

Background: RFC and Rad24-RFC are clamp loaders with different roles in DNA replication.
Results: RFC-PCNA binding is faster than PCNA opening, and PCNA opening by Rad24-RFC is about 10-fold weaker than by RFC.
Conclusion: RFC binds PCNA prior to opening PCNA, and the Rfc1 subunit missing in Rad24-RFC is important for PCNA opening.
Significance: These results provide insight into the mechanisms of these AAA+ protein complexes.
Keywords: ATPases, DNA Damage Response, DNA Replication, Enzyme Kinetics, Fluorescence, Protein-Protein Interactions, AAA+ ATPase
Abstract
Clamp loaders from all domains of life load clamps onto DNA. The clamp tethers DNA polymerases to DNA to increase the processivity of synthesis as well as the efficiency of replication. Here, we investigated proliferating cell nuclear antigen (PCNA) binding and opening by the Saccharomyces cerevisiae clamp loader, replication factor C (RFC), and the DNA damage checkpoint clamp loader, Rad24-RFC, using two separate fluorescence intensity-based assays. Analysis of PCNA opening by RFC revealed a two-step reaction in which RFC binds PCNA before opening PCNA rather than capturing clamps that have transiently and spontaneously opened in solution. The affinity of RFC for PCNA is about an order of magnitude lower in the absence of ATP than in its presence. The affinity of Rad24-RFC for PCNA in the presence of ATP is about an order magnitude weaker than that of RFC for PCNA, similar to the RFC-PCNA interaction in the absence of ATP. Importantly, fewer open clamp loader-clamp complexes are formed when PCNA is bound by Rad24-RFC than when bound by RFC.
Introduction
The proliferating cell nuclear antigen (PCNA)2 clamp is a ring-shaped trimer composed of three identical protein monomers (1). During DNA replication, PCNA encircles and slides freely along duplex DNA, anchoring DNA polymerases ϵ or δ to the template. The clamp loader, replication factor C (RFC), is part of the AAA+ family of ATPases and loads clamps onto DNA (reviewed in Ref. 2). The clamp loader uses ATP binding and hydrolysis to promote molecular interactions that catalyze the various steps of the clamp-loading reaction. RFC consists of five subunits arranged in a ring in which the subunits are held tightly together via interactions in the C-terminal domains (3). A gap between the N-terminal domains of two subunits allows DNA to enter the central chamber in the clamp loader and positions DNA into the open clamp (4). The clamp loader subunits and clamp adopt a spiral conformation that conforms to the DNA double helix. Electron microscopy studies of an archaeal RFC-PCNA-DNA-ATPγS complex (5) and structure-based modeling (6) suggest that PCNA forms an open lock-washer appearance that could allow the clamp to dock on the surface of the helical RFC.
Alternative RFC complexes exist in which the large RFC subunit (Rfc1) of the complex is replaced by another protein subunit such as Rad24 in Saccharomyces cerevisiae (Rad17 in humans), Ctf18, or Elg1 to form their respective RFC complexes, Rad24-RFC, Ctf18-RFC, and Elg1-RFC. Rad24-RFC functions as part of a DNA damage checkpoint pathway. This clamp loader loads an alternative clamp, the 9-1-1 complex, onto DNA. Loading of the 9-1-1 complex activates a DNA damage checkpoint via the protein kinase, ataxia telangiectasia mutated and Rad3-related protein (ATR) (reviewed in Refs. 7 and 8). Substitution of this single subunit, Rad24 for Rfc1, in the clamp loader complex alters the clamp specificity of the Rad24-RFC complex. Rad24-RFC can bind to PCNA and unload it from DNA, but it cannot productively load PCNA onto DNA (9, 10).
Clamp loading is a dynamic, multistep process that ultimately assembles PCNA onto DNA to allow for processive replication (reviewed in Refs. 11–13). Conformational changes in the clamp loader induced by ATP binding and hydrolysis modulate interactions of the clamp loader with the clamp and DNA to drive the clamp-loading reaction. This reaction can be divided into two phases based on ATP requirements of the clamp loader. First, ATP binding promotes clamp binding and opening, and second, ATP hydrolysis promotes release of the clamp on DNA. This study focuses on the first stage of the loading reaction, ATP-dependent clamp binding and opening.
EXPERIMENTAL PROCEDURES
Buffers
Assay buffer contained 30 mm HEPES (pH 7.5), 150 mm sodium chloride (NaCl), 8 mm magnesium chloride, 2 mm dithiothreitol (DTT), and the addition of 4% glycerol (v/v) where indicated. PCNA was stored in Buffer A, which contains 30 mm HEPES (pH 7.5), 0.5 mm EDTA, 150 mm NaCl, 2 mm DTT, and 10% glycerol (v/v). RFC was stored in Buffer A but with increased NaCl (300 mm). Buffer B is 20 mm Tris-HCl (pH 7.5), 0.5 mm EDTA, 50 mm NaCl, and 2 mm DTT with the addition of 10% glycerol (v/v) where indicated. Buffer C is 25 mm potassium phosphate (pH 7.0), 2 mm DTT, and 10% glycerol (v/v).
RFC and Rad24-RFC Purification
Rad24-RFC as described (10) and RFC containing a truncated Rfc1 subunit, missing 283 amino acids from the N terminus, were purified as described (14, 15). The concentrations of RFC and Rad24-RFC were determined by measuring the absorbance at 280 nm in 6 m guanidine hydrochloride and 66.7 mm sodium phosphate (pH 6.5) and using calculated extinction coefficients of 162,120 and 176,170 m−1 cm−1 for RFC and Rad24-RFC, respectively (16).
PCNA Expression and Purification
Cysteine residues 22, 62, and 81 were converted to Ser, Ile-111 and Ile-181 were converted to Cys (PCNA opening mutant), and Ser-43 was converted to Cys (PCNA binding mutant) by site-directed mutagenesis of the PCNA coding sequence using the QuikChange mutagenesis kit (Stratagene) as per the manufacturer's instructions. Wild-type PCNA and mutants were expressed in Escherichia coli BL21 (DE3). Cells were grown at 37 °C to an A600 of 0.6–0.7, the temperature was decreased to 15 °C, and protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mm.
PCNA was purified following a published protocol (17, 18) at 4 °C with minor modifications as described. Cells were lysed by French press, and the lysate was cleared by centrifugation. Ammonium sulfate (0.2 g/ml) was added to the clarified lysate, and the precipitate was discarded. Additional ammonium sulfate (0.15 g/ml) was added to the supernatant, and the precipitate was recovered by centrifugation. The pellet was resuspended and dialyzed in Buffer B plus 10% glycerol. The dialyzed protein was loaded onto two 5-ml HiTrap Q-Sepharose columns in tandem (GE Healthcare) and eluted with a linear gradient of 50–700 mm NaCl. The remaining two chromatographic steps on S-Sepharose and MonoQ columns were done as described previously (18). Purified PCNA was dialyzed against Buffer A and stored at −80 °C.
Covalent Labeling of PCNA with AF488 or MDCC
Purified PCNA-I111C/I181C or PCNA-S43C (65 nmol) was incubated with a solution of 1 mm tris(2-carboxyethyl) phosphine (TCEP) final concentration in 0.2 m Tris base (pH 8.0) for 5 min at room temperature. The PCNA/TCEP solution was incubated with either 1.9 μmol of Alexa Fluor 488 C5 maleimide (AF488)3 (Molecular Probes) or 0.98 μmol of N-(2-(1-maleimidyl)ethyl)-7-(diethylamino)coumarin-3-carboxamide (MDCC) (Molecular Probes) for 4 h at room temperature. Labeled protein was purified at 4 °C by removing excess fluorophore using a desalting column (Bio-Rad P6 DG) equilibrated with Buffer A minus glycerol (and minus DTT for PCNA-MDCC) followed by cation exchange chromatography on a 1-ml HiTrap Q-Sepharose (GE Healthcare) column. Labeled PCNA was dialyzed against Buffer A, aliquoted, and stored at −80 °C. The protein concentration of PCNA-AF4882 was calculated from the absorbance at 280 nm in 6 m guanidine hydrochloride and 66.7 mm sodium phosphate buffer (pH 6.5) (16) corrected for the contribution of AF488 at 280 nm (0.11 times AF488 absorbance at 495 nm) and using an extinction coefficient of 6,170 m−1 cm−1. The concentration of AF488 was calculated from the absorbance at 493 nm using an extinction coefficient of 73,000 m−1 cm−1. Based on these concentrations, 95–100% of PCNA is typically labeled by AF488. The protein concentration of PCNA-MDCC was calculated using a Bradford-type assay (Bio-Rad) with unlabeled WT PCNA standards. The concentration of MDCC was calculated from the absorbance at 436 nm using an extinction coefficient of 50,000 m−1 cm−1. Based on these concentrations, 73–100% of PCNA is typically labeled with MDCC.
Steady State Fluorescence Assays
Steady state fluorescence measurements were made on a QuantaMaster QM1 spectrofluorometer (Photon Technology International). Emission spectra for AF488 were measured from 505 to 605 nm using a 2.5-nm bandpass and excitation wavelength of 495 nm. Emission spectra for MDCC were measured from 450 to 550 nm using a 3.6-nm bandpass and excitation at 420 nm. Equilibrium binding assays were done by adding reagents sequentially to the cuvette starting with 72 μl of assay buffer containing ATP followed by 4 μl of PCNA-AF4882 or PCNA-MDCC, and finally, 4 μl of RFC or Rad24-RFC for a total reaction volume of 80 μl. Emission spectra were measured following each addition, and the intensities relative to free PCNA-AF4882 at 517 or PCNA-MDCC at 467 nm were plotted as a function of RFC concentration. Emission spectra were corrected for background by subtracting the signal for buffer. Dissociation constants (Kd) were calculated by fitting the observed intensity data (Iobs) to Equation 1 in which RFCo is the clamp loader concentration, PCNAo is the concentration of PCNA-AF4882 or PCNA-MDCC, Imin is the intensity of free PCNA-AF4882 or PCNA-MDCC, and Imax is the intensity of clamp loader-clamp complexes. Imax and Kd were fit as adjustable parameters by nonlinear regression using KaleidaGraph and Imin was set to 1.
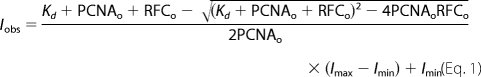 |
Competition binding assays were done by adding reagents sequentially to a cuvette starting with 68 μl of assay buffer with ATP followed by 4 μl of 400 nm PCNA-AF4882 and 4 μl of unlabeled WT PCNA, and finally, 4 μl of 400 nm RFC for a total reaction volume of 80 μl. The concentration of WT PCNA added for each measurement varied. Emission spectra were measured following each addition, and the relative intensity of AF488 at 517 nm was plotted as a function of WT PCNA concentration. Emission spectra were corrected for background by subtracting the signal for buffer. Data were fit to Equation 2 in which PCNAAF is the concentration of PCNA-AF4882 (20 nm), PCNAWT is the concentration of WT PCNA, Imax is the intensity in the absence of WT PCNA (set to a value of 1), and Imin is the signal at saturating concentrations of WT PCNA (fit as an adjustable parameter, calculated value of 0.757).
Pre-steady State Fluorescence Assays
Stopped-flow fluorescence measurements were made using a SX20MV stopped-flow fluorometer (Applied Photophysics, Ltd.). Single-mix reactions, in assay buffer containing 4% glycerol, were performed by mixing equal volumes (75–120 μl) of a solution of RFC or Rad24-RFC and ATP to a solution of PCNA-AF4882 or PCNA-MDCC and ATP. Data were collected for a total of 4–8 s at intervals of 0.5–0.8 ms, and six or more individual kinetic traces were averaged. Time courses were corrected for background by subtracting the signal for buffer. Fluorescence of AF488 was measured by exciting at 495 nm and using a 515-nm cut-on filter to collect emission, and MDCC fluorescence was measured by exciting at 420 nm and using a 455-nm cut-on filter to collect emission. The time courses were empirically fit using KaleidaGraph software to a single exponential (Equation 3)
in which a is the amplitude, kobs is the observed rate constant, and t is the reaction time.
RESULTS
Fluorescence Intensity-based Assay to Measure PCNA Opening
An intensity-based fluorescence assay was developed to measure PCNA opening. Three of the four naturally occurring Cys residues in S. cerevisiae PCNA were converted to Ser to allow for selective labeling of engineered Cys residues. The fourth Cys is buried in the protein and should not be accessible to extrinsic labeling reagents (3). Two new Cys residues were introduced by mutation of Ile-111 and Ile-181, which are located on opposite sides of monomer interfaces in PCNA (Fig. 1A, ribbon diagrams). Cys-111 and Cys-181 were labeled with AF488 in the presence of a reducing agent, TCEP, to prevent disulfide bond formation between the closely spaced cysteines. Each PCNA monomer of the mutant is labeled with two fluorophores, PCNA-AF4882. The homotrimer contains six fluorophores, a pair located at each of the three monomer interfaces.
FIGURE 1.

Physical separation of AF488 molecules relieves quench in fluorescence. A, emission spectra of 10 nm PCNA-AF4882 in the absence (− SDS, black) and presence (+ SDS, gray) of 0.5% SDS. Inset, top and edge views of PCNA in which each monomer is colored differently and the positions of Cys-111 and Cys-181 are indicated by green and yellow spheres, respectively. B, emission spectra of free PCNA-AF4882 before (gray trace) and after the addition of RFC (black trace) or RFC storage buffer (black dashed trace). Final concentrations were 10 nm PCNA-AF4882, 285 nm RFC (when present), and 0.5 mm ATP.
The α-carbon atoms of residues 111 and 181 are located within 5.5 Å of each other (3). At this distance, the two fluorophores should be able to physically interact and self-quench. Separation of these fluorophores, which occurs upon clamp opening, will remove stacking interactions and increase fluorescence. This “relief of quench” assay has been used to measure opening of the E. coli β-clamp (19). One mechanism to physically separate the fluorophores is by denaturation with a detergent. When a 0.5% sodium dodecyl sulfate (SDS) solution was added to PCNA-AF4882, the fluorescence increased. The fluorescence of the denatured PCNA-AF4882 in SDS was 8.6-fold greater than the fluorescence of AF488 in the native protein (Fig. 1A, gray and black traces, respectively). The clamp loader can physically separate fluorophores on either side of a monomer interface by opening the clamp. When RFC binds to PCNA-AF4882 in the presence of ATP, AF488 fluorescence increases due to the formation of an open clamp loader-clamp complex (Fig. 1B, solid black trace). At saturating concentrations of RFC, the intensity of AF488 is about 2-fold greater. As expected, the fluorescence intensity of AF488 in an open RFC-PCNA complex is less than the intensity for the denatured protein because interactions between fluorophores at only one of the three interfaces are expected to be disrupted on clamp opening. Environmental effects due to the presence of SDS and/or protein unfolding could also affect AF488 fluorescence under denaturing conditions.
PCNA Opening in Equilibrium Binding Reactions with RFC
To determine whether the introduction of site-directed mutations and/or the fluorophores affect interactions with RFC, PCNA opening was measured in equilibrium binding assays. The increase in AF488 fluorescence as a function of RFC concentration was measured at three different concentrations of PCNA-AF4882: 1 nm, 5 nm (Fig. 2A, red and blue circles, respectively), and 10 nm (Fig. 3A, filled circles). Dissociation constants (Kd) were calculated from three independent experiments at each concentration of PCNA-AF4882, and Kd values were 4.5 ± 0.1, 2.6 ± 0.1, and 3.0 ± 1.0 nm at 1, 5, and 10 nm PCNA-AF4882, respectively. These values are in agreement with each other and with a previously reported Kd value (1.3 nm) (20).
FIGURE 2.

PCNA opening in equilibrium binding assays with RFC. A, AF488 fluorescence was measured in solutions containing increasing concentrations of RFC and either 1 nm (red) or 5 nm (blue) PCNA-AF4882 and 0.5 mm ATP. B, AF488 fluorescence was measured in a competition binding assay containing 20 nm PCNA-AF4882, 20 nm RFC, 0.5 mm ATP and increasing concentrations of unlabeled PCNA. RFC, at a constant concentration, was added to the mixture of labeled and unlabeled PCNA. The average value of relative intensity of AF488 from three independent experiments is plotted with standard deviations.
FIGURE 3.

PCNA opening and binding in equilibrium assays with RFC and Rad24-RFC. A and B, PCNA opening (A) and binding (B) were measured in solutions containing increasing concentrations of either RFC (filled circles) or Rad24-RFC (filled squares), 0.5 mm ATP, and 10 nm PCNA-AF4882 (in opening assays) or 10 nm PCNA-MDCC (in binding assays). For comparison, PCNA opening and binding by RFC in the absence of ATP (open circles) is shown. Average values and standard deviations for three independent experiments are shown except for Rad24-RFC opening data, which is the result of two independent experiments. Solid lines through the data were generated by fitting to the quadratic equation (Equation 1, “Experimental Procedures”).
To verify that RFC binds the PCNA-AF4882 mutant with the same affinity as wild-type PCNA, binding was also measured in a competition assay. In this assay, RFC-PCNA-AF4882 binding was measured in the presence of increasing concentrations of unlabeled WT PCNA. As the concentration of unlabeled competitor PCNA increased, the amount of RFC bound to PCNA-AF4882 decreased with the interaction reaching mid-point at equal concentrations of labeled and unlabeled PCNA. The data can be fit by an equation (Equation 2) that simply takes into account the fraction of the total PCNA that is labeled. This demonstrates that RFC binds both PCNA-AF4882 and WT PCNA with the same affinity. Together, these binding experiments show that PCNA-AF4882 interacts with RFC in a manner similar to WT PCNA and that the introduction of mutations and fluorophores does not affect this interaction.
PCNA Opening by Rad24-RFC
Previous studies have shown that Rad24-RFC can remove or unload PCNA from a circular DNA plasmid but cannot load PCNA onto DNA (10). To determine what the molecular basis for this observation is, PCNA opening by Rad24-RFC was measured. The PCNA opening assay provides two pieces of information. 1) The clamp loader concentration dependence of the increase in AF488 fluorescence provides a measure of the binding interaction, and 2) the relative fluorescence intensity of AF488 at saturating concentrations of clamp loader provides a measure of the fraction of clamps in an open conformation. When PCNA-AF4882 was titrated with Rad24-RFC, there was very little increase in AF488 fluorescence. Opening data for assays containing 10 nm PCNA-AF4882 and either RFC (filled circles) or Rad24-RFC (filled squares) in the presence of 0.5 mm ATP are shown in Fig. 3A. For comparison, PCNA-AF4882 was titrated with RFC in the absence of ATP (Fig. 3A, open circles), which is required for efficient clamp opening. Based on the relative fluorescence of AF488 at high clamp loader concentrations, the clamp-opening activity of Rad24-RFC in the presence of ATP is similar to the weak clamp-opening activity of RFC in the absence of ATP.
The clamp-opening activity of Rad24-RFC could be weak because Rad24-RFC has a lower affinity for PCNA than RFC. To determine whether this is the case, PCNA binding was measured for both clamp loaders. A binding assay that has been used to measure the E. coli clamp loader binding to the clamp (21) was adapted for the S. cerevisiae system.4 Serine 43, which is located on the surface of PCNA to which RFC binds (3), was mutated to Cys in the triple Cys to Ser PCNA mutant. Cys-43 was covalently labeled with an environmentally sensitive fluorophore, MDCC, to form PCNA-MDCC. This PCNA mutant allows for incorporation of one MDCC moiety per monomer. When RFC (or Rad24-RFC) binds PCNA-MDCC in the presence of ATP, MDCC fluorescence intensity increases by just over 2-fold. Binding of RFC and Rad24-RFC to PCNA-MDCC (10 nm) was measured under equilibrium conditions (Fig. 3B, filled circles and filled squares, respectively). Kd values of 7.4 ± 1.2 and 36 ± 6 nm for RFC and Rad24-RFC, respectively, binding PCNA-MDCC were calculated by fitting data to Equation 1. These data show that the binding interaction between Rad24-RFC and PCNA is weaker than that for RFC and PCNA. However, at high (saturating) clamp loader concentrations, the fraction of PCNA bound by both RFC and Rad24-RFC is similar (Fig. 3B), but much less PCNA is in an open conformation at these concentrations in assays with Rad24-RFC than RFC (Fig. 3A). Therefore, the weak PCNA opening activity of Rad24-RFC is not simply the result of weak PCNA binding. It is formally possible that weak PCNA opening by Rad24-RFC could be due to weak ATP binding; however, a Kd value of 5 μm has been reported for ATP binding Rad24-RFC (22). Therefore, it is likely that Rad24-RFC is saturated with ATP at the 0.5 mm concentrations used in our experiments. Interestingly, when RFC binding to PCNA-MDCC was measured in the absence of ATP (Fig. 3B, open circles), a Kd value of 38 ± 8 nm was calculated that is similar to that for Rad24-RFC binding PCNA-MDCC in the presence of ATP. At saturating concentrations of RFC in the absence of ATP, the fluorescence of MDCC increases to a greater extent than in the presence of ATP. This likely reflects different interactions between the clamp loader and the clamp in the presence and absence of ATP.
Real-time PCNA Opening by RFC and Rad24-RFC
PCNA binding and opening by RFC were measured in real time by monitoring the increase in AF488 and MDCC fluorescence when a solution of RFC (200 nm final concentration) and ATP was added to a solution of labeled PCNA (20 nm final concentration) and ATP. The increase in MDCC fluorescence due to PCNA-MDCC binding PCNA-MDCC (Fig. 4A, gray trace) was much faster than the increase in AF488 fluorescence due to PCNA-AF4882 opening (Fig. 4A, black trace). Observed rates of binding and opening calculated from single exponential fits of the data were 41 and 2.3 s−1, respectively. The observed rate of PCNA opening in assays containing 20 nm PCNA-AF4882 and 25, 50, 100, 200, and 400 nm RFC was constant at a value of 2.2 ± 0.1 s−1. At these concentrations, the rate of clamp opening is limited by the slow opening reaction and not the rapid binding reaction.
FIGURE 4.

Rates of PCNA binding and opening by RFC and Rad24-RFC. A, time courses for RFC binding (gray) and opening (black) PCNA measured by the increase in fluorescence of MDCC or AF488, respectively, when a solution of RFC was added to a solution of PCNA. Final concentrations were 200 nm RFC, 0.5 mm ATP, and 20 nm PCNA-MDCC or PCNA-AF4882. B, time courses for PCNA opening were measured for Rad24-RFC (black trace) and RFC (gray trace) when a solution of clamp loader and ATP was added to a solution of PCNA-AF4882 and ATP. A control reaction in which buffer only was added to PCNA-AF4882, showing that the signal for AF488 remains constant in the absence of clamp loader, is shown. Final concentrations were 200 nm clamp loader, 20 nm PCNA-AF4882, and 0.5 mm ATP.
Because Rad24-RFC has a reduced ability to form stable open complexes with PCNA in the steady state as compared with RFC, we wanted to determine the rate at which Rad24-RFC opens PCNA. Stopped-flow binding assays were performed as in Fig. 4A in side-by-side experiments with Rad24-RFC and RFC in reactions that contained 20 nm PCNA-AF4882, 200 nm Rad24-RFC or RFC, and 0.5 mm ATP. The amplitude for the Rad24-RFC time course was small as expected from steady state assays; however, an exponential fit of the binding data yielded an observed rate of 1.5 s−1 (Fig. 4B, black trace). This is similar to the rate of 2.3 s−1 for RFC opening PCNA (Fig. 4B, gray trace) and suggests that the defect in PCNA opening by Rad24-RFC may stem from a decreased ability of this clamp loader to stabilize the open conformation (i.e. the PCNA closing rate is likely faster for Rad24-RFC).
DISCUSSION
Sliding clamps are assembled onto DNA by clamp loaders where they bind to DNA polymerases to increase the processivity of DNA synthesis and overall efficiency of replication in a process that is conserved from bacteria to humans (reviewed in Refs. 12 and 23). Although many of the structural and functional features of the replication machinery are conserved, studies using different organisms have shown that there is some divergence in the mechanistic details of the reactions. Here, we developed a fluorescence intensity-based assay to investigate the PCNA opening reaction catalyzed by RFC and the DNA damage checkpoint clamp loader, Rad24-RFC. The clamp-opening assay takes advantage of self-quenching fluorophores located within close proximity on either side of the PCNA monomer interfaces. Although this assay does not provide distance measurements that can be obtained from FRET-based assays, it is a robust and simple method for measuring the fraction of clamps that are open under a given set of conditions. Rates of PCNA opening measured in our relief of quench assay (2.2 ± 0.1 s−1) were identical to the rate of PCNA opening (2.13 ± 0.05 s−1) measured using a FRET-based assay (24).
Clamp loaders must hold clamps in an open conformation to load clamps onto DNA, and this opening reaction appears to differ between species. The bacteriophage T4 clamp loader binds clamps that have transiently opened in solution (25, 26), whereas the E. coli clamp loader binds closed clamps prior to opening them (19, 27, 28). To determine which mechanism is utilized by RFC, rates of PCNA binding and opening were measured under identical conditions (Fig. 4A), and the rate of RFC-PCNA binding was nearly 20-fold faster than the rate of PCNA opening. These kinetic data show that RFC binds closed PCNA prior to PCNA opening in a mechanism similar to the E. coli clamp loader. Interestingly, the rate of clamp opening by RFC is about 5-fold slower than the rate of clamp opening by the E. coli γ complex. The mechanistic basis for this difference is not clear, but the E. coli clamp loader may be a more robust clamp opener than RFC. Given the greater stability of the β-clamp (29), the E. coli clamp opener may actively pry the β-clamp open, whereas RFC may bind PCNA and wait for PCNA to open. Computational studies suggest that RFC simply stabilizes the open conformation of PCNA rather than destabilizing the closed conformation to crack PCNA open (30). Thus, the mechanism for clamp opening by RFC may be intermediate between the bacteriophage T4 clamp loader that captures open clamps in solution and the E. coli clamp loader that actively opens closed clamps.
ATP binding to clamp loaders increases the affinity of the clamp loaders for clamps. The binding affinity of the E. coli clamp loader for the clamp increases by 2–3 orders of magnitude in the presence of ATP (31, 32). There is a smaller difference in Kd values, about an order of magnitude, for RFC binding PCNA in the presence and absence of ATP (Fig. 3B). The smaller difference is due to stronger binding in the absence of ATP as Kd values for the E. coli γ complex-β-clamp (31) and RFC-PCNA interactions are about the same in the presence of ATP. Another interesting difference between the E. coli and S. cerevisiae clamp loaders is that there is no measurable formation of open clamp loader-clamp complexes in the absence of ATP for the E. coli γ complex (19). In contrast, based on the relative fluorescence of AF488 at saturating RFC concentrations, about 20% of the number of open RFC-PCNA complexes form in the absence of ATP as in the presence of ATP (Fig. 3A). Structural data and molecular modeling suggest that the contact area between clamp loaders and clamps is greater in the open than closed conformation (3, 6, 30). This would be consistent with the difference in clamp binding by the γ complex and RFC in the absence of ATP, where binding is stronger for RFC because RFC can form some open clamp loader-clamp complexes.
The PCNA binding and opening activities of Rad24-RFC are weaker than those for RFC. The Kd value for the Rad24-RFC-PCNA interaction in the presence of ATP is about the same as for the weaker RFC-PCNA interaction in the absence of ATP. The relative fluorescence of AF488 at saturating concentrations of Rad24-RFC is about 8% of that for RFC, showing that a much smaller fraction of clamps is open. The weaker clamp-opening activity of Rad24-RFC certainly contributes to the inability of Rad24-RFC to load PCNA on DNA but may not be the only factor involved. However, the weak opening activity may be sufficient to allow Rad24-RFC to unload PCNA from DNA, a reaction that may be catalyzed by a more transient interaction.
This work was supported, in whole or in part, by National Institutes of Health Grants GM082849 (to L. B. B.) and GM038839 (to M. O. D.).
Throughout this study, the designation AF488 indicates Alexa Fluor 488 C5 maleimide. The designation PCNA-AF4882 is used to indicate PCNA covalently labeled with AF488 on residues I111C and I181C, and PCNA-MDCC is used to indicate PCNA covalently labeled with MDCC on residue S43C.
M. R. Marzahn and L. B. Bloom, manuscript in preparation.
- PCNA
- proliferating cell nuclear antigen
- MDCC
- N-(2-(1-maleimidyl) ethyl)-7-(diethylamino) coumarin-3-carboxamide
- RFC
- replication factor C
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- TCEP
- tris(2-carboxyethyl) phosphine.
REFERENCES
- 1. Krishna T. S., Kong X. P., Gary S., Burgers P. M., Kuriyan J. (1994) Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 2. Erzberger J. P., Berger J. M. (2006) Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 [DOI] [PubMed] [Google Scholar]
- 3. Bowman G. D., O'Donnell M., Kuriyan J. (2004) Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429, 724–730 [DOI] [PubMed] [Google Scholar]
- 4. Simonetta K. R., Kazmirski S. L., Goedken E. R., Cantor A. J., Kelch B. A., McNally R., Seyedin S. N., Makino D. L., O'Donnell M., Kuriyan J. (2009) The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 137, 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyata T., Suzuki H., Oyama T., Mayanagi K., Ishino Y., Morikawa K. (2005) Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc. Natl. Acad. Sci. U.S.A. 102, 13795–13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kazmirski S. L., Zhao Y., Bowman G. D., O'donnell M., Kuriyan J. (2005) Out-of-plane motions in open sliding clamps: molecular dynamics simulations of eukaryotic and archaeal proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. U.S.A. 102, 13801–13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Navadgi-Patil V. M., Burgers P. M. (2011) Cell cycle-specific activators of the Mec1/ATR checkpoint kinase. Biochem. Soc. Trans. 39, 600–605 [DOI] [PubMed] [Google Scholar]
- 9. Bermudez V. P., Lindsey-Boltz L. A., Cesare A. J., Maniwa Y., Griffith J. D., Hurwitz J., Sancar A. (2003) Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1633–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yao N. Y., Johnson A., Bowman G. D., Kuriyan J., O'Donnell M. (2006) Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J. Biol. Chem. 281, 17528–17539 [DOI] [PubMed] [Google Scholar]
- 11. Bloom L. B. (2009) Loading clamps for DNA replication and repair. DNA Repair 8, 570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Indiani C., O'Donnell M. (2006) The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 7, 751–761 [DOI] [PubMed] [Google Scholar]
- 13. McHenry C. S. (2011) DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80, 403–436 [DOI] [PubMed] [Google Scholar]
- 14. Finkelstein J., Antony E., Hingorani M. M., O'Donnell M. (2003) Overproduction and analysis of eukaryotic multiprotein complexes in Escherichia coli using a dual-vector strategy. Anal. Biochem. 319, 78–87 [DOI] [PubMed] [Google Scholar]
- 15. Yao N., Coryell L., Zhang D., Georgescu R. E., Finkelstein J., Coman M. M., Hingorani M. M., O'Donnell M. (2003) Replication factor C clamp loader subunit arrangement within the circular pentamer and its attachment points to proliferating cell nuclear antigen. J. Biol. Chem. 278, 50744–50753 [DOI] [PubMed] [Google Scholar]
- 16. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 17. Ayyagari R., Impellizzeri K. J., Yoder B. L., Gary S. L., Burgers P. M. (1995) A mutational analysis of the yeast proliferating cell nuclear antigen indicates distinct roles in DNA replication and DNA repair. Mol. Cell Biol. 15, 4420–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bauer G. A., Burgers P. M. (1988) The yeast analog of mammalian cyclin/proliferating-cell nuclear antigen interacts with mammalian DNA polymerase δ. Proc. Natl. Acad. Sci. U.S.A. 85, 7506–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paschall C. O., Thompson J. A., Marzahn M. R., Chiraniya A., Hayner J. N., O'Donnell M., Robbins A. H., McKenna R., Bloom L. B. (2011) J. Biol. Chem. 286, 42704–42714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gomes X. V., Burgers P. M. (2001) ATP utilization by yeast replication factor C: I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem. 276, 34768–34775 [DOI] [PubMed] [Google Scholar]
- 21. Thompson J. A., Paschall C. O., O'Donnell M., Bloom L. B. (2009) A slow ATP-induced conformational change limits the rate of DNA binding but not the rate of β clamp binding by the Escherichia coli γ complex clamp loader. J. Biol. Chem. 284, 32147–32157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majka J., Chung B. Y., Burgers P. M. (2004) Requirement for ATP by the DNA damage checkpoint clamp loader. J. Biol. Chem. 279, 20921–20926 [DOI] [PubMed] [Google Scholar]
- 23. Johnson A., O'Donnell M. (2005) Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 74, 283–315 [DOI] [PubMed] [Google Scholar]
- 24. Zhuang Z., Yoder B. L., Burgers P. M., Benkovic S. J. (2006) The structure of a ring-opened proliferating cell nuclear antigen-replication factor C complex revealed by fluorescence energy transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alley S. C., Abel-Santos E., Benkovic S. J. (2000) Tracking sliding clamp opening and closing during bacteriophage T4 DNA polymerase holoenzyme assembly. Biochemistry 39, 3076–3090 [DOI] [PubMed] [Google Scholar]
- 26. Trakselis M. A., Alley S. C., Abel-Santos E., Benkovic S. J. (2001) Creating a dynamic picture of the sliding clamp during T4 DNA polymerase holoenzyme assembly by using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A. 98, 8368–8375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Indiani C., O'Donnell M. (2003) Mechanism of the δ wrench in opening the β sliding clamp. J. Biol. Chem. 278, 40272–40281 [DOI] [PubMed] [Google Scholar]
- 28. Jeruzalmi D., Yurieva O., Zhao Y., Young M., Stewart J., Hingorani M., O'Donnell M., Kuriyan J. (2001) Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 106, 417–428 [PubMed] [Google Scholar]
- 29. Yao N., Turner J., Kelman Z., Stukenberg P. T., Dean F., Shechter D., Pan Z. Q., Hurwitz J., O'Donnell M. (1996) Clamp loading, unloading and intrinsic stability of the PCNA, beta, and gp45 sliding clamps of human, E. coli, and T4 replicases. Genes Cells 1, 101–113 [DOI] [PubMed] [Google Scholar]
- 30. Tainer J. A., McCammon J. A., Ivanov I. (2010) Recognition of the ring-opened state of proliferating cell nuclear antigen by replication factor C promotes eukaryotic clamp-loading. J. Am. Chem. Soc. 132, 7372–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naktinis V., Onrust R., Fang L., O'Donnell M. (1995) Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 270, 13358–13365 [PubMed] [Google Scholar]
- 32. Snyder A. K., Williams C. R., Johnson A., O'Donnell M., Bloom L. B. (2004) Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp: II. Uncoupling the β and DNA binding activities of the γ complex. J. Biol. Chem. 279, 4386–4393 [DOI] [PubMed] [Google Scholar]