

Background: SALL4 is a critical transcription factor for stem cell character maintenance.
Results: SALL4 protein interacts with different DNA methyltransferases and associates with enzymatic activities. Overexpression of SALL4 induced increased DNA methylation in promoter regions of silenced genes.
Conclusion: SALL4 may form a large complex with epigenetic modifiers in suppressing gene expression.
Significance: A novel mechanism is provided by which stem gene SALL4 negatively regulates target genes.
Keywords: DNA Methylation, Gene Regulation, Gene Silencing, Histone Deacetylase, Histone Deacetylation, PTEN, Stem Cells, Transcription Repressor, HDAC1, HDAC2, Mi-2·NuRD Complex, SALL Family
Abstract
The stem cell protein SALL4 plays a vital role in maintaining stem cell identity and governing stem cell self-renewal through transcriptional repression. To explore SALL4-mediated mechanisms involved in transcriptional repression, we investigated DNA modifications underlying its regulatory activities. By a luciferase activity assay, we found that both histone deacetylase inhibitor valproic acid (VPA) and DNA methylation inhibitor 5-azacytidine (5-azaC) specifically reversed the repression effect of SALL4 on its own as well as other Sal gene promoter activities. Cotreatment of VPA with 5-azaC in cells almost completely blocked this repression effect. Further co-immunoprecipitation assay and enzyme activity analysis demonstrated that SALL4 protein directly interacted with different DNA methyltransferases (DNMTs) and purified DNMT enzymatic activities from nuclear extracts. In addition, SALL4 isoforms co-occupied the same regions of its own promoter as DNMT corepressors, and ectopic overexpression of SALL4 led to increased CpG island promoter methylation of silenced genes in various cell types. These included primary hematopoietic stem/progenitor cells, fibroblasts, and NB4 leukemic cells. In NB4 cells, treatment of cells with 5-azaC also caused decreased amounts of methylated alleles of SALL4 and PTEN and dramatically increased their mRNA expression. Our studies identify a new mechanism by which SALL4 represses gene expression through interaction with DNMTs. Furthermore, DNMTs and histone deacetylase repressors synergistically contribute to the regulatory effects of SALL4. These findings provide new insights into stem cell self-renewal mediated by SALL4 via epigenetic machinery.
Introduction
SALL4 is a zinc finger transcription factor that was originally cloned based on sequence homology to Drosophila spalt (sal) (1). In Drosophila, sal is a homeotic gene essential in the development of posterior-head and anterior-tail segments (2). Human SALL4 mutations are associated with the Duane-radial ray syndrome (Okihiro syndrome), a human autosomal dominant disease involving multiple organ defects (3–5). Sall4 homozygous knock-out mice die at an early embryonic stage (6, 7). We and others have reported that murine SALL4 plays a vital role in maintaining embryonic stem cell pluripotency and in governing the fate of the inner cell mass through transcriptional modulation of Oct4 and Nanog (7, 8). In embryonic stem cells, SALL4 can activate OCT4 and interact with Nanog, and the SALL4/OCT4/Nanog transcriptional network is essential for the maintenance of cell “stemness” (9–11). Recently, we have shown that SALL4 can serve as a robust stimulator for the expansion of hematopoietic stem/progenitor cells (12, 13). The mechanisms by which SALL4 affects hematopoietic stem/progenitor cell growth and identity include blocking differentiation and permitting proliferation of undifferentiated cells (12, 13).
As for detailed mechanisms of how SALL4 exerts its regulatory effects, it has been reported that SALL4 can suppress target genes through the epigenetic repressor Mi-2·NuRD complex. The SALL4-NuRD complex possesses histone deacetylase (HDAC)2 activity, which may participate in the repressing effects of SALL4 (14). We recently reported that in embryonic stem cells, SALL4 and OCT4 form a regulatory feedback loop (15). Importantly, SALL4 possesses a strong self-repressive effect, which seems to set a tight regulation for the proper expression of both genes (15). In the beginning of this study, we asked whether inhibition of the histone deacetylase can completely block the self-repression effect of SALL4. Our data showed that a HDAC inhibitor, valproic acid (VPA), did reverse the SALL4 self-repression effect to an extent of about 50%. We further extended this finding and demonstrated that, except for histone deacetylation modifications, SALL4 also interacts with different DNA methyltransferase proteins and brings about increased DNA cytosine methylation of silenced target genes in various cell systems. Our data define a previously unrecognized pathway for SALL4-mediated repression, supporting the notion that targeting of DNA methyltransferases by transcription factors is a wide mechanism within a cell. Moreover, DNA methylation and histone deacetylation mechanisms appear to contribute to the SALL4 regulatory activities synergistically.
EXPERIMENTAL PROCEDURES
Plasmid Constructions
The following plasmids have been described previously: pcDNA3-SALL4A-HA, pcDNA3-SALL4B-HA, pSALL1-Luc reporter, pSALL3-Luc reporter, pSALL4-Luc reporter, and pOCT4-Luc reporter (15). The primers used in generating SALL4 deletion mutants are described in supplemental Table S1. The PCR-amplified DNA was subcloned into pcDNA3.1/V5/His-TOPO (Invitrogen).
Cell Culture, Transfections, and Luciferase Assays
All cell cultures were maintained at 37 °C with 5% CO2. The human fibroblasts MIR90, foreskin fibroblast (FF) cells, the human embryonic kidney cell line HEK-293, and the acute promyelocytic leukemia cell line NB4 (all from ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin. Transfection of plasmids into cultured cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Luciferase assays were performed as described previously (15). Epigenetic drugs (valproic acid and 5-azacytidine) were purchased form Sigma. When used for luciferase assays, drugs were added at 4 h following transfection, and cells were collected after another 32 h.
Retrovirus Production and SALL4 Knockdown
Three short hairpin RNA-expressing plasmids (one control (pRS) and two SALL4-specific (numbers 7410 and 7412); all three from Origen)) were transfected into Phoenix packaging cells (Orbigen) using Lipofectamine 2000 (Invitrogen). Shed virus was harvested 48 h after transfection, and control or stable SALL4 knockdown 293 clones were obtained under puromycin (1.4 μg/ml) selection after 5 days.
Isolation of Murine Hematopoietic Stem/Progenitor Cells
Mouse bone marrow hematopoietic stem/progenitor cells (Lin−/Sca-1+/c-Kit+, LSK cells) were purified from 8–12-week-old C57Bl/6J mice as described previously (13).
Lentiviral Transduction and qRT-PCT Assays
Detailed transduction procedures for mouse bone marrow LSK cells were described previously (13). For NB4 cell transduction, SALL4 lentivirus particles at a multiplicity of infection of 10 or 20 were added to the cells at 37 °C. For controls, GFP-only lentivirus particles were added to the cells at similar multiplicities of infection. The cells were infected overnight for 12–15 h and then recovered in culture medium. qRT-PCR was performed as described previously (12). Primers are described in supplemental Table S1.
Immunoprecipitations and Western Blot Assays
HEK293 cells were transfected with expression plasmids and harvested after 48 h. Proteins were prepared with CelLyticTM MT cell lysis reagent (Sigma). Immunoprecipitations were performed using the Dynabeads® protein G immunoprecipitation kit (Invitrogen) according to the manufacturer's instructions. The eluted sample was loaded onto 7–10% SDS-polyacrylamide gels, and Western blots were performed with antibodies against HA (Bethyl Laboratories), HDAC1 (BioLegend), DNMT1 (Novus Biologicals), DNMT3L (Abcam), and DNMT3A, DNMT3B, and MBD2 (all from Epitomics).
DNA Methyltransferase Activity and HDAC Activity Assay
These experiments were carried out using the EpiQuik DNMT activity/inhibition assay Ultra kit and the Epigenase HDAC activity/inhibition direct assay kit (both from Epigentek) following the manufacturer's procedures. First, nuclear proteins were extracted from tested cells using the EpiQuikTM nuclear extraction kit (Epigentek). For immunoprecipitations preceding the assays, we used antibodies against HA or IgG control from Bethyl Laboratories.
Chromatin Immunoprecipitation (ChIP)
HEK293 cells were transiently transfected with expression plasmids or an empty vector. Cells were cross-linked with formaldehyde (Sigma) at room temperature for 10 min, lysed, and sonicated. Chromatin immunoprecipitations were conducted with specific antibodies (described above). For PCR detection, SALL4 primers within the −1100 bp region upstream of the ATG start codon were used to validate pull-down DNA fragments (15).
Bisulfite Genomic Sequencing
Briefly, genomic DNA (500 ng) from tested cells was subjected to sodium bisulfite modification by using BisulFlashTM DNA modification kit (Epigentek), and the promoter was PCR-amplified. The amplified product was subcloned into the pCR2.1 vector by TOPO TA cloning kit (Invitrogen) and sequenced via automated sequencing. Primers used in this study are described in supplemental Table S1.
MassARRAY Quantitative Methylation Analysis
Quantitative DNA methylation analysis at single CpG units was done by MassARRAY (Sequenom). Briefly, genomic DNA was treated with sodium bisulfite, PCR-amplified, in vitro transcribed, cleaved with RNase A, and subjected to matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Methylation standards (0, 20, 40, 60, 80, and 100% methylated genomic DNA) were used for data normalization.
Statistical Analysis
A Student's non-paired t test was used to determine the statistical significance where indicated.
RESULTS
Repression of Target Promoter Activities by SALL4 Is Reversed by Cotreatment of 5-AzaC and VPA
We recently reported the novel SALL4/OCT4 transcriptional feedback loop for embryonic stem cell pluripotency maintenance. In embryonic stem cells, SALL4 can repress the promoters of other SALL family members, such as SALL1 and SALL3, and competes with the activation effects of these two genes by OCT4. However, the precise molecular mechanisms of how SALL4 represses its own as well as other target genes are largely unknown. A recent study has shown that SALL4 can physically interact with the Mi-2·NuRD·HDAC complex and recruit it to specific sequence for gene silencing (14). First, we examined whether an inhibition of HDAC can completely block the transcriptional repression effect of SALL4. In a eukaryotic HEK293 cell line, full-length SALL4A- or SALL4B-expressing plasmid was cotransfected with a 2-kb SALL4-pGL3 promoter construct using a Lipofectamine reagent. The HDAC inhibitor, VPA (20 ng/ml), was administered at the same time to monitor the repression effect of promoter activity. Twenty-four hours post-transfection, the relative luciferase activity was analyzed using a luminometer. As seen in Fig. 1, a and b, the repression effect of the SALL4 promoter by either the SALL4A or SALL4B isoform was partially blocked to an extent of about 50% as judged by measuring relative luciferase units. Each experiment was repeated at least three times, and similar results were obtained. Thus, these data indicate that histone deacetylation only partially contributes to the SALL4 self-repression activity, and other regulatory mechanisms may exist and participate in this effect.
FIGURE 1.

Repression of target promoter activities by SALL4 is reversed by cotreatment of 5-azaC and VPA. A 2-kb SALL4-pGL3 promoter construct was cotransfected with SALL4A- or SALL4B-expressing plasmids in HEK293 cells. Luciferase activity assays were performed from lysates of cells that were untreated (a), treated with VPA only (b), treated with 5-azaC only (c), or cotreated (d). For comparison studies, other Sal gene (SALL1 and SALL3) (e) or OCT4 promoter constructs (f) were used to replace SALL4-pGL3, and repressive effects of both promoter activities by SALL4A were assessed after administration of 5-azaC. All numbers refer to -fold changes, and error bars represent S.D. values of three independent experiments. g, a representative Western blotting analysis shows expression of SALL4A-HA and SALL4B-HA proteins in transfected 293 cells. RLU, relative luciferase units.
Previously, it has been reported that HDAC1 and HDAC2 have the ability to bind the DNA methyltransferase DNMT1, and the immunoprecipitated complexes from HEK293 nuclear extracts bear both histone deacetylase and DNA methyltransferase activities (16, 17). Other DNMTs, including DNMT3A and DNMT3B, are also responsible for cytosine methylation in mammals and have a role in gene silencing. Involvement of DNMT proteins by other regulatory factors during transcriptional repression has been reported (18, 19). Because SALL4 interacts with the NuRD complex, and histone deacetylation only partially contributed to the SALL4-mediated repression activity, we speculate that an additional epigenetic mechanism, DNA methylation modification, may also participate in the repression effect of SALL4. To test this, 5-azacytidine (5-azaC), a DNA methylation inhibitor, was used to replace VPA, and SALL4A- or SALL4B-expressing plasmid was cotransfected with OCT4-pGL3 promoter plasmid as another control. As seen in Fig. 1, a and c, treatment of 5-azaC did reverse the self-repression effect of the SALL4 promoter, and the overall luciferase activity reactivated to ∼50%. Similarly, the repression effects of the SALL1 and SALL3 gene promoters by the SALL4A isoform were also differentially reversed by 5-azaC (Fig. 1e). By contrast, the activation of the OCT4 promoter by SALL4 isoforms was not affected (Fig. 1f), which is in agreement with the repression effect of active DNA methylation of gene promoters. To further confirm this effect, we treated cells with both 5-azaC and VPA in the third experiment and found that the self-repression of SALL4 promoter activity was almost completely blocked (Fig. 1, a and d). These results demonstrate that DNA methylation and histone deacetylation contribute to the repressing effect of SALL4 in a synergistic way.
SALL4 Interacts with Different DNMT Proteins and Associates with DNA Methyltransferase Activity
The above findings led us to study if SALL4 can actively recruit DNMT proteins to its target genes for gene silencing. First, we transiently transfected HEK293 cells with plasmids expressing SALL4 isoforms tagged with HA and analyzed the cell lysates by immunoprecipitation (IP) using an antibody against HA. We then performed Western blot analysis with antibodies against human DNMT proteins, including DNMT1, -3A, -3B, and -3L, and MBD2 (the methyl-CpG-binding domain 2 protein). As shown in Fig. 2, a and b, both SALL4A and SALL4B isoforms interacted with all of these proteins, whereas no precipitate was detected by using an unrelated IgG antibody. To confirm these observations and further define the regions that are responsible for SALL4-DNMT interactions, we produced a series of HA-tagged deletion mutants of SALL4 and performed similar IP experiments in 293 cells. Interestingly, deletion mutants containing the N-terminal 174-amino acid sequence (deletions 1 and 4) successfully precipitated all of the tested DNMT proteins, whereas other mutants that lack the N-terminal sequences (deletions 2 and 3) did not. It is notable that among the analyzed DNMTs, DNMT1 was precipitated less readily by SALL4 isoforms. These observations, that the N-terminal sequence of SALL4 isoforms is necessary for the interaction with DNMTs, prompt us to compare the known interaction of SALL4 isoforms with HDAC1. As shown in Fig. 2b, IP followed by Western blotting analysis clearly demonstrated that SALL4 isoforms and the N-terminal sequence-containing deletion mutants interacted with HDAC1.
FIGURE 2.

Interaction of SALL4 proteins with different DNMTs and HDAC1. a, SALL4 isoforms and mutant SALL4 deletions (Δ1–Δ4) are shown schematically. b, SALL4 isoforms and various HA-tagged SALL4 mutants were transfected into 293 cells in 60-mm culture dishes. After 20 h, cells were lysed, and about 100 μl of cell lysates per 5 μg of anti-HA antibody or IgG control were used for Dynabeads protein G immunoprecipitation. Anti-HA immunoprecipitates were analyzed by Western blotting with antibodies against HDAC1 or the indicated DNMT antibodies. Immunoprecipitation of SALL4 isoforms and different mutants from HEK293 nuclear extracts purified different amounts of DNMT (c) and HDAC (d) activities. Error bars, S.D.
Because SALL4 proteins physically interact with various DNMT proteins, we expected that SALL4-DNMT complex would have DNMT enzymatic activities. Using SALL4 isoforms as well as their deletion mutant-transfected cells, we evaluated each protein complex precipitated by antibodies against HA. As shown in Fig. 2c, both SALL4A- and SALL4B-containing protein complexes exhibited the highest amount of DNMT enzymatic activity (3.5- and 3.3-fold as compared with that of control). The complexes containing mutants 1 and 4 (which have the N-terminal sequence) showed a 2.3–2.8-fold higher level of DNMT activity, whereas mutant 2- or 3-containing protein complex exhibited a similar or slightly increased level of DNMT activities as compared with a control.
Next, by using an Epigenase HDAC activity/inhibition direct assay kit, we further examined HDAC enzymatic activities for the SALL4 isoform and each mutant-containing complex. Consistent with the IP and Western blot findings above, SALL4 proteins and their mutants containing the N-terminal 174 amino acids purified a much higher level of HDAC activities (Fig. 2d). Taken together, these data indicate that SALL4 binds to endogenous DNMT enzymes and that these complexes are associated with DNMT enzymatic activities in vivo. Moreover, the binding sites that are responsible for SALL4 interactions with DNMT proteins or HDAC1 are within its N-terminal region.
SALL4 Isoforms Co-occupy the Same Promoter Regions of SALL4 as DNMT Corepressors and HDAC1
As described above, SALL4 repressed its own promoter activity, and this repression activity was prevented by both DNMT and HDAC inhibitors. In addition, SALL4 was able to interact with DNMTs as well as HDAC1, a component of the Mi-2·NuRD complex that is known to be involved in transcriptional repression. These observations led us to investigate if SALL4 negatively regulated transcription by forming a complex with DNMTs and HDAC1. To test this, we used ChIP coupled with PCR detection (ChIP-PCR) to examine whether SALL4 and DNMT proteins share common binding site(s) within the SALL4 promoter. Four pairs of primers spanning SALL4 promoter region (−1.1 kb to +1 bp upstream of ATG site) were used in ChIP-PCR analysis, and PCR product intensities were normalized to those of amplified DNA fragments from the input. Overall, HDAC1-, DNMT1-, DNMT3A-, and DNMT3B-immunoprecipitated fragments showed significant enrichment of the P3 product (−550 to −250 bp upstream of the ATG site) as compared with the input or mock control (Fig. 3). SALL4A- and SALL4B-immunoprecipitated fragments also showed significant enrichment of the P3 product and slight enrichment of the P4 product (Fig. 3). These results illustrate that SALL4, HDAC1, and DNMT proteins co-occupy the same promoter sites of SALL4, and its own transcription may be down-regulated via recruitments of both DNA methylation and histone deacetylation enzymes.
FIGURE 3.
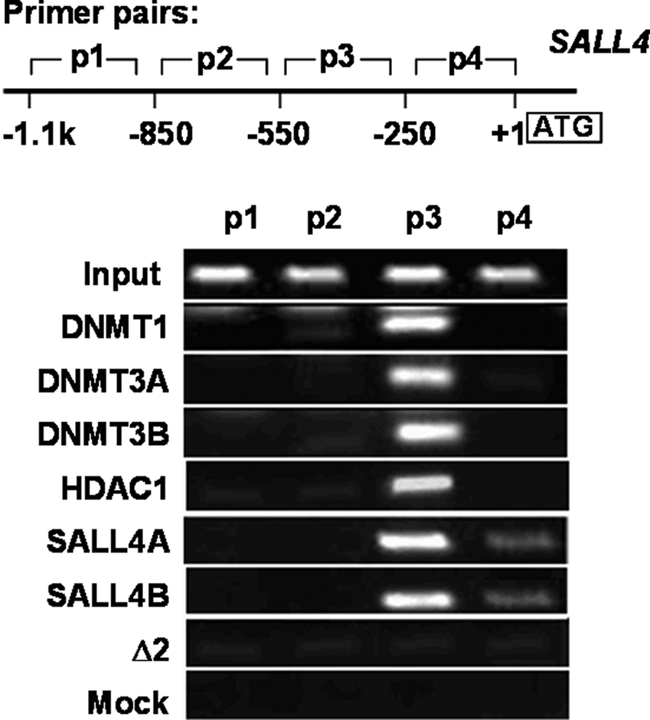
SALL4 isoforms co-occupy the same regions of its own promoter as HDAC1 and DNMT proteins. Top, diagram of SALL4 promoter. Bottom, agarose gel analysis of ChIP PCR products. Antibodies against HA (for SALL4A and SALL4B) or the indicated proteins were used to immunoprecipitate DNA fragments from HEK293 cells, and -fold enrichment was compared with input. One SALL4 mutant (Δ2) and mock (H2O) were used as control. Four pairs of primers spanning the SALL4 promoter region (−1.1 kb to +1 bp upstream of the ATG site) were used for ChIP-PCR analysis. There is a significant enrichment of the P3 product and slight enrichment of the P4 product for the SALL4 isoforms and for HDAC1 and DNMT protein purified fragments.
Forced Expression of SALL4 Isoforms Brings about Increased DNA Methylation Status of Silenced Genes in Various Cell Types
To find additional evidence that DNA methyltransferase activity does participate in SALL4-mediated transcriptional regulation, we performed bisulfite genomic sequencing to monitor changes of DNA methylation status on the SALL4 proximal promoter. In this set of experiments, we chose two distinct cell types, the HEK293 cell line and the primary human FF cells. The 293 cells stably express high amount of SALL4, whereas the FF cells have no detectable SALL4 expression. We chose the latter cell type also because it has been used for somatic reprogramming, and SALL4 plays positive roles in generating induced pluripotent stem cells (20). These two types of cells were transiently transfected with SALL4-expressing plasmid, and bisulfite genomic sequencing was conducted at 24 h after transfection. As seen in Fig. 4, a and b, in empty vector-treated FF cells, the frequency of methylcytosine residues in CpGs and GC sites was ∼58%. In SALL4-treated cells, expression of SALL4 isoforms significantly induced further methylation of this region to as high as 86%, confirming that the presence and/or overexpression of SALL4 is responsible, at least in part, for DNA methylation of its own promoter. In addition, SALL4 overexpression also induced increased DNA methylation within its promoter region in another fibroblast cell line, MIR90 (data not shown). In HEK293 cells, we failed to detect notable increase of SALL4 promoter methylation after forced protein expression; this may be due to the presence of relatively high levels of endogenous SALL4 (see below).
FIGURE 4.

Forced expression of SALL4 brings about increased DNA cytosine methylation of target genes in various cell types. a, human primary FF cells were transfected with SALL4A- expressing plasmid or vector control. Bisulfite sequencing shows that the SALL4 proximal promoter (100–720 bp upstream of the start codon) is differentially methylated at specific CpGs or GC-rich sites between the two groups of cells. Open circle, no methylation of cytosine; filled circle, methylated cytosine. b, chromatogram of one sequenced clone derived from bisulfite-treated FF cells that were transiently transfected with SALL4-expressing plasmids or pcDNA3 vector control. The arrows indicate differentially methylated sequences between the two cell types; unmethylated C converted to T upon bisulfite treatment. c, MassARRAY profiles showing methylation status of the PTEN promoter in NB4 leukemia cells 6 days after GFP control or SALL4-expressing lentivirus transduction. Percentages of methylation at each CpG site are indicated. The PTEN promoter region analyzed was chromosome 10, 40,427,070–40,427,349. d, 6 days after virus transduction, total RNA was extracted from the indicated group, and PTEN mRNA was measured by qRT-PCR. *, significant difference from the control group, p < 0.01. Error bars, S.E. (n = 3).
We next sought further evidence of SALL4-mediated DNA methylation modifications in human leukemia cells (NB4), in which SALL4 functions as a critical survival factor by suppressing PTEN expression (21). For this purpose, cells were transduced with GFP control or SALL4-expressing lentiviruses as described under “Experimental Procedures” and in Ref. 12. The ectopic expression of SALL4 protein was confirmed by Western blotting analysis as well as fluorescence microscopic examination (data not shown). Six days after virus transduction, we quantified the CpG methylation status of the promoters of PTEN and SALL4 using MassARRAY technology (Sequenom). Within the tested regions of the PTEN promoter, there was a generally increased percentage of methylation (a range of 1.2–2-fold) at various CpG sites (Fig. 4c). Notably, the SALL4 promoter was hypermethylated (an average of up to 95% methylation for each analyzed CpG site), and the methylation status was not greatly changed after SALL4 treatment (data not shown). This may be due to a reason similar to that described above. We further investigated the mRNA transcripts of PTEN, and qRT-PCR analysis showed a 1.4-fold decrease induced by SALL4 treatment (Fig. 4d). This result is consistent with our previous work from leukemia cell line as well as data obtained from leukemic bone marrow of SALL4B transgenic mice, in which PTEN expression was also suppressed (14, 21).
To further demonstrate that SALL4 recruits DNA methyltransferases to target gene promoters in other primary cells, we conducted similar experiments in isolated mouse bone marrow stem/progenitor cells. The LSK cells were isolated from 8–12-week-old mice and cultured and transducted with lentiviruses expressing SALL4A, SALL4B, or GFP control, and total cellular DNA was isolated on day 8 post-transduction. In this study, we have focused on the SALL4 gene as well as one of its target genes, EBF1, which was identified by our ChIP-chip assays from bone marrow stem cells (data not shown). As shown in Fig. 5, a and b, MassARRAY profiling showed that overexpression of SALL4 isoforms via lentiviral transduction induced a generally increased percentage of methylation at individual CpG sites of both endogenous genes' promoters (a range of 1.2–6-fold). In addition, the mRNA transcripts of EBF1 were also significantly reduced to about 63% as judged by qRT-PCR assays (Fig. 5c). We have hypothesized that SALL4 stimulates hematopoietic stem/progenitor cell expansion in part by inhibiting lineage differentiation genes. EBF1 is such a gene, which drives B cell lineage specification and commitment (22). The SALL4-induced hypermethylation of EBF1 promoter and subsequent gene suppression is supportive of the observed blood stem/progenitor cell expansion by treatment of SALL4 (12, 13).
FIGURE 5.

a and b, in mouse bone marrow LSK cells, MassARRAY DNA methylation profiles showing percentages of each methylated CpG site within the indicated promoter regions of the SALL4 gene (a) and EBF1 gene (b) after the indicated lentivirus transduction. c, 7 days after virus transduction, total RNA was extracted from the indicated group, and EBF1 mRNA was measured by qRT-PCR. Error bars, S.E. (n = 4). *, p < 0.05.
Down-regulation of SALL4 Led to Decreased DNA Methylation at Its Own Promoter
We have performed an RNA interference study to examine the impact of SALL4 knockdown on DNA methylation levels. In HEK293 cells, two short hairpin (shRNA) retroviral constructs that target different regions of the SALL4 mRNA, termed 7410 and 7412, were adopted, and their ability to knock down SALL4 mRNA was confirmed by qRT-PCR. Stably infected 293 cells were identified under puromycin selection for 5 days, and then the genomic DNA was extracted. We next examined the methylation levels of CpG islands and GC-rich sites by bisulfite sequencing. It was found that the methylation levels of the SALL4 promoter were significantly decreased in both 7410- and 7412-treated cells compared with levels in control shRNA-transfected cells (Fig. 6, a and b). This group data further suggest that SALL4 may silence target genes by recruitment of DNA methylation machinery. In other cell types, such as primary bone marrow cells and NB4 cells, the use of shRNA tools has been hampered by inefficient transfection protocols (data not shown).
FIGURE 6.

Down-regulation of SALL4 led to decreased DNA methylation at its own promoter. a, detection of shRNAs interferential efficiency by two SALL4-specific oligonucleotides. Relative reduction of SALL4 mRNA by 7410 and 7412 was 83.3 and 92.9%, respectively. b, DNA methylation status of the endogenous SALL4 promoter (from −965 through −845) was examined by bisulfite genomic sequencing after control shRNA (pRS) or SALL4-specific shRNA treatment. Filled and open circles, methylation and unmethylation, respectively.
Reexpression of SALL4 and PTEN Genes by 5-azaC Treatment in Leukemia Cells
To confirm whether demethylation of the CpG sequence in the promoter region resulted in re-expression of the SALL4 and PTEN genes, we cultured NB4 leukemic cells and treated with 5-azaC (200 nm) for 72 h with medium replacement every 24 h. 5-azaC has been known to deplete soluble DNMT proteins and thus lead to replication-dependent global demethylation and gene reactivation (23–25). In agreement with these reports, Western blot analyses showed that the protein levels of DNMT1 declined substantially after 72 h of treatment (Fig. 7a). Interestingly, bisulfite genomic sequencing revealed that the promoters of SALL4 and PTEN were highly methylated in untreated cells. By contrast, cells that were treated with 5-azaC exhibited dramatic decrease in DNA methylation of both gene promoters (Fig. 7b). Moreover, qRT-PCR analysis showed a striking increase in SALL4 and PTEN transcripts in the treated cells (Fig. 7c). These results strongly suggested that the SALL4 and PTEN genes in NB4 cells were hypermethylated in their putative promoter CpG sequences. These data were similar to a report that in another type of cancer cells (colorectal), the SALL4 promoter region was hypermethylated, and treatment of DNMT inhibitor 5-azaC notably reactivated its gene expression (26). In addition, these data are also in agreement with the above finding that the methylation status of the SALL4 promoter regions in NB4 cells was not significantly changed after SALL4 overexpression.
FIGURE 7.

Reexpression of SALL4 and PTEN genes by 5-azaC treatment. The NB4 cells were grown and treated with 5-azaC (200 nm; Sigma) for 72 h with medium replacement every 24 h. After 72 h of treatment, genomic DNA, total RNA, and proteins were isolated from the cells for DNA methylation and mRNA analysis. a, DNMT protein expression in the whole-cell lysates from treated and untreated NB4 cells. Identical amounts (50 μg) of protein were separated and subjected to Western blot analysis with antibodies specific for DNMT1. The membranes were reprobed with anti-β-actin antibody to show equal loading of the protein. b, DNA methylation status of the SALL4 and PTEN genes was examined by bisulfite genomic sequencing with or without 5-azaC treatment (3 days). Filled and open circles, methylation and unmethylation, respectively. c, total RNA was extracted from each group, and mRNA for the indicated genes was measured by qRT-PCR. Error bars, S.E. (n = 3). *, significant differences (p < 0.001) from the untreated group.
DISCUSSION
SALL4 has been characterized as an essential molecule in the regulation of stem cell self-renewal and developmental events. SALL4 bears dual functions and exerts its effect through both positively and negatively regulating transcriptions. Moreover, SALL4 can activate/silence gene by active recruitment of epigenetic regulators. Our early study on SALL4-mediated activation of Bmi-1 suggests that this process involves methylation of Lys-4 of histone H3 (27). In another study, SALL4 can act as a repressor that directly connects the Mi-2·NuRD repressor complex, which has histone deacetylase activity to its downstream target genes, including PTEN and SALL1 (14). Here, we report a new mechanism by which SALL4 silences genes through recruitment of the DNMT proteins, thereby leading to subsequent DNA methylation and gene suppression.
Methylation of cytosine residues in the context of CpG dinucleotides within DNA is one of the major epigenetic modifications in mammals. Furthermore, specialized roles of DNA methylation and methyltransferases in maintaining adult somatic stem cell function have been documented from recent works. These works suggest that further dissection of these mechanisms will shed new light on the complex nature of stem cell self-renewal (28, 29). For example, knock-out mice studies showed that DNMT1-deficient hematopoietic stem cells lost the ability of self-renewal and are unable to give rise to multilineage hematopoiesis (30). We have recently reported that SALL4 is a robust stimulator for hematopoietic stem/progenitor cell expansion (12, 31). It would be interesting to study the profound relationship between SALL4 and DNA methytransferases during stem cell proliferation and multipotency maintenance in the future. It is possible that SALL4 provides specific targets for DNA methyltransferases, thus resulting in the generation of specific DNA methylation patterns.
In luciferase activity assays, treatment of cells with either 5-azaC or VPA alone was unable to completely block the repression effect of SALL4. This indicates that both histone deacetylation and DNA methylation are required for the complete repression of SALL4 target genes. In addition, deletion analysis revealed that the N-terminal 174 amino acids of SALL4 interacted with HDAC1 and various DNMT proteins and associated with HDAC and DNMT enzymatic activities in vivo (Fig. 2). These findings raise the interesting question of whether the same domain bears different transcription repressors activities. In our studies, co-IP and ChIP-PCR analysis revealed a “common” site for interactions of SALL4 with HDAC1 and different DNMT proteins. This may indicate that SALL4 and epigenetic repressors (including DNMT1, DNMT3A, DNMT3B, DNMT3L, MBD2, HDAC1, HDAC2, and most likely others) form a large complex that cooperatively participates in regulation of downstream genes. It is reasonable to propose the following working model. SALL4 selectively recruits epigenetic modulators (DNMTs and HDACs) to specific DNA sequences of its downstream targets for gene suppression, whereas both classes of epigenetic inhibitor drugs may block this process and reactivate gene expression (Fig. 8). Nevertheless, based on accumulated studies over the past decades, it appears that DNMT1, DNMT3A, DNMT3B, DNMT3L, and MBD2 may together form a protein complex that interacts with histone deacetylases (HDAC1 and HDAC2) and other repressor proteins to maintain a precise level of DNA methylation and facilitate target gene silencing (16, 32–35).
FIGURE 8.

Hypothetical model. SALL4 selectively recruits epigenetic modulators (DNMT1, DNMT3A, DNMT3B, DNMT3L, MBD2, HDAC1, HDAC2, and most likely others) to specific DNA sequences of its downstream targets for gene suppression, whereas both classes of epigenetic drugs may block this process and reactivate gene expression.
In summary, our findings suggest that the presence of epigenetic repressors and/or enzymatic activities of DNMT1, DNMT3A, DNMT3B, DNMT3L, MBD2, and HDAC1 proteins are associated with the repressing activity of SALL4. Our studies have also provided novel mechanisms by which the SALL4 stem gene induces negative regulation of target genes.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant R01HL087948 (to Y. M.). This work was also supported in part by American Cancer Society-Institutional Research Grant 2011-2012 (to J. Y.) and Leukemia and Lymphoma Society special fellow award 3366-09 (to J. Y.).

This article contains supplemental Table S1.
- HDAC
- histone deacetylase
- VPA
- valproic acid
- FF
- foreskin fibroblast
- IP
- immunoprecipitation
- DNMT
- DNA methyltransferase
- qRT-PCR
- quantitative RT-PCR
- 5-azaC
- 5-azacytidine.
REFERENCES
- 1. Kohlhase J., Schuh R., Dowe G., Kühnlein R. P., Jäckle H., Schroeder B., Schulz-Schaeffer W., Kretzschmar H. A., Köhler A., Müller U., Raab-Vetter M., Burkhardt E., Engel W., Stick R. (1996) Isolation, characterization, and organ-specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics 38, 291–298 [DOI] [PubMed] [Google Scholar]
- 2. Kühnlein R. P., Schuh R. (1996) Dual function of the region-specific homeotic gene spalt during Drosophila tracheal system development. Development 122, 2215–2223 [DOI] [PubMed] [Google Scholar]
- 3. Al-Baradie R., Yamada K., St Hilaire C., Chan W. M., Andrews C., McIntosh N., Nakano M., Martonyi E. J., Raymond W. R., Okumura S., Okihiro M. M., Engle E. C. (2002) Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71, 1195–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kohlhase J., Heinrich M., Schubert L., Liebers M., Kispert A., Laccone F., Turnpenny P., Winter R. M., Reardon W. (2002) Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 11, 2979–2987 [DOI] [PubMed] [Google Scholar]
- 5. Borozdin W., Wright M. J., Hennekam R. C., Hannibal M. C., Crow Y. J., Neumann T. E., Kohlhase J. (2004) Novel mutations in the gene SALL4 provide further evidence for acro-renal-ocular and Okihiro syndromes being allelic entities and extend the phenotypic spectrum. J. Med. Genet. 41, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakaki-Yumoto M., Kobayashi C., Sato A., Fujimura S., Matsumoto Y., Takasato M., Kodama T., Aburatani H., Asashima M., Yoshida N., Nishinakamura R. (2006) The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation and cooperates with Sall1 in anorectal, heart, brain, and kidney development. Development 133, 3005–3013 [DOI] [PubMed] [Google Scholar]
- 7. Elling U., Klasen C., Eisenberger T., Anlag K., Treier M. (2006) Murine inner cell mass-derived lineages depend on Sall4 function. Proc. Natl. Acad. Sci. U.S.A. 103, 16319–16324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J., Tam W. L., Tong G. Q., Wu Q., Chan H. Y., Soh B. S., Lou Y., Yang J., Ma Y., Chai L., Ng H. H., Lufkin T., Robson P., Lim B. (2006) Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat. Cell Biol. 8, 1114–1123 [DOI] [PubMed] [Google Scholar]
- 9. Yang J., Chai L., Fowles T. C., Alipio Z., Xu D., Fink L. M., Ward D. C., Ma Y. (2008) Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 19756–19761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lim C. Y., Tam W. L., Zhang J., Ang H. S., Jia H., Lipovich L., Ng H. H., Wei C. L., Sung W. K., Robson P., Yang H., Lim B. (2008) Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell 3, 543–554 [DOI] [PubMed] [Google Scholar]
- 11. Wu Q., Chen X., Zhang J., Loh Y. H., Low T. Y., Zhang W., Zhang W., Sze S. K., Lim B., Ng H. H. (2006) Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J. Biol. Chem. 281, 24090–24094 [DOI] [PubMed] [Google Scholar]
- 12. Aguila J. R., Liao W., Yang J., Avila C., Hagag N., Senzel L., Ma Y. (2011) SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood 118, 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang J., Aguila J. R., Alipio Z., Lai R., Fink L. M., Ma Y. (2011) Enhanced self-renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J. Hematol. Oncol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu J., Jeong H. W., Kong N., Yang Y., Carroll J., Luo H. R., Silberstein L. E., Yupoma, Chai L. (2009) Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One 4, e5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J., Gao C., Chai L., Ma Y. (2010) A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One 5, e10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rountree M. R., Bachman K. E., Baylin S. B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277 [DOI] [PubMed] [Google Scholar]
- 17. Fuks F., Burgers W. A., Brehm A., Hughes-Davies L., Kouzarides T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 24, 88–91 [DOI] [PubMed] [Google Scholar]
- 18. Brenner C., Deplus R., Didelot C., Loriot A., Viré E., De Smet C., Gutierrez A., Danovi D., Bernard D., Boon T., Pelicci P. G., Amati B., Kouzarides T., de Launoit Y., Di Croce L., Fuks F. (2005) Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 24, 336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kundakovic M., Chen Y., Guidotti A., Grayson D. R. (2009) The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol. Pharmacol. 75, 342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsubooka N., Ichisaka T., Okita K., Takahashi K., Nakagawa M., Yamanaka S. (2009) Roles of Sall4 in the generation of pluripotent stem cells from blastocysts and fibroblasts. Genes Cells 14, 683–694 [DOI] [PubMed] [Google Scholar]
- 21. Yang J., Chai L., Gao C., Fowles T. C., Alipio Z., Dang H., Xu D., Fink L. M., Ward D. C., Ma Y. (2008) SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood 112, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maier H., Hagman J. (2002) Roles of EBF and Pax-5 in B lineage commitment and development. Semin. Immunol. 14, 415–422 [DOI] [PubMed] [Google Scholar]
- 23. Laurenzana A., Petruccelli L. A., Pettersson F., Figueroa M. E., Melnick A., Baldwin A. S., Paoletti F., Miller W. H., Jr. (2009) Inhibition of DNA methyltransferase activates tumor necrosis factor α-induced monocytic differentiation in acute myeloid leukemia cells. Cancer Res. 69, 55–64 [DOI] [PubMed] [Google Scholar]
- 24. Santi D. V., Norment A., Garrett C. E. (1984) Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U.S.A. 81, 6993–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghoshal K., Datta J., Majumder S., Bai S., Kutay H., Motiwala T., Jacob S. T. (2005) 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 25, 4727–4741 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Habano W., Sugai T., Jiao Y. F., Nakamura S. (2007) Novel approach for detecting global epigenetic alterations associated with tumor cell aneuploidy. Int. J. Cancer 121, 1487–1493 [DOI] [PubMed] [Google Scholar]
- 27. Yang J., Chai L., Liu F., Fink L. M., Lin P., Silberstein L. E., Amin H. M., Ward D. C., Ma Y. (2007) Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10494–10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bröske A. M., Vockentanz L., Kharazi S., Huska M. R., Mancini E., Scheller M., Kuhl C., Enns A., Prinz M., Jaenisch R., Nerlov C., Leutz A., Andrade-Navarro M. A., Jacobsen S. E., Rosenbauer F. (2009) DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 41, 1207–1215 [DOI] [PubMed] [Google Scholar]
- 29. Tadokoro Y., Ema H., Okano M., Li E., Nakauchi H. (2007) De novo DNA methyltransferase is essential for self-renewal but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 204, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trowbridge J. J., Snow J. W., Kim J., Orkin S. H. (2009) DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aguila J. R., Mynarcik D. C., Ma Y. (2011) SALL4. Finally an answer to the problem of expansion of hematopoietic stem cells? Expert. Rev. Hematol. 4, 479–481 [DOI] [PubMed] [Google Scholar]
- 32. Kim G. D., Ni J., Kelesoglu N., Roberts R. J., Pradhan S. (2002) Cooperation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 21, 4183–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Z. X., Mann J. R., Hsieh C. L., Riggs A. D., Chédin F. (2005) Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell Biochem. 95, 902–917 [DOI] [PubMed] [Google Scholar]
- 34. Fuks F., Burgers W. A., Godin N., Kasai M., Kouzarides T. (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 20, 2536–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Denslow S. A., Wade P. A. (2007) The human Mi-2/NuRD complex and gene regulation. Oncogene 26, 5433–5438 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.