

Background: β-Hydroxyacid dehydrogenases are ubiquitous enzymes, most of which remain uncharacterized.
Results: Biochemical, crystallographic, and mutational analyses identified uncharacterized Pseudomonas aeruginosa protein PA0743 as a l-serine dehydrogenase and characterized the molecular details of its active site.
Conclusion: PA0743 is the first NAD+-dependent l-serine dehydrogenase potentially involved in serine catabolism.
Significance: Our study provides molecular insights into the mechanism of β-hydroxyacid dehydrogenases.
Keywords: Dehydrogenase, Enzyme Catalysis, Enzyme Kinetics, Enzyme Mutation, Enzyme Structure, Enzymes, Pseudomonas aeruginosa
Abstract
The β-hydroxyacid dehydrogenases form a large family of ubiquitous enzymes that catalyze oxidation of various β-hydroxy acid substrates to corresponding semialdehydes. Several known enzymes include β-hydroxyisobutyrate dehydrogenase, 6-phosphogluconate dehydrogenase, 2-(hydroxymethyl)glutarate dehydrogenase, and phenylserine dehydrogenase, but the vast majority of β-hydroxyacid dehydrogenases remain uncharacterized. Here, we demonstrate that the predicted β-hydroxyisobutyrate dehydrogenase PA0743 from Pseudomonas aeruginosa catalyzes an NAD+-dependent oxidation of l-serine and methyl-l-serine but exhibits low activity against β-hydroxyisobutyrate. Two crystal structures of PA0743 were solved at 2.2–2.3-Å resolution and revealed an N-terminal Rossmann fold domain connected by a long α-helix to the C-terminal all-α domain. The PA0743 apostructure showed the presence of additional density modeled as HEPES bound in the interdomain cleft close to the predicted catalytic Lys-171, revealing the molecular details of the PA0743 substrate-binding site. The structure of the PA0743-NAD+ complex demonstrated that the opposite side of the enzyme active site accommodates the cofactor, which is also bound near Lys-171. Site-directed mutagenesis of PA0743 emphasized the critical role of four amino acid residues in catalysis including the primary catalytic residue Lys-171. Our results provide further insight into the molecular mechanisms of substrate selectivity and activity of β-hydroxyacid dehydrogenases.
Introduction
The β-hydroxyacid dehydrogenases represent a large, structurally conserved superfamily of enzymes that catalyze the NAD+- or NADP+-dependent oxidation of various β-hydroxy acids acting on the common 3-hydroxypropionate moiety of these substrates (1–4). The members of this family are found in all organisms and include 3-hydroxyisobutyrate (HIBA)2 dehydrogenase, 6-phosphogluconate dehydrogenase, d-phenylserine dehydrogenase, tartronate-semialdehyde reductase, and numerous uncharacterized homologues (5,594 sequences, IPR015815, InterPro database) (1, 3). The characterized β-hydroxyacid dehydrogenases have been shown to play important roles in the catabolism of amino acids, hepatic gluconeogenesis, the pentose phosphate pathway, glycerate biosynthesis, and nicotinate fermentation (3, 5, 6). Elevated levels of β-hydroxy acids (3-hydroxyisobutyrate, 2-methyl-3-hydroxyisobutyrate, and 3-hydroxyisovalerate) have been reported in several diseases (hydroxyisobutyric aciduria, brain disorders, and methylmalonic acidemia) that might be related to reduced activity of β-hydroxyacid dehydrogenases (7–9). These proteins are also considered to be promising industrial enzymes for various biocatalytic processes (10–12).
Among sequenced genomes, the number of β-hydroxyacid dehydrogenase genes can range from two to five in Archaea, 11 to 12 in rhizobia and Rhodococcus RHA1, and 16 in Arabidopsis thaliana, suggesting that these enzymes may have unique metabolic roles. β-Hydroxyacid dehydrogenases share low overall sequence similarity (15–30% identity), but they can be identified by the presence of four conserved sequence motifs (3). However, these sequence motifs only suggest the presence of dehydrogenase activity in these proteins, whereas the nature of the substrate used should be determined experimentally. Although β-hydroxyacid dehydrogenases exhibit some sequence similarity to short-chain dehydrogenases, they have been proposed to represent a distinct family of dehydrogenases (1, 3, 13, 14). The first β-hydroxyacid dehydrogenase was purified from pig kidney and biochemically characterized in 1957 (15). This enzyme has been shown to be highly specific to β-HIBA and catalyzed a reversible reaction of NAD-dependent oxidation of this substrate to methylmalonate semialdehyde. The HIBA dehydrogenases purified from Candida rugosa and rabbit liver were also found to be specific to HIBA and were active against both R- and S-HIBA isomers (12, 16). However, the HIBA dehydrogenases purified from Pseudomonas putida E23, rat, Escherichia coli, and Haemophilus influenzae were found to be active toward several substrates (in addition to HIBA) including l- or d-glycerate, 2-methyl-dl-serine, l-serine, phenylserine, d-threonine, hydroxybutyrate, and 3-hydroxypropionate (1, 17–19). Recent works have also revealed that some HIBA dehydrogenases can use NADP+ as a cofactor and identified novel substrates for these enzymes (succinate semialdehyde, 3-hydroxypropane sulfonate, methylglyoxal, and 3-hydroxypropionate) (11, 20). The structurally and biochemically characterized β-hydroxyacid dehydrogenases include the HIBA dehydrogenase TTHA0237 from Thermus thermophilus, 2-(hydroxymethyl)glutarate dehydrogenase Hgd from Eubacterium barkeri, and the sheep 6-phosphogluconate dehydrogenase (2, 4, 5). TTHA0237 is an NADP+-dependent HIBA dehydrogenase that produces methylmalonate semialdehyde as a product, whereas the E. barkeri Hgd catalyzes the NAD+-dependent conversion between (S)-2-(hydroxymethyl)glutarate and (S)-2-formylglutarate. Most of the 6-phosphogluconate dehydrogenases are NADP+-dependent enzymes, which oxidatively decarboxylate 6-phosphogluconate to give ribulose 5-phosphate. Crystal structures have revealed that the first two enzymes have tetrameric oligomerization, whereas the sheep 6-phosphogluconate dehydrogenase is a dimer (2, 4, 5). The protomers of these enzymes have two domains: the N-terminal α/β domain with the nucleotide binding region and the C-terminal helical catalytic domain. These domains are connected by one characteristic long α-helix (α7 in TTHA0237). The inhibitor, mutational, and structural studies with the sheep 6-phosphogluconate dehydrogenase and rat HIBA dehydrogenase demonstrated that a conserved Lys residue (Lys-183 and Lys-173, respectively) plays a critical role in catalysis (1, 3, 5). However, our knowledge of β-hydroxyacid dehydrogenases remains limited, and the vast majority of these enzymes are still uncharacterized.
The genome of the opportunistic pathogen Pseudomonas aeruginosa encodes six members of the β-hydroxyacid dehydrogenase superfamily: PA0743 (annotated as a probable HIBA dehydrogenase), PA1500 (a probable oxidoreductase), PA1576 (a probable HIBA dehydrogenase), PA2199 (a probable dehydrogenase), PA3312 (a probable HIBA dehydrogenase), and PA3569 (a HIBA dehydrogenase), which share 32–62% sequence identity with each other and remain biochemically uncharacterized. Based on the increased 3-hydroxyisobutyrate dehydrogenase activity of the E. coli cell-free extracts containing the PA3569 gene (mmsB) cloned into a pUC18 plasmid, it has been proposed that PA3569 is indeed a HIBA dehydrogenase (21). This is supported by the demonstration of this activity in the purified homologous protein PP4666 (MmsB) from P. putida (54.3% sequence identity) (17). Here, we present the results of biochemical and structural characterization of the P. aeruginosa β-hydroxyacid dehydrogenase PA0743. We have demonstrated that this protein is an NAD+-dependent l-serine dehydrogenase and determined the crystal structure of this protein both in the apoform and in complex with NAD+. The structure and site-directed mutagenesis of PA0743 suggest that several conserved residues including Lys-171 are critical for its enzymatic activity.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were purchased from Sigma. R-HIBA and S-HIBA was a kind gift of Dr. Naoki Kunishima (RIKEN Spring-8 Center, Harima Institute, Japan).
Gene Cloning, Overexpression, and Purification of PA0743
The PA0743 open reading frame was PCR-amplified using chromosomal DNA of the P. aeruginosa PAO1 strain and the cloning primers containing the restriction sites for BamHI and NdeI and was cloned into the modified pET15b vector (Novagen) in which the tobacco etch virus protease cleavage site replaced the thrombin cleavage site and a double stop codon was introduced downstream from the BamHI site (22). The overexpression plasmid was transformed into the E. coli BL21(DE3) Gold strain (Stratagene). PA0743 was overexpressed in E. coli and purified using metal chelate affinity chromatography on nickel affinity resin (Qiagen) with a high yield (>50 mg/liter of culture) and homogeneity (>95%) as described previously (22). Purified proteins were concentrated using a centrifugal membrane concentrator (Millipore), frozen as drops in liquid nitrogen, and stored at −80 °C.
Gel filtration analysis of the oligomeric state of PA0743 was performed with a Superdex 75 16/60 column (Amersham Biosciences) equilibrated with 10 mm HEPES-K (pH 7.5) and 0.2 m NaCl using ÄKTA FPLC (Amersham Biosciences). The column was calibrated with ribonuclease A (13.7 kDa), chymotrypsinogen (25 kDa), ovalbumin (43 kDa), albumin (67 kDa), and aldolase (158 kDa).
Enzymatic Assays
Dehydrogenase activity of PA0743 against various substrates was measured spectrophotometrically by following the increase of absorbance at 340 nm. The assays were performed at 37 °C in a reaction mixture (1 ml) containing 50 mm diethanolamine buffer (pH 11.0), 5 mm NAD+ (or NADP+), 5–15 mm substrate, and 1–10 μg of PA0743. The pH dependence of dehydrogenase activity of PA0743 with l-serine (5 mm) was determined using a polybuffer system described by Heering et al. (23). The formation of aldehydes as the products of the PA0743 reaction was analyzed using a colorimetric assay with 3-methyl-2-benzothiazolinone hydrazone as described previously (24), whereas the production of ammonia was tested using the phenol-hypochlorite method (25). For determination of the Km and Vmax of the wild-type and mutant PA0743, the assays contained substrates at concentrations of 0.025–50.0 mm. Kinetic parameters were determined by nonlinear curve fitting from the Lineweaver-Burk plot using GraphPad Prism (version 4.00 for Windows, GraphPad Software, San Diego, CA).
Site-directed Mutagenesis of PA0743
Site-directed mutagenesis was performed using the QuikChangeTM site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The amino acids selected to be mutated were all changed to alanine. DNA encoding wild-type PA0743 cloned into the modified pET15b was used as a template for mutagenesis. The standard PCR mixture contained 50–100 ng of template DNA and 150–250 ng of each mutagenizing primer. The methylated plasmid was digested with DpnI, and 4 μl of each reaction were used to transform competent DH5α cells. Plasmid was purified from the resulting ampicillin-resistant colonies using the Qiaprep Spin Mini Prep kit (Qiagen), and all mutations were verified by DNA sequencing. Plasmids containing the desired mutations were transformed into the E. coli BL21(DE3) strain, and the mutant PA0743 proteins were overexpressed and purified in the same manner as the wild-type PA0743.
Growth Experiments
The P. aeruginosa wild-type (PAO1) and PA0743 deletion (PW2350) strains were obtained from the P. aeruginosa PAO1 transposon mutant library (University of Washington Genome Center) (26). The strains were grown aerobically at 37 °C (250 rpm) on MOPS minimal medium supplemented with glucose (0.2%) (27) and containing l-serine or other substrates as nitrogen sources (leucine, valine, uracil, and thymine; 1 g/liter). In other growth experiments, the MOPS medium was supplemented with NH4Cl as a nitrogen source (9.5 mm; no glucose), and l-serine or the above mentioned nitrogen compounds were added as carbon sources. The culture growth was followed by measuring the absorbance at 600 nm.
Protein Crystallization
Crystals of PA0743 were grown at 21 °C by the hanging drop vapor diffusion method with 2 μl of protein sample mixed with an equal volume of the reservoir buffer as described previously (28). The crystals of the wild-type PA0743 grew after 1 week in the presence of 4 m ammonium acetate and 0.1 m sodium acetate (pH 5.4). The crystals of the PA0743 complex with NAD+ were obtained by soaking the PA0743 crystals (obtained as described above) in 10 mm NAD+. For diffraction studies, the crystals were stabilized with the crystallization buffer supplemented with 12% ethylene glycol as a cryoprotectant and flash frozen in liquid nitrogen.
Data Collection and Structure Determination
Crystals of selenomethionine-enriched PA0743 protein (both alone and in complex with NAD+) were used to collect diffraction data at a wavelength 0.97921 Å at the APS beamline 19-ID of the Structural Biology Center of the Advanced Photon Source (29). Diffraction data were integrated and scaled using HKL3000 (30). Positions of heavy atoms were found using SHELXD (31) followed by phase improvement with MLPHARE (32) and density modification using DM (33), and an initial model was automatically built using ARP/wARP (34) within the HKL3000 program suite (30, 35). Alternate cycles of manual building and water picking were performed using COOT (36), and restrained refinement against a maximum likelihood target with 5% of the reflections randomly excluded as an Rfree test set was carried out using REFMAC in the CCP4 program suite (37). Additional refinement using translation/libration/screw (TLS) parameterization within CCP4 (38, 39), and Phenix refine from the PHENIX crystallography package (40) was used to further improve the model. The final models were refined to an Rwork and Rfree of 19.7 and 24.8% for the apostructure and 19.1 and 26.2% for the structure of PA0743-NAD complex, respectively. The Ramachandran plot generated by PROCHECK (41) showed good stereochemistry overall with 99.6 and 100% in the most favored and additional allowed regions for the apostructure and PA0743-NAD complex structures, respectively. Data collection and structure refinement statistics for the PA0743 structures are summarized in Table 2.
TABLE 2.
Crystallographic data collection and model refinement statistics
Values in parentheses are for the highest resolution shell. R.m.s., root mean square.
| PA0743 (apo) (Protein Data Bank code 3OBB) | PA0743 (+NAD+) (Protein Data Bank code 3Q3C) | |
|---|---|---|
| Data collection | ||
| Space group | P6222 | P6222 |
| Cell dimensions (Å) (a, c) | 92.6, 124.9 | 92.7, 126.5 |
| Wavelength | 0.97921 | 0.97921 |
| Resolution (Å) | 30.32-2.2 (2.28-2.2) | 50-2.3 (2.38-2.3) |
| Rmerge | 0.116 (0.747) | 0.096 (0.743) |
| I/σI | 32.6 (3.6) | 48.7 (2.2) |
| Completeness (%) | 99.7 (100.0) | 99.2 (94.0) |
| Redundancy | 10.5 (10.7) | 15.2 (6.7) |
| Refinement | ||
| Resolution (Å) | 30.32-2.20 | 37.39-2.3 |
| Number of reflectionsa | 15,695/837 | 14,766/750 |
| Rwork/Rfree | 0.197/0.243 | 0.191/0.262 |
| Number of atoms | 2,251 | 2,200 |
| Protein | 2,142 | 2,125 |
| Major ligand (NAD) | 44 | |
| Solvent | 70 | 31 |
| Other | 31 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.015 | 0.008 |
| Bond angles (°) | 1.65 | 1.33 |
| Ramachandran plot | ||
| Most favored (%) | 91.6 | 91.0 |
| Additionally allowed (%) | 7.9 | 9.0 |
| Disallowed (%) | 0.0 | 0.0 |
a Number of reflections overall and in the test set.
Protein Data Bank Accession Codes
Coordinates and structure factors have been deposited under accession codes 3OBB (apostructure) and 3Q3C (NAD+ complex).
RESULTS AND DISCUSSION
Dehydrogenase Activity of PA0743
Sequence analysis of PA0743 revealed the presence of four conserved sequence motifs of β-hydroxyacid dehydrogenases (3) involved in dinucleotide cofactor binding (GLGXMG; motif-1), substrate binding (DAPVSGG; motif-2), catalysis (GXXGXGXXXKXXN; motif-3) with a catalytic Lys (Lys-171 in PA0743), and cofactor-binding (KDL; motif-4) (Fig. 1). PA0743 shares 32.9–38.2% sequence identity with the biochemically characterized HIBA dehydrogenases from rat and T. thermophilus (Fig. 1). In addition, PA0743 shows rather high similarity (62% sequence identity) to PA3569, which has been proposed to be a HIBA dehydrogenase in P. aeruginosa (Fig. 1) (21). This level of sequence similarity between PA0743 and PA3569 suggests that PA0743 might be another HIBA dehydrogenase in P. aeruginosa.
FIGURE 1.

Structure-based sequence alignment of PA0743 and several β-hydroxyacid dehydrogenases. The secondary structure elements of PA0743 and human HIBA dehydrogenase are shown above and below the alignment, respectively. Residues conserved in all aligned β-hydroxyacid dehydrogenases are boxed. The residues comprising the four characteristic β-hydroxyacid dehydrogenase sequence motifs (3) are highlighted in different colors and labeled below the alignment. The PA0743 residues mutated to Ala in this work are marked with an asterisk above the alignment and numbered. The proteins compared are PA0743 (UniProTKB Q9I5I6), PA3569 (P28811), rat HIBA dehydrogenase (P29266), the HIBA dehydrogenase TTHA0237 from T. thermophilus (Q5SLQ6), 2-(hydroxymethyl)glutarate dehydrogenase Hgd from E. barkeri (Ebar) (Q0QLF5), and the predicted human HIBA dehydrogenase (HIBADH) (P31937).
To determine the substrate specificity of PA0743, its gene was overexpressed in E. coli, and recombinant PA0743 was affinity-purified with a high yield to over 95% homogeneity as assessed by SDS-PAGE gels (not shown). Purified PA0743 was tested for the presence of dehydrogenase activity against both S-HIBA and R-HIBA using NAD+ or NADP+ as the electron acceptor and the reaction mixture described for the HIBA dehydrogenase TTHA0237 (4). PA0743 showed low NAD+-dependent activity with both substrates (Fig. 2), and therefore it was tested for dehydrogenase activity against a broader range of substrates described in previous works on β-hydroxyacid dehydrogenases. With these substrates, PA0743 demonstrated significant NAD+-dependent dehydrogenase activity against l-serine, methyl-dl-serine, methyl-(S)-(+)-3-hydroxy-2-methylpropionate, d-glycerate, tert-butyl-3-hydroxypropionate, and methyl-2,2-dimethyl-3-hydroxypropionate as well as detectable activity toward several other substrates (Fig. 2). Previously, a number of HIBA dehydrogenases have been shown to be active against several substrates including l-serine, but their activity or affinity toward l-serine was lower than those with HIBA (1, 2, 4, 17).
FIGURE 2.

Substrate profile of PA0743. Dehydrogenase activity of purified PA0743 against l-serine and other substrates measured in the presence of 5 mm NAD+ (as described under “Experimental Procedures”) is shown. The following substrates were also used for screening and produced negative results: (S)-l-β-hydroxybutyric acid, (R)-(−)-3-hydroxybutyric acid, 2-hydroxyisobutyric acid, methyl-2-hydroxyisobutyric acid, ethyl-2-hydroxyisobutyric acid, 2-hydroxybutyric acid, dl-malic acid, and dl-homoserine. M(R)-3H-2M-Propionate, methyl-(R)-3-hydroxy-2-methylpropionic acid; M-2,2dM-3H-Propionate, methyl-2,2-dimethyl-3-hydroxypropionic acid; tertB-3H-Propionate, tert-butyl-3-hydroxypropionic acid; M(S)-3H-2M-Propionate, methyl-(S)-3-hydroxy-2-methylpropionic acid. Each bar represents an average of the results from at least two independent determinations, with S.D. indicated by error bars (in all figures).
Like the β-hydroxyacid dehydrogenases from rat, rabbit liver, and T. thermophilus (1, 4, 16), PA0743 exhibited maximal activity at alkaline pH (pH 10–11). PA0743 can also use NADP+ as a cofactor for the oxidation of l-serine, but this activity was significantly lower than that with NAD+ (4–6%). This is consistent with the presence of an Asp (Asp-31) downstream of the motif-1 in the PA0743 sequence, which is known to determine the preference of HIBA dehydrogenases for NAD+, whereas the NADP+-dependent enzymes usually contain an Arg at this position (as in TTHA0237) (Fig. 1). The biochemical analysis of the PA0743 reaction mixture after incubation with l-serine revealed no presence of free ammonium, suggesting that PA0743 is not a deaminating dehydrogenase and, like other β-hydroxyacid dehydrogenases (3), this enzyme catalyzes a cofactor-dependent oxidation of a hydroxy acid substrate to a corresponding semialdehyde. This was confirmed by the detection of an aldehyde-containing product in the PA0743 reaction mixture using a hydrazone-based spectrophotometric assay for aldehydes (data not shown). Thus, with l-serine or methyl-l-serine as substrates, the proposed reaction product of PA0743 will be 2-aminomalonate semialdehyde or 2-aminomethylmalonate semialdehyde (Fig. 3). With all substrates, PA0743 exhibited classical, hyperbolic saturation kinetics with the lowest Km to l-serine and methyl-dl-serine (Table 1). The catalytic efficiency of PA0743 with l-serine as substrate is comparable with that reported for other β-hydroxyacid dehydrogenases (4, 17). Compared with the biochemically characterized NADP+-dependent l-serine dehydrogenases from the short-chain dehydrogenase/reductase superfamily (Agrobacterium tumefaciens ICR1600 and E. coli YdfG) (42–44), PA0743 has a lower Km and higher catalytic efficiency toward l-serine (Table 1).
FIGURE 3.
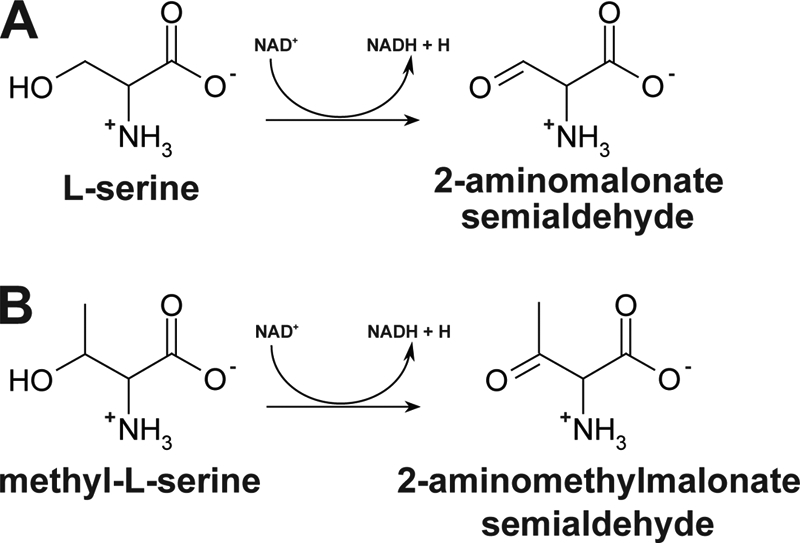
Proposed reactions catalyzed by PA0743. The enzyme catalyzes the NAD+-dependent dehydrogenation of l-serine (A) or methyl-l-serine (B) at C3, producing 2-aminomalonate semialdehyde or 2-aminomethylmalonate semialdehyde, respectively, and NADH.
TABLE 1.
Kinetic parameters of PA0743 with various substrates
| Protein and variable substrate | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| mm | μmol/min mg protein | s−1 | m−1s−1 | |
| Wild type | ||||
| l-Serine | 2.5 ± 0.1 | 18.7 ± 0.3 | 10.4 ± 0.2 | 0.4 × 104 |
| Methyl-dl-serine | 2.4 ± 0.2 | 17.2 ± 0.4 | 9.6 ± 0.2 | 0.4 × 104 |
| dl-Glyceric acid | 19.8 ± 0.4 | 10.4 ± 0.1 | 5.8 ± 0.1 | 0.3 × 103 |
| MDHPa | 17.4 ± 1.2 | 20.9 ± 0.7 | 11.6 ± 0.4 | 0.7 × 103 |
| NAD+ | 3.4 ± 0.2 | 2.8 ± 0.1 | 1.6 ± 0.1 | 0.5 × 103 |
| T96A | ||||
| l-Serine | 12.3 ± 1.8 | 2.6 ± 0.2 | 1.4 ± 0.1 | 0.1 × 103 |
| S122A | ||||
| l-Serine | 10.6 ± 0.5 | 0.8 ± 0.02 | 0.4 ± 0.01 | 0.4 × 102 |
| N175A | ||||
| l-Serine | 4.2 ± 0.2 | 4.9 ± 0.1 | 2.7 ± 0.1 | 0.6 × 103 |
| Y219A | ||||
| l-Serine | 11.0 ± 2.4 | 1.4 ± 0.1 | 0.8 ± 0.1 | 0.7 × 102 |
a Methyl-2,2-dimethyl-3-hydroxypropionate.
Crystal Structure of PA0743
PA0743 was crystallized using the hanging drop vapor diffusion protocol, and its crystal structure was solved to 2.2-Å resolution using the single wavelength anomalous diffraction method (Table 2). The structure revealed a two-domain protein with the N-terminal Rossmann fold domain (α1–α8 and β1–β9; residues 1–162) and the C-terminal all-α domain (α10–α15; residues 198–296) connected by the long α9 helix (residues 166–195) (Fig. 4A). The N-terminal domain interacts with the short fragment of the long α9 helix, whereas the C-terminal domain is wrapped around this helix, covering almost half of its length at three sides and creating a large cleft between the two domains (Fig. 4A). Analysis of the crystal contacts in the final model using the quaternary prediction server PISA suggests that PA0743 forms a tetramer with four protomers arranged as a dimer of dimers (Fig. 4). This is consistent with the results of gel filtration experiments that showed that PA0743 exists as a tetramer in solution (120.2 ± 7 kDa; predicted monomer molecular mass, 30.8 kDa). In the PA0743 dimer, two protomers are connected by the extensive non-polar and polar interactions between the side chains of residues located mainly on the long α9 helix as well as on the α13, α14, α15, α10 (Arg-208), and α11 helices (Fig. 4B). Two dimers of PA0743 are connected by polar and non-polar interactions between the α-helices (α13, α14, and α15 of the C-terminal domain creating a butterfly-like tetrameric structure (Fig. 4C)). A Dali search (45) for the PA0743 structural homologs identified several β-hydroxyacid dehydrogenases including the predicted human HIBA dehydrogenase (Protein Data Bank codes 2GF2 and 2I9P; Z-score, 40.8–41.4; root mean square deviation, 1.2–1.4 Å), tartronate semialdehyde reductase GarR from Salmonella typhimurium (Protein Data Bank codes 1VPD and 1TEA; Z-score, 37.1; root mean square deviation, 2.1 Å), and the 2-(hydroxymethyl)glutarate Hgd from E. barkeri (Protein Data Bank code 3CKY; Z-score, 35.8; root mean square deviation, 1.9 Å). The high Z-scores with relatively low sequence similarity (30–40% sequence identity) suggest strong evolutionary conservation of this structural fold.
FIGURE 4.

Crystal structure of PA0743: overall structure of protomer, dimer, and tetramer. A, the protomer structure is shown in three orientations (related by 90° rotation) with the N-terminal domain colored orange, the C-terminal domain in cyan, and the long α9 helix connecting the two domains in green. The N and C termini are labeled (N and C). B, the dimer structure is shown in two orientations (related by 90° rotation) with the protomers colored in orange and green. C, the PA0743 tetramer with four protomers shown in different colors (cyan, magenta, orange, and green).
The apostructure of PA0743 also revealed an additional weaker electron density located in the interdomain cleft. We have modeled this density as being partially occupied (occupancy of 0.5) by a weakly bound HEPES molecule based on its shape and presence in the crystallization solution (Fig. 5, A and C). In the HIBA dehydrogenase TTHA0237, the interdomain cleft accommodates the protein active site with the NADP cofactor and sulfate ion bound near the catalytic Lys-165 (4). In the structure of the E. coli 6-phosphogluconate dehydrogenase complex with 6-phosphogluconate (Protein Data Bank code 3FWN), the substrate is also bound near the catalytic Lys-183, and the long axis of the substrate is oriented almost perpendicular to the long α8 helix (46). In the PA0743 structure (Fig. 6A), the modeled HEPES molecule is bound deeply in the interdomain cleft with its sulfonic group located near the predicted catalytic Lys-171 (3.5–3.9 Å) and conserved Asn-175 (also motif-3; 2.4–3.2 Å) and Asp-247 (motif-4; 3.1 Å), whereas its hydroxyl group is close to the conserved Tyr-219 (3.4 Å). The modeled HEPES molecule likely mimics the position of a substrate (l-serine or methyl-l-serine) bound in the PA0743 substrate-binding site, which is formed by the residues from two protomers forming a dimer (Figs. 5A and 6A). With l-serine or methyl-l-serine bound in the PA0743 active site, the substrate amino, carboxyl, and methyl groups can be easily accommodated in the substrate-binding site and can potentially interact with side chains of conserved Ser-122 (motif-2), Ser-210 (from another subunit), Lys-246 (motif-4), or Phe-239 or with the Gly-123 and Gly-124 main chain groups (motif-2) (2.9–5.7 Å to the HEPES sulfonic group). The conserved Phe-239 restricts binding of larger substrates on one side, and the motif-2 residues (Ser-122, Gly-123, and Gly-124) confine the active site on another side. The structure of the PA0743-HEPES complex suggests that homologous dehydrogenases (HIBA-like) bind their substrates differently from 6-phosphogluconate dehydrogenases with the substrate long axes positioned parallel to the long α7 helix and oriented toward the Tyr-219 side chain. Thus, our results together with the structure of TTHA0327 (4) have revealed the location of the substrate-binding site and suggest a possible orientation of a bound substrate in HIBA-like dehydrogenases. The relatively large size of the substrate-binding site is in line with the observed substrate promiscuity of β-hydroxyacid dehydrogenases.
FIGURE 5.

Close-up view of PA0743 structure with bound HEPES or NAD+. A and B, surface presentation of the PA0743 active site with the modeled HEPES or bound NAD+. The substrate-binding site (A) is formed by the residues from two protomers (from one dimer) shown in different colors (gray and tan), whereas the cofactor-binding site (B) is made of the residues from one protomer. Both ligands are almost completely buried in the PA0743 active site. The location of the cofactor- and substrate-binding sites is labeled (NAD site and Substrate site with an arrow, respectively). C and D, electron density maps showing the positions of bound HEPES (C) or NAD+ (D) and selected residues (labeled). The omit map was generated by omitting the HEPES (C) and NAD+ (D) from the model. Cyan-colored density represents the resulting Fo − Fc map contoured at 3.0 σ. Ligand atoms and selected PA0743 residues are labeled and shown as a stick representation with magenta carbon atoms representing the ligand and green carbon atoms representing the selected PA0743 residues.
FIGURE 6.

Close-up stereoview of PA0743 active site. A, substrate-binding site with the modeled HEPES molecule. B, cofactor-binding site with the bound NAD+. C, the relative orientation of NAD+ and HEPES near the catalytic Lys-171 in the active site. Two PA0743 structures (Protein Data Bank codes 3OBB and 3Q3C) were superimposed, and the PA0743 ribbon from 3OBB is shown with the bound NAD+ and HEPES. The amino acid side chains and ligands are shown as sticks and labeled along with the protein ribbon of two protomers colored in gray and tan.
Crystal Structure of PA0743 in Complex with NAD+
To obtain a structure of a PA0743-NAD+ complex, the wild-type protein crystals were soaked in the reservoir solution containing 10 mm NAD+, and the structure was solved using the single wavelength anomalous diffraction method. The structure revealed the presence of the cofactor in the active site located in the interdomain cleft (Fig. 5, B and D, and Table 2). The NAD+ molecule is coordinated by the interactions with side chains and main chain groups of conserved residues located in both protein domains of one protomer. Its adenosine moiety is located in the shallow pocket of the N-terminal domain and interacts mainly with the hydrophobic side chains of Ile-7 (3.5 Å), Phe-30 (3.7 Å), Leu-32 (3.3 Å), and Leu-65 (3.7 Å) (Fig. 6B). The ribose 2′- and 3′-hydroxyls of adenosine interact with the side chain carboxyl oxygens of Asp-31 (2.4 and 2.7 Å), explaining the observed preference of PA0743 for NAD+ over NADP+ (Fig. 6B). The NADP+-dependent HIBA dehydrogenase TTHA0237 contains an Arg residue (Arg-31) at this position (4) (Fig. 1). The 3′-hydroxyl is also close to the Gly-10 main chain amide (3.5 Å). The oxygen atoms of the pyrophosphate moiety of NAD+ are coordinated by interactions with the main chain amido groups of Gly-10 (3.0 Å), His-11 (2.9 Å), and Met-12 (2.7 Å) (Fig. 6B), which are located on the β1-α1 loop containing HIBA dehydrogenase motif-1 GXGXMG (Fig. 1).
The nicotinamide moiety of NAD+ is mostly buried in the interdomain cleft of the PA0743 protomer (Figs. 5B and 6B). Two ribose hydroxyl groups of the nicotinamide mononucleotide interact with the side chain of the conserved Lys-246 (2.5 and 3.0 Å). The ϵ-amino group of Lys-246 is coordinated by the interaction with the side chain carboxyl oxygen of the conserved Asp-247 (3.0 Å) (Fig. 6B). These two conserved residues comprise sequence motif-4 of β-hydroxyacid dehydrogenases (Fig. 1). It is known that carboxylate-lysine interactions stabilize the protonated form of lysine (by increasing their pKa), which can be required to bind a substrate (47). Another carboxyl oxygen of Asp-247 is close to the side chain of the conserved Asn-175 from motif-3 (2.6 Å) (Fig. 6B). The nicotinamide ring of NAD+ is coordinated by van der Waals stacking with the side chains of the conserved Met-12 (3.7 Å) and Phe-239 (3.9 Å) (Fig. 6B). Because the nicotinamide ring is in the syn conformation (its amido group is close to the pyrophosphate moiety), the hydride ion transfer can occur only on the si-face of the nicotinamide ring where the predicted catalytic Lys-171 is located. This suggests that like the sheep 6-phosphogluconate dehydrogenase and TTHA0237 (4, 5) PA0743 is a pro-S (B) dehydrogenase.
In several dehydrogenases including the E. coli 6-phosphogluconate dehydrogenase and ketopantoate reductase, binding of a redox cofactor has been shown to induce a large conformational change producing a closed conformation of the active site (46, 48). The superposition of the PA0743 apostructure with the structure of the NAD+ complex (generated using the SSM server) revealed no significant structural rearrangements with a pairwise root mean square deviation value of the main chain Cα atoms of 0.43 Å for the superposition of residues 2–296. No considerable cofactor-induced changes have been found in the structure of the HIBA dehydrogenase TTHA0237 either (4). In Fig. 6C, the superimposed PA0743 structures show the active site with the bound NAD+ and HEPES molecules illustrating the relative arrangement of the cofactor and substrate-like ligand near the catalytic Lys-171. In the superimposed model of the PA0743 active site, the C2 atom of HEPES is located 3.8 Å away from the C4 atom of the nicotinamide ring and likely mimics the location of the l-serine C3 atom positioned for catalytic reaction (hydride transfer). In the structures of PA0743, TTHA0237, and E. coli 6-phosphogluconate dehydrogenase, the binding sites for cofactor and substrate do not appear to be overlapping and likely function independently (Fig. 6C) (4, 46). This differs from the binding of cofactor and substrate in short-chain dehydrogenases and homoserine dehydrogenases whose cofactor-binding sites are partially occluded by substrate-binding sites. In these enzymes, the cofactor should bind to the active site first, and then the substrate binds on top of the cofactor nicotinamide ring, locking it in the active site (49, 50).
Mutational Studies of PA0743 and Potential Catalytic Mechanism
To identify the residues of PA0743 important for its catalytic activity, we mutated eight conserved and semiconserved residues to Ala and determined the catalytic activity of purified mutant proteins against l-serine. Site-directed mutagenesis revealed an important role of the conserved Asn-175 of PA0743 motif-3 (4.4–5.0 Å to Lys-171) (Fig. 7 and Table 1). In the apostructure of PA0743, the Asn-175 side chain is involved in the coordination of the modeled HEPES sulfonic group, suggesting that it can potentially coordinate the substrate (l-serine or methyl-l-serine) hydroxyl group (Fig. 6A). Alanine replacement of conserved residues from PA0743 motif-2 and motif-4 (Ser-122, Lys-246, and Asp-247) also resulted in proteins with very low activity (Fig. 7). The structure of PA0743 shows that Lys-246 is directly involved in the cofactor coordination (nicotinamide ribose oxygens), suggesting that the side chains of Ser-122 and Asp-247 might contribute to substrate binding (Fig. 6B). The substrate-binding site of PA0743 accommodates two semiconserved aromatic residues, Trp-214 and Tyr-219, whose replacement to Ala also had a strong negative effect on PA0743 activity (Fig. 7). We propose that the Tyr-219 side chain hydroxyl might contribute to substrate coordination (interaction with the serine carboxyl group), whereas the role of Trp-214 in the activity of PA0743 remains unclear.
FIGURE 7.

Alanine replacement mutagenesis of PA0743. Dehydrogenase activity of purified mutant proteins with l-serine as substrate (in the presence of 5 mm NAD+) is shown.
As expected, the replacement of the predicted catalytic Lys-171 by Ala produced essentially inactive protein, confirming its critical role in PA0743 activity (Fig. 7). We propose that, as in TTHA0237 and E. barkeri Hgd, the PA0743 Lys-171 functions as a catalytic base, accepting the proton from the substrate hydroxyl. After binding of l-serine to the PA0743 active site, the substrate C3 hydride is transferred to the C4 of the NAD+ nicotinamide ring, whereas its hydroxyl proton is transferred to the unprotonated side chain of Lys-171. The developing negative charge on the carbonyl group of malonate semialdehyde can be stabilized by the side chain of the conserved Asn-175. The Lys-171 side chain is positioned close to the side chain of the semiconserved Thr-96 (2.8 Å; replaced by a Ser in some HIBA dehydrogenases), and the T96A mutant protein also exhibited a greatly reduced activity (Fig. 7 and Table 1). This suggests that the interaction of the catalytic Lys-171 with a hydroxyl side chain (Thr or Ser) is important for the activity of PA0743 (probably through the maintenance of the proper orientation and/or unprotonated state of Lys-171). Overall, the results of site-directed mutagenesis of PA0743 support a catalytic role of Lys-171 and identify other residues important for activity.
Implications for Potential role of PA0743 in P. aeruginosa
Thus, biochemical characterization of PA0743 has revealed that this protein exhibits an NAD+-dependent dehydrogenase activity with substrate preference for l-serine and methyl-l-serine. An NADP+-dependent serine dehydrogenase has been purified from A. tumefaciens and is proposed to function in an unknown biosynthetic pathway (42). NAD+-dependent dehydrogenases appear to function primarily in catabolic reactions (51), suggesting that PA0743 might be involved in serine/methylserine degradation. A role in serine metabolism has been proposed previously for the HIBA dehydrogenase from P. putida E23 (17). In addition, several Pseudomonas species have been shown to be able to grow on l-serine as a sole nitrogen source and to actively degrade this amino acid (52–54). However, our growth experiments with various nitrogen and carbon sources (including l-serine) revealed no difference between the P. aeruginosa wild-type and PA0743 deletion strains, suggesting that this organism might contain other (complementing) serine dehydrogenases. In P. aeruginosa, the gene encoding the HIBA dehydrogenase PA3569 (mmsB) is linked in an operon with the methylmalonate-semialdehyde dehydrogenase gene mmsA, and these enzymes are proposed to function together in the distal pathway of valine catabolism (21). Moreover, the rat liver methylmalonate-semialdehyde dehydrogenase (EC 1.2.1.27) has also been shown to be active against malonate semialdehyde (55). One hypothesis is that in vivo the expected reaction products of PA0743 (2-aminomalonate semialdehyde and 2-aminomethylmalonate semialdehyde; Fig. 3) are deaminated by a presently unknown deaminase, producing methylmalonate or malonate semialdehydes, which are substrates for a methylmalonate-semialdehyde dehydrogenase. Thus, the activity of the l-serine dehydrogenase PA0743 might provide a link between serine degradation and the distal pathway of valine catabolism. Other, presently uncharacterized β-hydroxyacid dehydrogenases from P. aeruginosa are also expected to produce semialdehydes as reaction products. Biochemical and structural characterization of these proteins will reveal novel biochemical reactions and provide a greater understanding of the molecular mechanisms of substrate specificity and catalysis by β-hydroxyacid dehydrogenases.
Acknowledgments
We thank all members of the Structural Proteomics in Toronto (SPiT) Centre, the Structural Biology Center at the Advanced Photon Source, and Midwest Center for Structural Genomics for help in conducting these experiments and discussions. We are grateful to Dr. Naoki Kunishima (RIKEN Spring-8 Center, Japan) for providing (R)- and (S)-3-hydroxyisobutyrate. Use of the Advanced Photon Source was supported by the United States Department of Energy, Basic Energy Sciences, Office of Science, and use of the Structural Biology Center beamlines was supported by the Office of Biological and Environmental Research under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grant GM074942 from the Protein Structure Initiative (to the Midwest Center for Structural Genomics). This work was also supported by the Government of Canada through Genome Canada and Ontario Genomics Institute Grant 2009-OGI-ABC-1405.
The atomic coordinates and structure factors (codes 3OBB and 3Q3C) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- HIBA
- 3-hydroxyisobutyric acid.
REFERENCES
- 1. Hawes J. W., Harper E. T., Crabb D. W., Harris R. A. (1996) Structural and mechanistic similarities of 6-phosphogluconate and 3-hydroxyisobutyrate dehydrogenases reveal a new enzyme family, the 3-hydroxyacid dehydrogenases. FEBS Lett. 389, 263–267 [DOI] [PubMed] [Google Scholar]
- 2. Reitz S., Alhapel A., Essen L. O., Pierik A. J. (2008) Structural and kinetic properties of a beta-hydroxyacid dehydrogenase involved in nicotinate fermentation. J. Mol. Biol. 382, 802–811 [DOI] [PubMed] [Google Scholar]
- 3. Njau R. K., Herndon C. A., Hawes J. W. (2001) New developments in our understanding of the beta-hydroxyacid dehydrogenases. Chem. Biol. Interact. 130–132, 785–791 [DOI] [PubMed] [Google Scholar]
- 4. Lokanath N. K., Ohshima N., Takio K., Shiromizu I., Kuroishi C., Okazaki N., Kuramitsu S., Yokoyama S., Miyano M., Kunishima N. (2005) Crystal structure of novel NADP-dependent 3-hydroxyisobutyrate dehydrogenase from Thermus thermophilus HB8. J. Mol. Biol. 352, 905–917 [DOI] [PubMed] [Google Scholar]
- 5. Adams M. J., Ellis G. H., Gover S., Naylor C. E., Phillips C. (1994) Crystallographic study of coenzyme, coenzyme analogue and substrate binding in 6-phosphogluconate dehydrogenase: implications for NADP specificity and the enzyme mechanism. Structure 2, 651–668 [DOI] [PubMed] [Google Scholar]
- 6. Letto J., Brosnan M. E., Brosnan J. T. (1986) Valine metabolism. Gluconeogenesis from 3-hydroxyisobutyrate. Biochem. J. 240, 909–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Congdon P. J., Haigh D., Smith R., Green A., Pollitt R. J. (1981) Hypermethioninaemia and 3-hydroxyisobutyric aciduria in an apparently healthy baby. J. Inherit. Metab. Dis. 4, 79–80 [DOI] [PubMed] [Google Scholar]
- 8. Landaas S. (1975) Accumulation of 3-hydroxyisobutyric acid, 2-methyl-3-hydroxybutyric acid and 3-hydroxyisovaleric acid in ketoacidosis. Clin. Chim. Acta 64, 143–154 [DOI] [PubMed] [Google Scholar]
- 9. Ko F. J., Nyhan W. L., Wolff J., Barshop B., Sweetman L. (1991) 3-Hydroxyisobutyric aciduria: an inborn error of valine metabolism. Pediatr. Res. 30, 322–326 [DOI] [PubMed] [Google Scholar]
- 10. van Maris A. J., Konings W. N., van Dijken J. P., Pronk J. T. (2004) Microbial export of lactic and 3-hydroxypropanoic acid: implications for industrial fermentation processes. Metab. Eng. 6, 245–255 [DOI] [PubMed] [Google Scholar]
- 11. Yao T., Xu L., Ying H., Huang H., Yan M. (2010) The catalytic property of 3-hydroxyisobutyrate dehydrogenase from Bacillus cereus on 3-hydroxypropionate. Appl. Biochem. Biotechnol. 160, 694–703 [DOI] [PubMed] [Google Scholar]
- 12. Hasegawa J. (1981) Purification, crystallization and some properties of beta-hydroxyisobutyrate dehydrogenase from Candida rugosa IFO 0750. Agric. Biol. Chem. 45, 2805–2814 [Google Scholar]
- 13. Hawes J. W., Crabb D. W., Chan R. M., Rougraff P. M., Harris R. A. (1995) Chemical modification and site-directed mutagenesis studies of rat 3-hydroxyisobutyrate dehydrogenase. Biochemistry 34, 4231–4237 [DOI] [PubMed] [Google Scholar]
- 14. Rougraff P. M., Zhang B., Kuntz M. J., Harris R. A., Crabb D. W. (1989) Cloning and sequence analysis of a cDNA for 3-hydroxyisobutyrate dehydrogenase. Evidence for its evolutionary relationship to other pyridine nucleotide-dependent dehydrogenases. J. Biol. Chem. 264, 5899–5903 [PubMed] [Google Scholar]
- 15. Robinson W. G., Coon M. J. (1957) The purification and properties of beta-hydroxyisobutyric dehydrogenase. J. Biol. Chem. 225, 511–521 [PubMed] [Google Scholar]
- 16. Rougraff P. M., Paxton R., Kuntz M. J., Crabb D. W., Harris R. A. (1988) Purification and characterization of 3-hydroxyisobutyrate dehydrogenase from rabbit liver. J. Biol. Chem. 263, 327–331 [PubMed] [Google Scholar]
- 17. Chowdhury E. K., Nagata S., Misono H. (1996) 3-Hydroxyisobutyrate dehydrogenase from Pseudomonas putida E23: purification and characterization. Biosci. Biotechnol. Biochem. 60, 2043–2047 [DOI] [PubMed] [Google Scholar]
- 18. Njau R. K., Herndon C. A., Hawes J. W. (2000) Novel beta-hydroxyacid dehydrogenases in Escherichia coli and Haemophilus influenzae. J. Biol. Chem. 275, 38780–38786 [DOI] [PubMed] [Google Scholar]
- 19. Packdibamrung K., Misono H., Harada M., Nagata S., Nagasaki S. (1993) An inducible NADP(+)-dependent D-phenylserine dehydrogenase from Pseudomonas syringae NK-15: purification and biochemical characterization. J. Biochem. 114, 930–935 [DOI] [PubMed] [Google Scholar]
- 20. Saito N., Robert M., Kochi H., Matsuo G., Kakazu Y., Soga T., Tomita M. (2009) Metabolite profiling reveals YihU as a novel hydroxybutyrate dehydrogenase for alternative succinic semialdehyde metabolism in Escherichia coli. J. Biol. Chem. 284, 16442–16451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steele M. I., Lorenz D., Hatter K., Park A., Sokatch J. R. (1992) Characterization of the mmsAB operon of Pseudomonas aeruginosa PAO encoding methylmalonate-semialdehyde dehydrogenase and 3-hydroxyisobutyrate dehydrogenase. J. Biol. Chem. 267, 13585–13592 [PubMed] [Google Scholar]
- 22. Zhang R. G., Skarina T., Katz J. E., Beasley S., Khachatryan A., Vyas S., Arrowsmith C. H., Clarke S., Edwards A., Joachimiak A., Savchenko A. (2001) Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure 9, 1095–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heering H. A., Weiner J. H., Armstrong F. A. (1997) Direct detection and measurement of electron relays in a multicentered enzyme: voltammetry of electrode-surface films of E. coli fumarate reductase, an iron-sulfur flavoprotein. J. Am. Chem. Soc. 119, 11628–11638 [Google Scholar]
- 24. Anthon G. E., Barrett D. M. (2004) Comparison of three colorimetric reagents in the determination of methanol with alcohol oxidase. Application to the assay of pectin methylesterase. J. Agric. Food Chem. 52, 3749–3753 [DOI] [PubMed] [Google Scholar]
- 25. Solorzano L. (1969) Determination of ammonia in waters by the phenolhypochlorite method. Limnol. Oceanogr. 14, 799–801 [Google Scholar]
- 26. Jacobs M. A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R., Chun-Rong L., Guenthner D., Bovee D., Olson M. V., Manoil C. (2003) Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 100, 14339–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neidhardt F. C., Bloch P. L., Smith D. F. (1974) Culture medium for enterobacteria. J. Bacteriol. 119, 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimber M. S., Vallee F., Houston S., Necakov A., Skarina T., Evdokimova E., Beasley S., Christendat D., Savchenko A., Arrowsmith C. H., Vedadi M., Gerstein M., Edwards A. M. (2003) Data mining crystallization databases: knowledge-based approaches to optimize protein crystal screens. Proteins 51, 562–568 [DOI] [PubMed] [Google Scholar]
- 29. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., Walsh M. A., Westbrook E., Joachimiak A. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 31. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta. Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 32. Otwinowski Z. (1991) in Isomorphous Replacement and Anomalous Scattering (Wolf W., Evans P. R., Leslie A. G. W., eds) p. 80, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 33. Cowtan K. (1994) ‘dm’: an automated procedure for phase improvement by density modification. Newslett. Prot. Crystallogr. 31, 34–38 [Google Scholar]
- 34. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 38. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 39. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 40. Afonine P. V., Grosse-Kunstleve R. W., Adams P. D. (2005) CCP4 Newsletter, Contribution 8, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 41. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK–a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 42. Chowdhury E. K., Higuchi K., Nagata S., Misono H. (1997) A novel NADP(+)-dependent serine dehydrogenase from Agrobacterium tumefaciens. Biosci. Biotechnol. Biochem. 61, 152–157 [DOI] [PubMed] [Google Scholar]
- 43. Fujisawa H., Nagata S., Misono H. (2003) Characterization of short-chain dehydrogenase/reductase homologues of Escherichia coli (YdfG) and Saccharomyces cerevisiae (YMR226C). Biochim. Biophys. Acta 1645, 89–94 [DOI] [PubMed] [Google Scholar]
- 44. Yamazawa R., Nakajima Y., Mushiake K., Yoshimoto T., Ito K. (2011) Crystal structure of serine dehydrogenase from Escherichia coli: important role of the C-terminal region for closed-complex formation. J. Biochem. 149, 701–712 [DOI] [PubMed] [Google Scholar]
- 45. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Y. Y., Ko T. P., Chen W. H., Lo L. P., Lin C. H., Wang A. H. (2010) Conformational changes associated with cofactor/substrate binding of 6-phosphogluconate dehydrogenase from Escherichia coli and Klebsiella pneumoniae: Implications for enzyme mechanism. J. Struct. Biol. 169, 25–35 [DOI] [PubMed] [Google Scholar]
- 47. Gutteridge A., Thornton J. M. (2005) Understanding nature's catalytic toolkit. Trends Biochem. Sci. 30, 622–629 [DOI] [PubMed] [Google Scholar]
- 48. Ciulli A., Chirgadze D. Y., Smith A. G., Blundell T. L., Abell C. (2007) Crystal structure of Escherichia coli ketopantoate reductase in a ternary complex with NADP+ and pantoate bound: substrate recognition, conformational change, and cooperativity. J. Biol. Chem. 282, 8487–8497 [DOI] [PubMed] [Google Scholar]
- 49. DeLaBarre B., Thompson P. R., Wright G. D., Berghuis A. M. (2000) Crystal structures of homoserine dehydrogenase suggest a novel catalytic mechanism for oxidoreductases. Nat. Struct. Biol. 7, 238–244 [DOI] [PubMed] [Google Scholar]
- 50. Carius Y., Christian H., Faust A., Zander U., Klink B. U., Kornberger P., Kohring G. W., Giffhorn F., Scheidig A. J. (2010) Structural insight into substrate differentiation of the sugar-metabolizing enzyme galactitol dehydrogenase from Rhodobacter sphaeroides D. J. Biol. Chem. 285, 20006–20014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith E. L., Austen B. M., Blumenthal K. M., Nyc J. F. (1975) in The Enzyme (Boyer P.D., ed) pp. 293–367, Academic Press, New York [Google Scholar]
- 52. Wong H. C., Lessie T. G. (1979) Hydroxy amino acid metabolism in Pseudomonas cepacia: role of l-serine deaminase in dissimilation of serine, glycine, and threonine. J. Bacteriol. 140, 240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Keune H., Sahm H., Wagner F. (1976) Production of l-serine by the methanol-utilizing bacterium Psuedomonas 3ab. Eur. J. Appl. Microbiol. 2, 2175–2184 [Google Scholar]
- 54. Heptinstall J., Quayle J. R. (1970) Pathways leading to and from serine during growth of Pseudomonas AM1 on C1 compounds or succinate. Biochem. J. 117, 563–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goodwin G. W., Rougraff P. M., Davis E. J., Harris R. A. (1989) Purification and characterization of methylmalonate-semialdehyde dehydrogenase from rat liver. Identity to malonate-semialdehyde dehydrogenase. J. Biol. Chem. 264, 14965–14971 [PubMed] [Google Scholar]