathy: report of 12 cases and a review of the literature. Br. J. Cancer. 2000;82:291–294. doi: 10.1054/bjoc.1999.0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C, Hongwan D, Kupfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol. 2000;56:247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- Pfaffendorf M, Bruning TA, Batnik HD, van Zwieten PA. The interaction between methylene blue and the cholinergic system. Br. J. Pharmacol. 1997;122:95–98. doi: 10.1038/sj.bjp.0701355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier GL, Amin E, Good MA, Aggleton JP. Early-onset dysfunction of retrosplenial cortex precedes overt amyloid plaque formation in Tg2576 mice. Neuroscience. 2011;174:71–83. doi: 10.1016/j.neuroscience.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppers PJ, Mastri AR, Lebeaux M, Covino BG. The effect of methylene blue on neural tissue. Anesthesiology. 1970;33:335–340. doi: 10.1097/00000542-197009000-00014. [DOI] [PubMed] [Google Scholar]
- Poremba A, Jones D, Gonzalez-Lima F. Metabolic effects of blocking tone conditioning on the rat auditory system. Neurobiol. Learn. Mem. 1997;68:154–171. doi: 10.1006/nlme.1997.3792. [DOI] [PubMed] [Google Scholar]
- Poremba A, Jones D, Gonzalez-Lima F. Classical conditioning modifies cytochrome oxidase activity in the auditory system. Eur. J. Neurosci. 1998;10:3035–3043. doi: 10.1046/j.1460-9568.1998.00304.x. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, Gonzalez-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol. Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rainer M, Kraxberger E, Haushofer M, Mucke HA, Jellinger KA. No evidence for cognitive improvement from oral nicotinamide adenine dinucleotide (NADH) in dementia. J. Neural Transm. 2000;107:1475–1481. doi: 10.1007/s007020070011. [DOI] [PubMed] [Google Scholar]
- Ramsay RR, Dunford C, Gillman PK. Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br. J. Pharmacol. 2007;152:946–951. doi: 10.1038/sj.bjp.0707430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein V, Baysang G, Rao S, Meier F, Bonert A, Muller-Spahn F, et al. Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol. Neurobiol. 2009;29:1063–1071. doi: 10.1007/s10571-009-9398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur. J. Pharmacol. 2005;511:151–158. doi: 10.1016/j.ejphar.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Riha PD, Rojas JC, Colorado RA, Gonzalez-Lima F. Animal model of posterior cingulate cortex hypometabolism implicated in amnestic MCI and AD. Neurobiol. Learn. Mem. 2008;90:112–124. doi: 10.1016/j.nlm.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha PD, Rojas JC, Gonzalez-Lima F. Beneficial network effects of methylene blue in an amnestic model. Neuroimage. 2011;54:2623–2634. doi: 10.1016/j.neuroimage.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Gonzalez-Lima F. Mitochondrial optic neuropathy: In vivo model of neurodegeneration and neuroprotective strategies. Eye and brain. 2010;2:21–37. doi: 10.2147/eb.s9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Gonzalez-Lima F. Low-level light therapy of the eye and brain. Eye and brain. 2011;3:49–67. doi: 10.2147/EB.S21391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, John JM, Lee J, Gonzalez-Lima F. Methylene blue provides behavioral and metabolic neuroprotection against optic neuropathy. Neurotox. Res. 2009a;15:260–273. doi: 10.1007/s12640-009-9027-z. [DOI] [PubMed] [Google Scholar]
- Rojas JC, Simola N, Kermath BA, Kane JR, Schallert T, Gonzalez-Lima F. Striatal neuroprotection with methylene blue. Neuroscience. 2009b;163:877–889. doi: 10.1016/j.neuroscience.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaris SC, Babbs CF, Voorhees WD., 3rd Methylene blue as an inhibitor of superoxide generation by xanthine oxidase. A potential new drug for the attenuation of ischemia/reperfusion injury. Biochem. Pharmacol. 1991;42:499–506. doi: 10.1016/0006-2952(91)90311-r. [DOI] [PubMed] [Google Scholar]
- Scott A, Hunter FE., Jr. Support of thyroxine-induced swelling of liver mitochondria by generation of high energy intermediates at any one of three sites in electron transport. J. Biol. Chem. 1966;241:1060–1066. [PubMed] [Google Scholar]
- Schneider F, Lutun P, Hasselmann M, Stoclet JC, Tempe JD. Methylene blue increases systemic vascular resistance in human septic shock. Preliminary observations. Intensive Care Med. 1992;18:309–311. doi: 10.1007/BF01706481. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Miclescu A, Wiklund L. Cardiac arrest-induced regional blood-brain barrier breakdown, edema formation and brain pathology: a light and electron microscopic study on a new model for neurodegeneration and neuroprotection in porcine brain. J. Neural Transm. 2011;118:87–114. doi: 10.1007/s00702-010-0486-4. [DOI] [PubMed] [Google Scholar]
- Shumake J, Gonzalez-Lima F. Brain systems underlying susceptibility to helplessness and depression. Behav. Cogn. Neurosci. Rev. 2003;2:198–221. doi: 10.1177/1534582303259057. [DOI] [PubMed] [Google Scholar]
- Smith RP, Thron CD. Hemoglobin, methylene blue and oxygen interactions in human red cells. J. Pharmacol. Exp. Ther. 1972;183:549–558. [PubMed] [Google Scholar]
- Sweet G, Standiford SB. Methylene-blue-associated encephalopathy. J. Am. Coll. Surg. 2007;204:454–458. doi: 10.1016/j.jamcollsurg.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, et al. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- Teichert J, Hermann R, Ruus P, Preiss R. Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers. J. Clin. Pharmacol. 2003;43:1257–1267. doi: 10.1177/0091270003258654. [DOI] [PubMed] [Google Scholar]
- Vaiserman AM. Radiation hormesis: historical perspective and implications for low-dose cancer risk assessment. Dose Response. 2010;8:172–191. doi: 10.2203/dose-response.09-037.Vaiserman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Yaari R, Wolf AB, Kusne Y, Beach TG, Roher AE, et al. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 2010;22:307–313. doi: 10.3233/JAD-2010-100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visarius TM, Stucki JW, Lauterburg BH. Stimulation of respiration by methylene blue in rat liver mitochondria. FEBS Lett. 1997;412:157–160. doi: 10.1016/s0014-5793(97)00767-9. [DOI] [PubMed] [Google Scholar]
- Visarius TM, Stucki JW, Lauterburg BH. Inhibition and stimulation of long-chain fatty acid oxidation by chloroacetaldehyde and methylene blue in rats. J. Pharmacol. Exp. Ther. 1999;289:820–824. [PubMed] [Google Scholar]
- Volke V, Wegener G, Vasar E, Rosenberg R. Methylene blue inhibits hippocampal nitric oxide synthase activity in vivo. Brain Res. 1999;826:303–305. doi: 10.1016/s0006-8993(99)01253-6. [DOI] [PubMed] [Google Scholar]
- Vutskits L, Briner A, Klauser P, Gascon E, Dayer AG, Kiss JZ, et al. Adverse effects of methylene blue on the central nervous system. Anesthesiology. 2008;108:684–692. doi: 10.1097/ALN.0b013e3181684be4. [DOI] [PubMed] [Google Scholar]
- Wainwright M, Crossley KB. Methylene Blue--a therapeutic dye for all seasons? J. Chemother. 2002;14:431–443. doi: 10.1179/joc.2002.14.5.431. [DOI] [PubMed] [Google Scholar]
- Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011;286:16504–15515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley M, Guo A, Bachman NJ, Lomax MI. Human COX6A1 gene: promoter analysis, cDNA isolation and expression in the monkey brain. Gene. 2000;247:63–75. doi: 10.1016/s0378-1119(00)00121-9. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Nie F, Hevner R, Liu S. Brain cytochrome oxidase: Functional significance and bigenomic regulation in the CNS. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. Plenum Press; New York: 1998. pp. 1–54. [Google Scholar]
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann. Emerg. Med. 1999;34:646–656. doi: 10.1016/s0196-0644(99)70167-8. [DOI] [PubMed] [Google Scholar]
- Wrubel KM, Barrett D, Shumake J, Johnson SE, Gonzalez-Lima F. Methylene blue facilitates the extinction of fear in an animal model of susceptibility to learned helplessness. Neurobiol. Learn. Mem. 2007a;87:209–217. doi: 10.1016/j.nlm.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Wrubel KM, Riha PD, Maldonado MA, McCollum D, Gonzalez-Lima F. The brain metabolic enhancer methylene blue improves discrimination learning in rats. Pharmacol. Biochem. Behav. 2007b;86:712–717. doi: 10.1016/j.pbb.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox. Res. 2006;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]