

Abstract
CRAC channel-mediated Ca2+ entry plays a crucial role in T lymphocyte activation. Activated T cells display enhanced Ca2+ signaling compared with resting T cells; this is partially attributed to activation-induced upregulation of CRAC channel expression. Orai and Stim family genes encode CRAC channel structural elements and regulatory proteins, respectively, but studies of their expression in T cells have led to controversial results. We re-examined Orai and Stim gene expression in resting, activated and Jurkat T cells. Levels of Orai1 transcripts, encoding the human T cell CRAC channel subunit, were not significantly different between resting and activated T cells. The total amount of all Orai transcripts was 2-fold higher in activated T cells than in resting T cells. Orai1 and total Orai transcript levels were significantly higher in Jurkat T cells than those in resting T cells. Stim expression did not vary significantly among cell types. Maximal whole-cell CRAC current amplitudes were 1.4-fold and 2.3-fold higher in activated and Jurkat T cells, respectively, than in resting T cells. Due to the small size of resting T cells, the surface CRAC channel density was 2.5-fold and 1.6-fold higher in resting T cells than in activated and Jurkat T cells, respectively. Predicted the rates of cytosolic Ca2+ elevation calculated using the average values of CRAC channel currents and cell volumes showed that <2-fold increase in the functional CRAC channel expression level cannot account for the enhanced rate of store-operated Ca2+ entry in activated T cells compared with resting T cells.
Key words: human T lymphocytes, T cell activation, CRAC channels, Orai gene, Stim gene
Introduction
Naïve and memory T cells, commonly referred to as resting T cells, bind an antigen displayed on the surface of antigen-presenting cells. An initial response of resting T cells induced by cross-linking of surface T cell receptors (TCR) with an antigen is called activation and is tightly regulated.1,2 TCR engagement causes sustained or oscillatory elevation in cytosolic Ca2+ concentration ([Ca2+]i), which drives transformation of resting T cells into activated T cells by inducing or suppressing the expression of multiple genes.2–10 Activated T cells proliferate, produce a variety of cytokines, and subsequently differentiate into effector T cells committed to secrete specific cytokines that modulate immune response.
Calcium influx through CRAC channels activated by intracellular Ca2+ store depletion induced by TCR stimulation is a major source for [Ca2+]i elevation in human T cells.11 The crucial role of CRAC channels in regulation of T cell functions is underscored by the fact that reduction in CRAC channel activity causes a decrease in T cell Ca2+ responses and development of immunodeficiencies.12 In response to TCR engagement or direct store depletion, activated T cells display enhanced store-operated Ca2+ entry compared with resting T cells13–15 that may be required for T cell effector functions. Augmentation of store-operated Ca2+ entry in activated T cell has been partially attributed to overexpression of intermediate conductance Ca2+-activated (KCa3.1) or voltage-gated (Kv1.3) K+ channels, which hyperpolarize the cell membrane and enhance driving forces for Ca2+ entry via CRAC channels.16–19 In addition, one study reported that enhancement of store-operated Ca2+ influx in activated human T cells correlated with upregulation of the expression of Orai family genes Orai1, Orai2 and Orai3.14 Orai1 upregulation is of particular importance because this gene encodes a pore-forming subunit of human T cell CRAC channel.20 It was also reported that TCR activation stimulated expression of Stim1, a gene encoding CRAC channel-associated endoplasmic reticulum Ca2+ sensor.14 These results suggested that an increase in the number of functional CRAC channels might contribute to the enhanced Ca2+ signaling in activated T cells. However, another study found no changes in Orai1 or Stim1 expression following T cell activation.21 None of the previous studies have directly addressed the issue concerning CRAC channel functional expression by performing a comparative analysis of CRAC channel activity in resting and activated T cells. Thus, it remains unclear whether CRAC channel expression is regulated during T cell activation and whether it contributes to the augmentation of Ca2+ influx in activated T cells.
To resolve these issues, we reexamined Orai and Stim gene expression in relation to two stably expressed house-keeping genes (HKGs) in resting and in vitro-activated human T cells using the real-time quantitative reverse transcription PCR (RT-qPCR) method. We also determined the levels of CRAC channel functional expression in resting and activated T cells by measuring whole-cell CRAC currents using the patch-clamp technique. For comparison, gene expression assays and CRAC current measurements were also performed in Jurkat cells, a human lymphoblastic leukemia T cell line, which is extensively used in CRAC channel studies.
Results
Orai and Stim family gene expression in resting, activated and Jurkat T cells.
Resting CD3+ T cells were freshly isolated from the peripheral blood mononuclear cells of healthy volunteers. Activated T cells were prepared by stimulating resting T cells with anti-CD3 and anti-CD28 monoclonal antibodies (anti-CD3/CD28 mAb), which cross-link TCR. A proliferation assay demonstrated that at day four after stimulation, about 80% of the total T cell population was composed of cells that had undergone at least one round of cell division (Fig. 1A; n = 4), confirming that stimulation with anti-CD3/CD28 mAb transformed the quiescent resting T cells into a proliferating activated T cell population.
Figure 1.
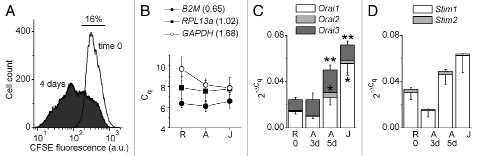
Orai and Stim family gene expression profiles in resting, activated and Jurkat T cells. (A) Representative fluorescence profiles of CFSE-loaded resting T cells (time 0) and 4-day activated T cells from the same donor. The horizontal line and number above it indicate an estimated fraction of undivided cells in activated T cell population. (B) Raw average Cq values for B2M (filled circles), RPL13a (filled squares) and GAPDH (open circles) in resting (R, n = 8), activated primary human T cells (A, n = 8; 3-day and 5-day activated T cell samples were combined) and Jurkat T cells (J, n = 7). Error bars show standard deviation (SD) in each group of samples; numbers in the parentheses indicate Cq SD values for B2M, RPL13a and GAPDH in all samples. (C) Linearized Orai1 (open bars), Orai2 (light grey bars) and Orai3 (dark grey bars) Cq values normalized to the geometric average of B2M and RPL13a Cq values in resting T cells (R, n = 8), 3-day and 5-day activated primary human T cells (A 3d, n = 3; and A 5d, n = 6) and Jurkat T cells (J, n = 7). Data presented as mean ± SE. (*) indicates that mean amount of transcripts of a specific Orai homolog is significantly different from that in resting T cells (independent Student's t test, p < 0.05). (**) indicates that mean cumulative amount of all Orai transcripts is significantly different from that in resting T cells (independent Student's t rest, p < 0.05). (D) Linearized Stim1 (open bars) and Stim2 (light grey bars) Cq values normalized to the geometric average of B2M and RPL13a Cq values in resting T cells (R, n = 8), 3-day and 5-day activated primary human T cells (A 3d, n = 3; and A 5d, n = 6) and Jurkat T cells (J, n = 7). Data presented as mean ± SE. n, number of samples. Each primary resting T cell sample was obtained from a different donor. Activated primary T cell samples are from the same donors as resting T cell samples.
Because quantitative assessment of target gene expression requires normalization to the amount of reference gene transcripts, we first explored whether there were variations among T cell types in the expression of three HKGs, beta-2 microglobulin (B2M), ribosomal protein L13a (RPL13a) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), previously shown to be stably expressed in T cells.22,23 Comparative quantification cycle (Cq), also known as threshold cycle (Ct), method analysis of RT-qPCR assays showed that standard deviations (SD) of the raw Cq values of B2M and RPL13 in all samples were 0.65 and 1.0, respectively (Fig. 1B), whereas Pearson correlation coefficient was 0.81. These results indicate that according to the established criteria,22,24,25 both B2M and RPL13a were stably expressed in resting, activated and Jurkat T cells. For GAPDH, the SD of raw Cq values was 1.68 in all samples and its expression increased >2-fold in activated and Jurkat T cells compared with resting, which indicated a lack of stability. Based on these results, we used B2M and RPL13a as reference genes, whereas GAPDH was excluded from further consideration.
Using a geometric average of B2M and RPL13a raw Cq values for normalization, we determined the relative abundance of Orai1, Orai2, Orai3, Stim1 and Stim2 transcripts in resting, 3-day and 5-day activated primary human T cells and Jurkat T cells (Fig. 1C and D). In all primary human T cell samples, the amounts of Orai2 transcripts were 6-fold to 20-fold lower than those of Orai1 or Orai3 (Fig. 1C). A comparison of expression levels of each Orai homolog between primary human T cell samples revealed a significant 5-fold increase in the amount of Orai2 transcripts in 5-day activated T cells compared with that in resting T cells. Although the relative amounts of each of Orai1 or Orai3 transcripts were 1.8- and 3-fold, respectively, higher in 5-day activated T cells than those in resting T cells, the differences between means were not statistically significant. Nevertheless, the total amounts of Orai1 and Orai3 transcripts were significantly (2-fold) higher in 5-day activated T cells than that in resting T cells. On average, the total amount of all Orai transcripts (Orai1, Orai2 and Orai3) increased by a factor of 2 in 5-day activated primary human T cells, compared with that in resting T cells. The levels of Orai transcripts in 3-day activated T cells were not different from those in resting T cells. In Jurkat cells, the levels of Orai1 transcripts and the total amount of all Orai transcripts were 3.9-fold and 2.9-fold, respectively, higher than those in primary human resting T cells (Fig. 1C). The differences in the expression of any Orai homolog or total Orai transcript levels between primary human activated T cells and Jurkat cells were insignificant. The Stim1 transcripts were 10-fold more abundant than Stim2 transcripts in all samples. Neither the total amount of all Stim transcripts nor levels of any Stim homolog transcript were significantly different between samples (Fig. 1D). These data indicate that TCR crosslinking weakly stimulates Orai but not Stim family gene expression. We next performed a functional assay to determine whether the number of functional CRAC channels changes after TCR activation.
CRAC channel current (ICRAC) measurements in resting, activated and Jurkat T cells.
CRAC channels were activated in nominally Ca2+-free extracellular solution by depleting the store with thapsigargin, an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPase and inositol-1,4,5-trisphosphate, an activator of Ca2+ release from the endoplasmic reticulum. Calcium current through CRAC channels (Ca2+-ICRAC), was evoked by adding 20 mM Ca2+ to the bath solution (Fig. 2A). A divalent cation-free (DVF) bath solution was subsequently applied to evoke a larger amplitude Na+ current through the CRAC channels (Na+-ICRAC). In all cells tested, application of 20 mM Ca2+-containing or DVF solutions produced measurable currents in both resting and activated T cells. The recorded currents were identified as Ca2+-ICRAC and Na+-ICRAC based on the delayed response to the extracellular Ca2+ application resulting from Ca2+-dependent potentiation (Fig. 2A), rapid current inactivation in DVF bath solution (Fig. 2A), and inwardly rectifying current-voltage relationships displaying the reversal potentials expected for Ca2+ and Na+ currents (Fig. 2B and C). Under our experimental conditions, voltage-gated Ca2+ currents were not detectable in resting or activated primary human T cells, or in Jurkat cells.
Figure 2.

CRAC currents in resting, activated and Jurkat T cells. (A) Representative time courses of Ca2+-ICRAC and Na+-ICRAC recorded at −100 mV in resting (R, open circles) and activated (A, filled circles) primary human T cells. The Ca2+-free (0 Ca), 20 mM Ca2+-containing (20 Ca) and divalent cation-free (DVF) solutions were applied as indicated. Cm values for each cell are indicated in parentheses. (B and C) Ca2+-ICRAC (B) and Na+-ICRAC (C) evoked by voltage ramp from −120 mV to +100 mV at time points indicated with arrowheads and arrows in (A). (D) Transmitted light images of primary human resting (left part) and activated (right part) T cells. White arrows show where measurements of cell diameters were performed. Bars, 5 µm. (E–G) Average values for Cm (E) and surface densities of Ca2+-ICRAC (F) and Na+-ICRAC (G) at −100 mV membrane potential in resting (R, open bars), activated (A, black bars), and Jurkat (J, grey bars) T cells. An asterisk (*) indicates that differences between means are significant (p < 0.01, independent t test). n, number of cells. Cells were from two male and two female donors.
On average, the maximal amplitudes of Ca2+-ICRAC and Na+-ICRAC measured at a membrane potential of −100 mV were 1.4-fold and 2.3-fold higher in activated and Jurkat T cells, respectively, than in resting T cells (Fig. 2A–C, Table 1 and Sup. Fig.), indicating that activated and Jurkat T cells expressed a larger number of functional CRAC channels per cell than resting T cells. However, activated and Jurkat T cells were larger in size than resting T cells (Fig. 2D). Consequently, the average value of cell capacitance (Cm), which is proportional to the cell surface area, of activated or Jurkat T cells was ∼3.7-fold larger than that of resting T cells (Fig. 2E). Normalization of the ICRAC values to the corresponding Cm values revealed that Ca2+-ICRAC and Na+-ICRAC surface densities were significantly lower in activated and Jurkat T cells compared with those in resting T cells (Fig. 2F and G). An important question that arises from these findings is whether a larger number of CRAC channels in activated T cells than in resting T cells provide sufficient Ca2+ entry to compensate for the activation-induced increase in cell size. We addressed this question by estimating the rates of Ca2+ accumulation per cell volume per unit time in intact resting, activated and Jurkat T cells using average values of CRAC channel currents, cell volumes and a number of assumptions based on the results of previous studies.
Table 1.
Maximal CRAC channel currents, CRAC channel densities, and morphometric parameters of resting, activated, and Jurkat T cells
| T cell type | Ca2+-ICRAC maximal amplitude at −100 mV (pA) | Na+-ICRAC maximal amplitude at −100 mV (pA) | Number of channels per cell* | Cell surface area (µm2)** | Channel surface density (channels/µm2) | Cell diameters (µm)*** | Cell volume (fL)**** |
| Resting | −5.3 ± 0.8 (n = 24) | −26.1 ± 3.0 (n = 19) | ∼1,400 | 198.6 ± 8.8 (n = 24) | ∼7 | 6.4 ± 0.03 (n = 101) | 137.2 ± 2.2 (n = 101) |
| Activated | −7.6 ± 0.8 (n = 32) | −52.0 ± 6.4 (n = 29) | ∼2,000 | 741.1 ± 26.1 (n = 32) | ∼2.7 | 11.8 ± 0.1 (n = 122) | 894 ± 34.9 (n = 122) |
| Jurkat | −12.5 ± 1.3 (n = 25) | −62.4 ± 7.0 (n = 21) | ∼3,300 | 744.2 ± 37.2 (n = 25) | ∼4.4 | 12.3 ± 0.16 (n = 143) | 1049.7 ± 38.3 (n = 143) |
Average ± SE are presented; n is number of cells.
Calculated using an estimated value of unitary CRAC channel amplitude of 3.8 fA at −110 mV in 20 mM Ca2+ Ringer solution.36
Calculated from Cm values assuming the cell membrane specific capacitance of 0.01 pF µm−2.
Measured from transmitted light images as shown in Figure 2D.
Calculated from cell diameters measured in transmitted light images.
Estimated rates of initial [Ca2+]i elevation following CRAC channel activation in resting, activated and Jurkat T cells.
We assumed that the membrane potential during CRAC channel-mediated Ca2+ influx was −50 mV in intact resting T cells26 and −90 mV in intact activated and Jurkat T cells.27–29 Membrane hyperpolarization in activated and Jurkat T cells is caused by overexpression of Ca2+-activated KCa1.3 or KCa2.2 channels, respectively.16,30 We calculated the total charge (Q) that entered a cell within the first 60 s after Ca2+-ICRAC activation by integrating the average Ca2+-ICRAC recorded at −50 mV or −90 mV in 20 mM Ca2+-containing solution in resting, activated and Jurkat T cells (Sup. Information). Then, we estimated the total charge that would enter the cell at a physiologically relevant concentration of extracellular Ca2+ (2 mM) by scaling down the Q value by a factor of 0.1. From the adjusted Q values we determined that the average rates of total Ca2+ accumulation per cell would be 80 amolmin−1cell−1, 260 amolmin−1cell−1 and 350 amolmin−1cell−1, in resting, activated and Jurkat T cells, respectively.
Micrivilli and raffles on T cell surface dramatically increase the cell surface area without significant increase in the cell volume,31 thus the T cell volume cannot be accurately calculated from Cm measurements. Therefore, we measured average cell diameters in transmitted light images so that cell protrusions and microvilli were excluded from consideration (Fig. 2D). Assuming cells are spherical, the average total cell volumes calculated from the measurements of cell diameters were 137 fL, 894 fL and 1,050 fL, in resting, activated and Jurkat T cells, respectively (Table 1), which are comparable with previously reported values of 142 fL and 520 fL for resting and activated T cells, respectively, calculated from transmitted electron microscopic images.32 Using the values of cell volume determined from the transmitted light cell images and the values of total cell surface area determined from Cm values (Table 1), we calculated the surface-area-to-volume ratios to be 1.44 µm2µm−3, 0.82 µm2µm−3 and 0.71 µm2µm−3 in resting, activated and Jurkat T cells, respectively.
Assuming that 85% of the total cell volume is occupied by the cytosol and nucleus,32,33 and that buffering capacity of the cytosol is 100,33,34 we estimated that rates of [Ca2+]i rise during Ca2+ entry via maximally activated CRAC channels were 110 nM/s, 57 nM/s and 65 nM/s in resting, activated and Jurkat T cell, respectively. Although this is a rough estimate given that many parameters used for this calculation are uncertain, it indicates that the average rate of [Ca2+]i rise in resting T cells should be ∼2-fold higher than that in activated or Jurkat T cells.
Discussion
Here we have shown that the total amount of homologous Orai transcripts increased by factor of two in 5-day activated T cells relative to that in resting T cells, which is comparable with a previously reported ∼1.5-fold increase in Orai1, Orai2 and Orai3 transcript levels in 3-day activated T cells.14 However, we did not detect significant differences in transcript levels of Orai1, a gene encoding human T cell CRAC channel pore-forming subunit,35 between resting and activated primary human T cells. This is consistent with a previous report showing that Orai1 expression did not change significantly after T cell activation.21 It is notable that relative abundance of Stim transcripts did not change significantly after activation, indicating that genes encoding key regulators of CRAC channel gating are stably expressed in resting and activated T cells. The significance of 5-fold increase in Orai2 expression following activation is not clear because the contribution of Orai2 protein in store-operated Ca2+ influx remains undetermined.20 An increase in the total amount of Orai homologous transcripts following T cell activation may result in formation of hetero-multimeric channels with properties distinct from those of the canonical CRAC channel.20 Taken together, our data indicate that expression of homologous Orai genes is upregulated after activation but this upregulation is weak compared with activation-induced upregulation of other channel genes. For example, KCa3.1 transcript levels increased 10-fold in mitogen-activated human T cells,17 whereas levels of TRPV1 and TRPC3 transcripts increased 6-fold and 8-fold, respectively, in anti-CD3/CD28 mAb-activated T cells21 compared with those in resting T cells.
Consistent with the weak upregulation of the Orai gene expression, our analysis of CRAC channel functional expression revealed that, on average, maximal ICRAC amplitudes were only 1.4-fold and 2.4-fold higher in primary human activated T cells and Jurkat cells, respectively, compared with those in resting T cells. Using an estimated value of unitary CRAC channel amplitude of 3.8 fA at −110 mV in 20 mM Ca2+ Ringer solution,36 we calculated that maximal numbers of functional CRAC channels per cell were 1,400 and 2,000 in resting and activated primary human T cells, respectively. In Jurkat cells, an average estimated number of CRAC channels per cell was 3,300 (ranging from 1,300 to 6,000 channels per cell), which is in a reasonable agreement with a previous estimation of 5,000–10,000 CRAC channels per Jurkat cell.36
The less than 2-fold increase in the number of functional CRAC channels per cell observed upon activation is much smaller than the previously reported 50-fold increase in the number of KCa3.1 channels per cell in activated T cells compared with resting T cells.16 Moreover, despite the fact that resting T cells had a lowest number of CRAC channels per cell, the CRAC channel surface density in resting T cells was 2.5-fold and 1.6fold higher than that in activated and Jurkat T cells, respectively, due to the larger surface area of activated and Jurkat T cells (Table 1). This finding differs from our previous report that CRAC channel surface density increased after activation.13 The apparent discrepancy is due to the fact that under experimental conditions used in the previous study, the Mg2+-inhibited cation currents surpassed CRAC channel currents36 causing an overestimation of the CRAC channel number in activated T cells.
Calculations based on the average values of ICRAC amplitude, cell volume and expected values of membrane potential showed that the initial rate of [Ca2+]i elevation caused by Ca2+ entry via CRAC channels in resting T cells should be 2-fold higher than that in activated and Jurkat T cells. This result is inconsistent with previous studies that reported a 1.6-fold to 4-fold increase in the initial rate of [Ca2+]i elevation following activation of the store-operated Ca2+ entry in activated T cells compared with that in resting T cells.13,14 Thus, these results strongly indicate that an increase in the number of CRAC channels alone cannot account for the enhanced Ca2+ signaling in activated T cells compared with resting T cells.
Other mechanisms differentially expressed in resting and activated T cells that modulate Ca2+ influx via CRAC channels are likely to be responsible for activation-induced strengthening of Ca2+ responses. For example, a recent study reported that hydrogen peroxide suppresses store-operated Ca2+ entry, presumably via modulation of Orai1-mediated current, in naïve but not in activated T cells, indicating that CRAC channel activity may be suppressed by reactive oxygen species in resting but not activated T cells.37 Consistent with the idea that CRAC channel activity may be suppressed in resting T cells under certain conditions, we found that the rate of total Ca2+ accumulation in resting T cells under whole-cell patch-clamp conditions was 2-fold higher than previously reported uptake rate of 45Ca2+ in mitogen-stimulated intact resting T cells6 (80 amolmin−1cell−1 versus 40 amolmin−1cell−1, respectively).
CRAC channel activity can be also modulated by protein kinases,38 [Ca2+]i levels in the vicinity of CRAC channels,39–41 and Ca2+ levels within the store,42 which depends on activity of intracellular Ca2+ release channels.43,44 In addition, human T cells express a number of Ca2+-permeable transient receptor potential (TRP) channels, some of which are significantly upregulated after activation.21,45 TCR stimulation or CRAC channel activation following store depletion may stimulate Ca2+ influx via TRP channels in activated T cells by multiple mechanisms, including enhancing driving forces for Ca2+ due to activation of KCa channels and consequent membrane hyperpolarization, elevating [Ca2+]i, or activating intracellular signaling cascades linked to TRP channels.16,46,47 It is likely that upregulation of Ca2+ signaling requires a combination of several factors that modulate CRAC and/or TRP channel activity in activated T cells in the absence of marked upregulation of CRAC channel expression. Because activated T cells exist in multiple functional states, a future challenge will be to identify those factors in each T cell subset, which may lead to identifying molecular targets for controlled manipulation of effector T cell functions and immune response.
Materials and Methods
T cell cultures and chemicals.
Peripheral blood samples were collected from healthy human subjects of both genders and different ethnic backgrounds. All procedures involving human subjects were approved by UC Davis Internal Review Board. Venous blood was collected by venipuncture into sodium heparin-containing collection tubes (Becton-Dickinson, Franklin Lakes, NJ). CD3+ resting T cells were purified from the whole blood by a negative selection method using the RosetteSep® Human T Cell Enrichment Cocktail (StemCell Technologies, Vancouver, BC, Canada) and RosetteSep® Density Medium (StemCell Technologies) according to the manufacturer's instructions. After isolation, resting T cells were kept in cell culture medium at the density of 0.5 × 106 cells/ml for 2–4 h before the experiment. A fraction of resting T cells was activated with 5–10 µg/ml anti-CD3 mAb (Miltenyi Biotech, Auburn, CA) coated on cell culture dishes and 1–5 µg/ml soluble anti-CD28 mAb and incubated for 3–5 days before analysis. Jurkat cells (clone E6-1) were purchased from ATCC (Manassas, VA) and maintained in culture according to the ATCC's recommendations. Cell culture medium contained RPMI-1640 supplemented with 12.5% HEPES (Lonza BioWhittaker, Basel, Switzerland), 10% FBS (Omega Scientific, Tarzana, CA), 2% GlutaMAX™-I (Invitrogen, Carlsbad, CA), 1% RPMI 1640 vitamin solution, 1% RPMI 1640 amino acids solution, 1% sodium pyruvate and 0.03% β-mercaptoethanol. Cells were kept at 37°C in a humidified cell culture incubator containing 5% CO2. Unless otherwise indicated, all chemicals were from Sigma-Aldrich (St. Louis, MO).
Cell proliferation assay.
A cell division track assay was performed using the carboxyfluorescein diacetate succinimidyl ester (CFSE) proliferation kit (Invitrogen) as previously described in reference 43. Briefly, resting T cells were washed, resuspended in a phosphate-buffered saline (PBS) containing 4 µM CFSE at the density of 1 × 106 cells/ml and incubated at 37°C for 10 min. Labeling was quenched by adding five volumes of cold RPMI 1640 culture medium containing 10% FBS. After washing three times with RPMI 1640 + 10% FBS, a fraction of CFSE-labeled cells was pelleted down, fixed with 1% paraformaldehyde in PBS, and then analyzed by flow cytometry to establish the CFSE fluorescence profile of undivided cells (time 0). The remaining CFSE-labeled cells were activated using anti-CD3/CD28 mAb. After four days of activation, cells were harvested, washed with PBS and fixed with 1% paraformaldehyde in PBS. CFSE was excited at 488 nm using an argon ion laser and emitted fluorescence intensity was collected using a FACScan flow cytometer and CellQuest software (BD Biosciences, Mountain View, CA). The data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
RT-qPCR analyses.
Five hundred microliters of stabilization solution (1x TransPrep, nucleic acid purification lysis buffer; Applied Biosystems, Foster City, CA) was added to each sample containing from 2 × 106 to 4 × 106 cells and stored at −20°C. Proteinase K (Invitrogen) and two grinding beads (4-mm diameter, stainless steel beads; SpexCertiprep, Metuchen, NJ) were added to the samples and the samples were homogenized in a GenoGrinder 2000 (SpexCertiprep) for 2 min at 1,000 strokes/min. The resulting lysate was allowed to stand for at least 1 hour at −20°C to reduce foam, and then the protein was digested at 56°C for 30 min. Total RNA was extracted from 200 µl of lysate of each sample using a 6100 Nucleic Acid PrepStation (Applied Biosystems) according to the manufacturer's instructions. RNA concentrations ranged from 90–200 ng/µl as determined by measuring absorbance at 260 nm.
First-strand cDNA was generated using the QuantiTect Reverse transcription kit (Qiagen, Valencia, CA) as follows. A mixture of 1 µl genomic DNA Wipeout Buffer, 10 µl RNA and 1 µl RNase-free water was added to each well in a 384-well plate and incubated at 42°C for 2 min then briefly centrifuged. Each sample was digested with DNase and a 1-µl aliquot from each sample was analyzed by qPCR with a reference gene assay designed to detect genomic DNA and cDNA to confirm that all genomic DNA had been digested. Reverse transcription was performed by incubating 0.5 µl Quantitect Reverse Transcriptase, 2 µl 5x Quantitect RT buffer, 0.5 µl RT Primer Mix, 0.5 µl 20 pM Random Primers (Invitrogen), and 4.5 µl RNase free water with each RNA sample at 42°C for 40 minutes; the reaction was inactivated at 95°C for 3 min.
All samples were pre-amplified using Advantage 2 PCR Enzyme System (Clontech, Mountain View, CA) using the conditions described by Dolganov and colleagues.48 Dilutions of 1:100 and 1:1,000 of the pre-amplified material were used for the RT-qPCR analyses. The human RT-qPCR gene expression assays used for B2M, (Hs99999907_m1) RPL13a (Hs01926559_g1), GAPDH (HS99999905_m1), Orai1 (Hs0038567_m1), Orai2 (Hs00259863_m1) Orai3 (Hs00743683_s1), Stim1 (Hs00963373_m1) and Stim2 (Hs00372712_m1) were obtained from Applied Biosystems.
For quantitative RT-PCR, 5 µl of the diluted cDNA sample was added to a TaqMan Fast Universal PCR Master Mix (Applied Biosystems) and TaqMan Gene Expression Assay primer/probe mixes to achieve a final reaction volume of 12 µl according to the manufacturer's instructions. Negative controls were performed using sterile water instead of cDNA templates. The samples were placed in wells of a 384-well plate and amplified in an automated fluorometer ABI PRISM 7900 HTA FAST Real-time PCR System (Applied Biosystems). Amplification conditions used were: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C. Fluorescence signals were collected during the annealing temperature and Cq values were exported with a threshold of 0.1 and a baseline of 3–10 for the genes of interest (GOI) and a range of 1–4 for the HKGs. The comparative Cq method49 was used to calculate linearized levels of each gene of interest relative to the geometric average of HKG, using the formulas:
Linearized levels of GOI relative to HKGs = 2−ΔCq, where
CRAC channel currents measurements.
Whole-cell currents were recorded from resting T cells on the day of isolation and from 5-day activated T cells using an EPC-10 patch-clamp amplifier (HEKA Instruments, Bellmore, NY) and Pulse acquisition software (HEKA Instruments) as described previously in reference 50. Briefly, the recording electrodes were pulled from borosilicate glass (Sutter Instrument, Novato, CA), coated with HIPEC® R6101 Semiconductor Protective Coating (Dow Corning, Midland, MI), and fire-polished. Cells were plated onto glass-bottom recording chambers coated with poly-L-lysine. Experiments were performed in whole-cell voltage-clamp recording configuration at room temperature. Prior to the gigaseal formation, cells were preincubated with 0.5 µM thapsigargin for 8–10 min in nominally Ca2+-free bath solution to deplete the store and activate CRAC channels. After whole-cell contact with a cell was established, the cell was kept for 1 min in Ca2+free bath solution to allow for intracellular solution exchange and “leak” current recording. A liquid junction potential of −13 mV was corrected before each experiment. To augment ICRAC amplitude, the Ca2+-free solution was substituted with 20 mM Ca2+-containing bath solution. Cells were stimulated with voltage ramps from −120 to +100 mV of 50 ms in duration applied every 0.5–2 s from +30 mV holding potential. Currents were sampled at 40 kHz and filtered at 2.9 kHz with a 3-pole Bessel filter. CRAC currents were recorded in 20 mM Ca2+-containing or divalent cation-free bath solutions. “Leak” current traces were averaged and subtracted from all other recorded current traces prior to data analysis.
Solutions were as follows: (1) nominally Ca2+-free bath solution: 140 mM sodium methanesulfonate, 3 mM MgCl2, 10 mM Na-HEPES, 2 mM NaCl; 10 mM glucose, pH 7.4 (adjusted with acetic acid); (2) 20 mM Ca2+-containing bath solution: 115 mM sodium methanesulfonate, 1 mM MgCl2, 10 mM Na-HEPES, 4 mM NaCl, 20 mM Ca(OH)2, 10 mM glucose, pH 7.4 (adjusted with acetic acid); (3) divalent cation-free (DVF) bath solution: 125 mM sodium methanesulfonate, 10 mM Na-HEPES, 5 mM NaCl, 10 mM N-(2-hydroxyethyl) ethylenediamine triacetic acid (HEDTA), 1 mM EDTA, 10 mM glucose, pH 7.4 (adjusted with NaOH); and (4) pipette solution: 125 mM aspartic acid, 15 mM HEPES, 12 mM 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 5 mM MgCl2, 2 mM MgSO4, 20 µM inositol-1,4,5-trisphosphate, pH 7.2 (adjusted with CsOH). BAPTA and inositol-1,4,5-trisphosphate were included in pipette solution to expedite store depletion and prevent Ca2+-dependent CRAC channel inactivation; Mg2+ was included to prevent development of Mg2+-inhibited cation current.
Cell volume calculation from transmitted light images.
Cells were plated onto glass-bottom cell culture dishes coated with poly-L-lysine in Hank's buffered salt solution and allowed to attach to the coverslips for 20 min at room temperature. Transmitted light images of living cells were acquired using LSM 510 confocal scanning imaging system (Carl Zeiss, Gottingen, Germany) and a 63/1.4 oil immersion Zeiss objective. Cell diameters were measured along the perpendicular lines drawn across a cell as shown in Figure 2D using Zeiss LSM Image Browser software. Assuming that cells were spherical, the value of cell volume (V) was calculated for each cell from the average cell diameter using the equation:
where D is an average cell diameter and π = 3.14.
Data analysis.
Statistical analyses were performed using Origin 7 software (OriginLab, Northampton, MA). Unless indicated otherwise, all data are presented as the mean ± standard error of the mean (SE); n = number of experiments. Hypothesis testing was performed using the Student's independent t and Wilcoxon signed-rank nonparametric tests. Differences between means were accepted as statistically significant at p < 0.05.
Acknowledgements
We are highly indebted to the Department of Physiology and Membrane Biology for providing all the facilities for the study of ion channels in human T lymphocytes. We would like to thank the staff of the UC Davis Lucy Whittier Molecular & Diagnostic Core Facility for the help with RT-qPCR experiments, Dr. Liudmila Zakharova for performing the proliferation assay, Dr. Sepehr Dadsetan for collecting samples from human subjects and Drs. Heike Wulff and Jose Puglisi for critical reading and comments on the manuscript.
Abbreviations
- CRAC
Ca2+ release-activated Ca2+
- TCR
T cell receptor
- [Ca2+]i
cytosolic Ca2+ concentration
- RT-qPCR
realtime quantitative polymerase chain reaction
- KCa3.1
intermediate conductance Ca2+-activated potassium channel
- KCa2.2
small conductance Ca2+-activated potassium channel
- Kv1.3
voltage-gated potassium channel
- mAb
monoclonal antibody
- HKGs
housekeeping genes
- B2M
beta-2 microglobulin
- RPL13a
ribosomal protein L13a
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GOI
genes of interest
- ICRAC
CRAC current
- Ca2+-ICRAC
Ca2+ current through CRAC channels
- Na+-ICRAC
Na+ current through CRAC channels
- DVF
divalent cation-free
- Q
charge
- TRP
transient receptor potential
- FBS
fetal bovine serum
- HEDTA
N-(2-hydroxyethyl) ethylenediamine triacetic acid
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- PBS
phosphate buffered saline
- SD
standard deviation
- SE
standard error
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Germain RN, Stefanova I. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Ann Rev Immunol. 1999;17:467–522. doi: 10.1146/annurev.immunol.17.1.467. [DOI] [PubMed] [Google Scholar]
- 2.Favero J, Lafont V. Effector pathways regulating T cell activation. Biochem Pharmacol. 1998;56:1539–1547. doi: 10.1016/s0006-2952(98)00213-5. [DOI] [PubMed] [Google Scholar]
- 3.Killeen N, Irving BA, Pippig S, Zingler K. Signaling checkpoints during the development of T lymphocytes. Curr Opin Immunol. 1998;10:360–367. doi: 10.1016/s0952-7915(98)80176-3. [DOI] [PubMed] [Google Scholar]
- 4.Swain SL. Helper T cell differentiation. Curr Opin Immunol. 1999;11:180–185. doi: 10.1016/s0952-7915(99)80030-2. [DOI] [PubMed] [Google Scholar]
- 5.Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–322. doi: 10.1038/nri798. [DOI] [PubMed] [Google Scholar]
- 6.Metcalfe JC, Pozzan T, Smith GA, Hesketh TR. A calcium hypothesis for the control of cell growth. Biochem Soc Symp. 1980;45:1–26. [PubMed] [Google Scholar]
- 7.Crabtree GR. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 8.Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 9.Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao A. NF-ATp: A transcription factor required for the co-ordinate induction of several cytokine genes. Immunol Today. 1994;15:274–281. doi: 10.1016/0167-5699(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 11.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Ann Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 12.Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fomina AF, Fanger CM, Kozak JA, Cahalan MD. Single channel properties and regulated expression of Ca2+ release-activated Ca2+ (CRAC) channels in human T cells. J Cell Biol. 2000;150:1435–1444. doi: 10.1083/jcb.150.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, et al. Orai1 and Stim1 move to the immunological synapse and are upregulated during T cell activation. Proc Natl Acad Sci USA. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hess SD, Oortgiesen M, Cahalan MD. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J Immunol. 1993;150:2620–2633. [PubMed] [Google Scholar]
- 16.Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, et al. Upregulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- 18.Grissmer S, Nguyen AN, Cahalan MD. Calcium-activated potassium channels in resting and activated human T lymphocytes. Expression levels, calcium dependence, ion selectivity and pharmacology. J Gen Physiol. 1993;102:601–630. doi: 10.1085/jgp.102.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annual review of immunology. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenning AS, Neblung K, Strauss B, Wolfs MJ, Sappok A, Hoth M, et al. TRP expression pattern and the functional importance of TRPC3 in primary human T-cells. Biochim Biophys Acta. 2011;1813:412–423. doi: 10.1016/j.bbamcr.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 22.Mane VP, Heuer MA, Hillyer P, Navarro MB, Rabin RL. Systematic method for determining an ideal housekeeping gene for real-time PCR analysis. J Biomol Tech. 2008;19:342–347. [PMC free article] [PubMed] [Google Scholar]
- 23.Banda M, Bommineni A, Thomas RA, Luckinbill LS, Tucker JD. Evaluation and validation of housekeeping genes in response to ionizing radiation and chemical exposure for normalizing RNA expression in real-time PCR. Mutat Res. 2008;649:126–134. doi: 10.1016/j.mrgentox.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 25.de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, et al. Evidence based selection of housekeeping genes. PLoS One. 2007;2:898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pahapill PA, Schlichter LC. Modulation of potassium channels in intact human T lymphocytes. J Physiol. 1992;445:407–430. doi: 10.1113/jphysiol.1992.sp018931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verheugen JA, Vijverberg HP, Oortgiesen M, Cahalan MD. Voltage-gated and Ca2+-activated K+ channels in intact human T lymphocytes. Noninvasive measurements of membrane currents, membrane potential and intracellular calcium. J Gen Physiol. 1995;105:765–794. doi: 10.1085/jgp.105.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grissmer S, Lewis RS, Cahalan MD. Ca2+-activated K+ channels in human leukemic T cells. J Gen Physiol. 1992;99:63–84. doi: 10.1085/jgp.99.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fanger CM, Rauer H, Neben AL, Miller MJ, Wulff H, Rosa JC, et al. Calcium-activated potassium channels sustain calcium signaling in T lymphocytes. Selective blockers and manipulated channel expression levels. J Biol Chem. 2001;276:12249–12256. doi: 10.1074/jbc.M011342200. [DOI] [PubMed] [Google Scholar]
- 30.Desai R, Peretz A, Idelson H, Lazarovici P, Attali B. Ca2+-activated K+ channels in human leukemic Jurkat T cells. Molecular cloning, biochemical and functional characterization. J Biol Chem. 2000;275:39954–39963. doi: 10.1074/jbc.M001562200. [DOI] [PubMed] [Google Scholar]
- 31.Majstoravich S, Zhang J, Nicholson-Dykstra S, Linder S, Friedrich W, Siminovitch KA, et al. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich Syndrome protein (WASp) for their morphology. Blood. 2004;104:1396–1403. doi: 10.1182/blood-2004-02-0437. [DOI] [PubMed] [Google Scholar]
- 32.Konwinski M, Kozlowski T. Morphometric study of normal and phytohemagglutinin-stimulated lymphocytes. Z Zellforsch Mikrosk Anat. 1972;129:500–507. doi: 10.1007/BF00316745. [DOI] [PubMed] [Google Scholar]
- 33.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donnadieu E, Bismuth G, Trautmann A. Calcium fluxes in T lymphocytes. J Biol Chem. 1992;267:25864–25872. [PubMed] [Google Scholar]
- 35.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 36.Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:24. doi: 10.1126/scisignal.2000672. [DOI] [PubMed] [Google Scholar]
- 38.Kawasaki T, Ueyama T, Lange I, Feske S, Saito N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J Biol Chem. 2010;285:25720–25730. doi: 10.1074/jbc.M109.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J Biol Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 41.Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dadsetan S, Zakharova L, Molinski TF, Fomina AF. Store-operated Ca2+ influx causes Ca2+ release from the intracellular Ca2+ channels that is required for T cell activation. J Biol Chem. 2008;283:12512–12519. doi: 10.1074/jbc.M709330200. [DOI] [PubMed] [Google Scholar]
- 44.Grupe M, Myers G, Penner R, Fleig A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium. 2010;48:1–9. doi: 10.1016/j.ceca.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwarz EC, Wolfs MJ, Tonner S, Wenning AS, Quintana A, Griesemer D, et al. TRP channels in lymphocytes. Handb Exp Pharmacol. 2007:445–456. doi: 10.1007/978-3-540-34891-7_26. [DOI] [PubMed] [Google Scholar]
- 46.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 47.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 48.Dolganov GM, Woodruff PG, Novikov AA, Zhang Y, Ferrando RE, Szubin R, et al. A novel method of gene transcript profiling in airway biopsy homogenates reveals increased expression of a Na+-K+-Cl− cotransporter (NKCC1) in asthmatic subjects. Genome Res. 2001;11:1473–1483. doi: 10.1101/gr.191301. [DOI] [PubMed] [Google Scholar]
- 49.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 50.Thakur P, Fomina AF. Whole-cell recording of calcium release-activated calcium (CRAC) currents in human T lymphocytes. J Vis Exp. 2010 doi: 10.3791/2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.