

Abstract
The amyloid cascade hypothesis suggests that the aggregation and deposition of amyloid-β protein is an initiating event in Alzheimer's disease (AD). Using amyloid imaging technology, such as the positron emission tomography (PET) agent Pittsburgh compound-B (PiB), it is possible to explore the natural history of preclinical amyloid deposition in people at high risk for AD. With this goal in mind, asymptomatic (n = 5) and symptomatic (n = 5) carriers of presenilin-1 (PS1) mutations (C410Y or A426P) that lead to early-onset AD and noncarrier controls from both kindreds (n = 2) were studied with PiB–PET imaging and compared with sporadic AD subjects (n = 12) and controls from the general population (n = 18). We found intense and focal PiB retention in the striatum of all 10 PS1 mutation carriers studied (ages 35–49 years). In most PS1 mutation carriers, there also were increases in PiB retention compared with controls in cortical brain areas, but these increases were not as great as those observed in sporadic AD subjects. The two PS1 mutation carriers with a clinical diagnosis of early-onset AD did not show the typical regional pattern of PiB retention observed in sporadic AD. Postmortem evaluation of tissue from two parents of PS1C410Y subjects in this study confirmed extensive striatal amyloid deposition, along with typical cortical deposition. The early, focal striatal amyloid deposition observed in these PS1 mutation carriers is often is not associated with clinical symptoms.
Keywords: Alzheimer's disease, positron emission tomography, amyloid-β, diagnosis, Pittsburgh Compound-B, striatum
Introduction
Amyloid-β (Aβ) plaque deposition is a pathological hallmark of Alzheimer's disease (AD), and altered metabolism of Aβ and the β-amyloid precursor protein (APP) in the brain has been postulated to be the primary cause of AD (Beyreuther et al., 1992; Hardy and Higgins, 1992; Joachim and Selkoe, 1992). If this “amyloid cascade hypothesis” is correct, amyloid deposits in the brains of those destined to develop AD should be present before the onset of clinical symptoms. Several postmortem studies suggest that this is true (Morris et al., 1996; Haroutunian et al., 1998; Price and Morris, 1999), but it is impossible to define precisely the natural history of amyloid deposition in AD by the use of postmortem studies alone.
Several groups have reported in vivo amyloid imaging studies in cognitively normal controls and subjects with mild cognitive impairment (MCI) (Petersen, 2004; Gauthier et al., 2006) or AD using the positron emission tomography (PET) tracer Pittsburgh compound-B (PiB) (Klunk et al., 2004; Verhoeff et al., 2004; Buckner et al., 2005; Lopresti et al., 2005; Price et al., 2005; Archer et al., 2006; Drzezga et al., 2006; Edison et al., 2006; Engler et al., 2006; Fagan et al., 2006; Kemppainen et al., 2006; Mintun et al., 2006; Villemagne et al., 2006; Zhou et al., 2006; Ziolko et al., 2006; Rabinovici et al., 2007). Other PET tracers have been used for in vivo amyloid imaging as well (Shoghi-Jadid et al., 2002; Verhoeff et al., 2004; Small et al., 2006). PiB is selectively retained in brain areas known to have heavy postmortem amyloid deposition in AD. This suggests that PiB–PET imaging is well suited to answer questions such as those regarding the natural history of amyloid deposition within individuals over time and the relationship of asymptomatic amyloid deposition to the eventual development of clinical AD.
Despite the availability of in vivo amyloid imaging technology, natural history studies of preclinical amyloid deposition remain difficult in the general population because there is currently no clinical method that can be used to accurately identify those asymptomatic individuals who are destined to develop AD. However, the predictable course of AD in the relatively rare [∼1% of all AD (Pastor and Goate, 2004)] individuals who develop early-onset familial AD with an autosomal dominant inheritance pattern (eoFAD) offers an opportunity to learn about the natural history of amyloid deposition (Tanzi et al., 1996; St George-Hyslop, 1999). Known eoFAD mutations occur in the presenilin-1 (PS1) gene on chromosome 14, the PS2 gene on chromosome 1, and the APP gene on chromosome 21 (Hardy et al., 1998). The chromosome 14 PS1 mutations are the most common known mutations and account for ∼75% of all eoFAD (Pastor and Goate, 2004). The PS1 gene codes for a protein that is strongly implicated to be an essential component of the “γ-secretase” enzyme complex responsible for C-terminal cleavage of Aβ from its precursor APP (Xia et al., 2000). More than 150 different pathogenic PS1 mutations have been described previously (Cruts and Van Broeckhoven, 1998) with mean age of onset across families from 32 to 56 years (for review, see Tanzi et al., 1996; St George-Hyslop, 1999). Most of these genes have 100% penetrance before age 60 (Tanzi et al., 1996; St George-Hyslop, 1999). Therefore, in these eoFAD families, genetic testing can identify those individuals destined to develop AD with great certainty decades before the expected onset of symptoms.
In this study, we used PiB–PET amyloid imaging to study both asymptomatic and mildly symptomatic individuals from two unrelated pedigrees who carry different mutations in the PS1 gene [i.e., a cysteine to tyrosine mutation at position 410 of the PS1 protein (C410Y) (Campion et al., 1995; Sherrington et al., 1995) or an alanine to proline mutation at position 426 (A426P) (Poorkaj et al., 1998)]. This baseline data are intended to serve as the initial time point in a longitudinal natural history study.
Materials and Methods
Human subjects
Table 1 lists the demographic, clinical, genetic, and cognitive characteristics of the subjects included in this study. From the C410Y kindred, we studied five subjects who carried the PS1C410Y mutation (CY1–CY5; 35–45 years of age) and one control subject who was a cousin of the PS1C410Y carriers but did not carry the mutation (CY-C; 35 years of age). None of these subjects were symptomatic, but the average age for onset of symptoms in this C410Y kindred has been reported to be 48 years (range, 42–56 years) (Mann et al., 2001; Moonis et al., 2005). From the A426P kindred, we studied five siblings who carried the PS1A426P mutation (AP1–AP5; 35–49 years of age) and one control sibling who did not carry the mutation (AP-C; age of 47 years). All five PS1A426P mutation carriers were at least mildly symptomatic (i.e., MCI) with ages of onset from 35–44 years. All subjects were studied in Pittsburgh, except CY4 and CY5, who were studied at Massachusetts General Hospital/Harvard Medical School (MGH).
Table 1.
Clinical, genetic, and cognitive characteristics of subjects
| Clinical characteristic or cognitive testa | Sporadic ADb mean (SD) | Controlsb mean (SD) | Subject code |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CY-Cc (35) | CY1c (35) | CY2c (37) | CY3c (38) | CY4c (42)d | CY5c (45)d | AP-Ce (47) | AP1e (35) | AP2e (43) | AP3d (45) | AP4e (46) | AP5e (49) | |||
| Clinical diagnosis | Normal | Normal | Normal | Normal | Normal | Normal | Normal | MCI | MCI | AD | MCI | AD | ||
| ApoE genotype | 3/4 | 3/3 | 3/4 | 3/4 | 2/3 | 2/3 | 2/3 | 2/3 | 2/3 | 2/3 | 2/3 | 3/3 | ||
| MMSE | 21.7 (3.7) | 28.4 (1.5) | 29 | 29 | 27 | 28 | 30 | 29 | 28 | 22 | 27 | 21 | 25 | 16 |
| CDR | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | ||
| CERAD EPSf | 0 | 0 | 0 | 4 | 2 | 4 | 0 | 6 | ||||||
| UPDRSg | 0 | 2 | 4 | |||||||||||
| CERAD WLL delayed recall (max of 10) | 0.6 (1.2) | 8.1 (1.7) | 7 | 7 | 8 | 9 | 10 | 5 | 5 | 3 | 2 | 0 | ||
| Digit span forward | 6.3 (1.2) | 6.8 (1.3) | 6 | 8 | 9 | 7 | 7 | 4 | 8 | 6 | 7 | 6 | 6 | 6 |
| Digit span backward | 3.4 (1.4) | 5.3 (1.1) | 5 | 7 | 8 | 5 | 7 | 6 | 7 | 4 | 5 | 4 | 4 | 3 |
| Trail Making Test A (s) | 80.7 (65.3) | 32.0 (8.8) | 25 | 10 | 18 | 20 | 24 | 27 | 19 | 28 | 43 | 38 | 27 | 78 |
| Trail Making Test B (s) | 235.3 (64.5) | 75.2 (34.3) | 32 | 24 | 37 | 50 | 52 | 57 | 59 | 71 | 118 | 300 | 73 | 300 |
| Letter fluency (FAS total) | 31.5 (12.7) | 47.3 (17.0) | 58 | 65 | 45 | 61 | 40 | 33 | 31 | 22 | 26 | 7 | 43 | 16 |
| Category fluency (animals) | 11.8 (5.7) | 20.3 (4.9) | 27 | 30 | 28 | 18 | 22 | 20 | 23 | 15 | 15 | 14 | 18 | 13 |
| Boston Naming Test (Saxton 30-item version) | 23.3 (4.6) | 28.6 (2.6) | 27 | 29 | 28 | 29 | 30 | 28 | 26 | 21 | 26 | 16 | 26 | 14 |
| Modified Rey-Osterrieth Figure copy (max of 24) | 17.4 (5.6) | 21.9 (1.1) | 21 | 24 | 20 | 22 | 20 | 23 | 17 | 15.5 | 23 | 8.5 | ||
| FCSRT free recall (max of 48) | 7.4 (5.6)h | 32.8 (6.6)i | 37 | 31 | ||||||||||
| Benton VFDT (max of 32) | 17.5 (9.3)h | 30.3 (2.9)i | 30 | 26 | ||||||||||
aSee Materials and Methods for abbreviations and descriptions. Bold values are abnormal, i.e., more than 1.5 SDs away from the control norm.
bThe means listed are from the 12 AD and 18 control patients who participated in this study in Pittsburgh except when indicated.
cC410Y mutation kindred (Campion et al., 1995; Sherrington et al., 1995). CY-C indicates the noncarrier cousin control subject. Ages are shown in parentheses.
dSubjects studied at MGH.
eA426P mutation kindred (Poorkaj et al., 1998). AP-C indicates the noncarrier sibling control subject. Ages are shown in parentheses.
fExtrapyramidal symptom total score according to the Consortium to Establish a Registry for Alzheimer's Disease (Morris et al., 1989).
gTotal score from the Unified Parkinson's disease rating scale (Fahn and Elton, 1987).
hHistorical mean (SD) from AD patients studied at MGH (n = 20; mean age, 69.7 ± 11.0).
iHistorical mean (SD) from control subjects studied at MGH (n = 55; mean age, 71.3 ± 8.7).
To put the findings from the PS1 mutation carriers into context, we compared their PiB–PET data with that of 12 sporadic AD subjects (68.5 ± 8.6 years) and 18 cognitively normal controls (70.3 ± 15.3 years), none of whom had any family history of eoFAD. Four relatively young controls (39, 45, 45, and 48 years) were included among these 18 controls because of the young ages of the PS1 mutation carriers (35–49 years). PiB–PET data for six of these AD and six control subjects have been published previously (Lopresti et al., 2005; Price et al., 2005) and were presented again here in Figure 5 along with additional new AD (n = 6) and control (n = 12) data solely for comparison with the data from the PS1 mutation carriers (none of which has been published previously).
Figure 5.

Quantitative assessment of PiB retention in controls, AD subjects, and PS1 mutation carriers. Rectangles show the range of PiB retention for cognitively normal controls (red), AD subjects (blue), and PS1 mutation carriers (white). The young control (ages of 39–48 years) range is shown as a darker red box behind the full control range. PiB retention in individual subjects are shown within each rectangle for controls (red diamonds), AD subjects (blue triangles), and PS1 mutation carriers with normal cognition (white diamonds studied at Pittsburgh; black diamonds studied at MGH), mutation carriers with MCI (white circles), and mutation carriers with AD (white triangles). The filled red diamond represents NC+ from Figure 1 (shown for comparison but not included in the statistical analysis). PiB retention is expressed as the SUVR with pons as a reference averaged over 40–60 min after injection of PiB for all data. The means ± SD are shown with error bars within each rectangle. p values for the difference between controls and PS1 mutation carriers are indicated in red along the bottom, and p values comparing PS1 mutation carriers with AD patients are indicated in blue along the top (ns, not significant; *p < 0.005, **p < 0.00005, ***p < 0.0000005). Brain areas included were the frontal cortex (FRC), precuneus (PRC), parietal cortex (PAR), anterior–ventral striatum (AVS), occipital cortex (OCC), mesial temporal cortex (MTC), thalamus (THL), and cerebellum (CER).
Cognitively normal controls occasionally show clear evidence of PiB retention from visual inspection of the PiB image and quantitative analysis of the scan data (Mintun et al., 2006). These “PiB-positive controls” were not included here for the purpose of defining the range of PiB retention in amyloid-negative control brains. However, one such PiB-positive control (NC+, in which NC indicates “normal control”) with early evidence of amyloid deposition is shown as an example in Figures 1 and 5 but was not included in any of the quantitative analyses.
Figure 1.

PiB–PET and MRI images from sporadic AD and two older controls. Transaxial PiB–PET SUVRpons images (40–60 min) from a mild AD patient (77 years old; MMSE of 27), a normal elderly control (NC−: 72 years old; MMSE of 30), an elderly PiB-positive normal control (NC+) at baseline (MMSE of 30) and 2 years later (MMSE of 29), along with the MRI of the NC+ subject. The age of the NC+ subject was in the mid-70s and is not specified for reasons of confidentiality and blinding. A tracing of the location of the striatum from each subject's MRI scan is superimposed on the respective PiB image, but only the MRI for the NC+ subject is shown (far right).
All subjects were evaluated with standard neurological, psychiatric, and neuropsychological examinations through the University of Pittsburgh Alzheimer Disease Research Center (ADRC) as described previously (Lopez et al., 2000; Price et al., 2005), or with an equivalent evaluation at MGH (CY4 and CY5). The neuropsychological test battery is described below. All subjects and their caregivers (when appropriate) provided informed consent for both the ADRC clinical examination and the PET imaging protocol. This study was approved by the Human Use Subcommittee of the Radioactive Drug Research Committees and the Institutional Review Boards of the University of Pittsburgh and Massachusetts General Hospital. The C410Y subjects were initially recruited at the University of Massachusetts and participated in a CSF Aβ and tau study there (Moonis et al., 2005).
Neuropsychological evaluation
The neuropsychological test battery was designed to assess those cognitive domains known to be impaired in AD and also to be sensitive to MCI (Petersen, 2004; Gauthier et al., 2006) in younger individuals and included tests of attention, memory, visuoconstructive/spatial ability, language, and executive functioning. The test batteries included the Mini Mental State Exam (MMSE) (Folstein et al., 1975), Clinical Dementia Rating Scale (CDR) (Morris, 1993), the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Word List Learning (WLL) Delayed Recall Test (Welsh-Bohmer et al., 1994), the Wechsler Adult Intelligence Scale III Digit Span forward and backward (Wechsler, 1997), Trail Making Parts A and B (Reitan, 1958), letter fluency (words beginning with the letters “F,” “A,” or “S”) (Benton, 1968), category fluency (animals) (Borkowski et al., 1967), Boston Naming Test (Saxton 30-item version) (Saxton et al., 2000), and Modified Rey-Osterrieth Figure copy (maximum of 24) (Becker et al., 1987). The two subjects studied at MGH had the Free and Cued Selective Reminding Test (FCSRT free recall) (Buschke, 1984) substituted for the CERAD WLL and the Benton Visual Form Discrimination Test (VFDT) (Benton et al., 1994) substituted for the Modified Rey-Osterrieth Figure copy.
PET studies
Tracer and acquisition.
High specific-activity PiB (carbon-11 labeled) was produced as described previously (Wilson et al., 2004). The PIB–PET data were acquired as described recently by Lopresti et al. (2005). PET imaging was conducted using a Siemens (Munich, Germany) CTI ECAT HR+ scanner (three-dimensional mode; 15.2 cm field of view; 63 image planes; reconstructed image resolution of ∼6 mm). The subject's head was immobilized to minimize head motion during the scan. PIB was injected intravenously (14.4 ± 1.9 mCi, over 20 s; specific activity, 1.5±.7 Ci/μmol). For the 10 subjects studied in Pittsburgh, dynamic PET scanning was performed over 90 min (34 time frames). The two subjects studied at MGH were scanned on a GE Scanditronix (Uppsala, Sweden) PC4096WB scanner (two-dimensional mode; 9.7 cm axial field of view; 15 image planes), and PiB–PET data were acquired over 60 min (69 frames). Subjects studied in Pittsburgh also underwent magnetic resonance imaging (MRI) scans. Before the PET imaging sessions in Pittsburgh, a spoiled gradient recalled MR scan was obtained for each subject for MR-PET image coregistration and anatomical volume-of-interest (VOI) definition as described previously (Lopresti et al., 2005; Price et al., 2005). Unlike previous studies (Lopresti et al., 2005; Price et al., 2005), MRI-based partial volume correction of the Pittsburgh data were not performed, to be comparable with the MGH data (for which no MRI data were available). VOIs were defined on the coregistered MR image as described previously (Price et al., 2005) and included the following: frontal cortex, precuneus/posterior cingulate gyrus, parietal cortex, anterior–ventral striatum, occipital cortex, mesiotemporal cortex, thalamus, cerebellum, and pons. The mesiotemporal cortex region included the amygdala, hippocampus, entorhinal cortex, and other portions of the parahippocampal gyrus. Equivalent VOIs were generated for the PET images of the MGH subjects without the use of MRI. Additional areas covering a large portion of the brain also were analyzed as described previously (Price et al., 2005), but the findings in these additional areas correspond closely to the representative sample discussed here.
Choice of reference region.
Analysis of the PiB–PET data used the standardized uptake value (SUV) approach (Klunk et al., 2004). This was chosen over the previously validated and commonly used Logan graphical analysis approach (Price et al., 2005) for several reasons. First, because arterial blood data were not available for 7 of the 10 subjects, the Logan distribution volume (DV) using an arterial input function could not be determined (Price et al., 2005). Second, the applicability of the cerebellar reference tissue approach to the Logan graphical method (Lopresti et al., 2005; Mintun et al., 2006) could not be fully justified for the eoFAD subjects because postmortem studies of PS1 mutation carriers have shown significantly greater Aβ plaque densities in the cerebellum compared with sporadic AD and controls (Lippa et al., 1996; Mann et al., 2001). This is consistent with the finding that PiB retention in the cerebellum of the eoFAD subjects, when expressed as the SUV, appeared to increase with age in the eoFAD subjects (r = 0.826; p = 0.003). PiB retention with age was not observed in sporadic AD cases. However, because there was no significant age-related increase of PiB retention in the pons of the eoFAD cases, the pons was used as the reference tissue for this study. The use of pons as an amyloid-free reference is supported by studies showing an absence of pontine Aβ plaques (Thal et al., 2002) and cerebrovascular amyloid (Kyriakides et al., 1994) in the vast majority of sporadic AD cases. Minoshima et al. (1995) have previously used the pons as a reference region for glucose metabolic PET studies in AD for similar reasons. In subjects without amyloid, the pharmacokinetics of PiB differs in the pons (and subcortical white matter) compared with cortical areas (Price et al., 2005). Therefore, PiB pharmacokinetics in the pons may not adequately represent the cortical tissue kinetics of nonspecifically bound and free PiB. As a result, PiB retention in the pons was not used as an input function for the Logan reference tissue method (Logan et al., 1996). As a compromise, the SUV ratio using pons as the reference tissue (SUVRpons) was chosen as the preferred method of analysis for this eoFAD study.
Choice of analysis window.
Although 90 min of data were collected for the subjects studied in Pittsburgh, the SUVRpons values were averaged over 40–60 min because the MGH data were collected for only 60 min. SUVR data averaged over 40–60 min has been shown previously to compare well with Logan DV ratio (DVR) data collected over 90 min (Lopresti et al., 2005). In this study, the 40–60 min SUVRpons values in the control and sporadic AD cases were highly comparable with the Logan DVR data using an input function derived from cerebellar tissue radioactivity [referred to as the CER90 method by Lopresti et al. (2005)] (e.g., for anterior–ventral striatum, r = 0.95; p < 0.000001; slope, 1.07). Furthermore, we saw no significant difference in the pons SUV in control (1.44 ± 0.26), AD (1.43 ± 0.33), and eoFAD groups (1.50 ± 0.20), making this a good reference region. Separate analyses were run using the CER90 method and yielded equivalent results.
The quantitative analysis (see Fig. 5) was performed over the 40–60 min epoch for all Pittsburgh and MGH cases. However, the MGH images shown in Figure 2 represent data averaged over a longer time period (i.e., 20–60 min) to minimize noise in the images and facilitate the comparison with the Pittsburgh images. The increased noise in the MGH images was a result of differences in scanner type, two-dimensional versus three-dimensional acquisition, short frame durations, and image processing. All other images, except the two MGH images in Figure 2, represent parametric SUVRpons data collected over 40–60 min.
Figure 2.
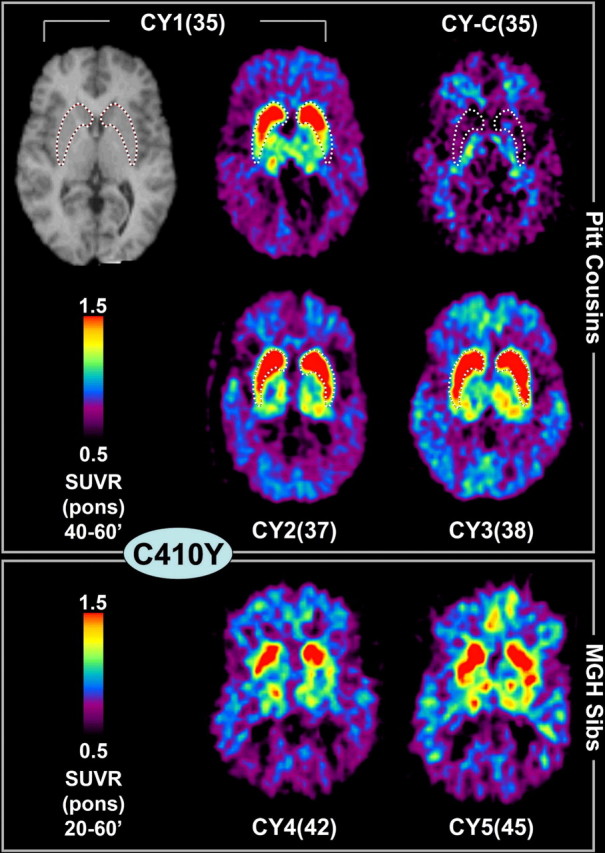
PS1C410Y mutation carriers: PiB–PET and MRI images. Transaxial PiB–PET SUVRpons images from five cousins who carry the PS1C410Y mutation and a cousin without the mutation, CY-C(35). All are cognitively normal. The top row shows the MRI and PiB–PET scan of CY1(35) and the PiB–PET of the noncarrier cousin. The middle row shows the two other PS1C410Y carrier cousins studied in Pittsburgh. The bottom row shows two PS1C410Y carrier sibs (cousins of the Pittsburgh subjects) that were imaged at MGH (averaged over 20–60 min to facilitate comparison with the Pittsburgh images; see Materials and Methods). A tracing of the location of the striatum from each Pittsburgh subject's MRI scan is superimposed on the respective PiB image, but only the MRI for the CY1(35) subject is shown (top left).
Statistical methods
The statistical significance of group differences was tested for each VOI using the Wilcoxon's rank-sum test (α = 0.05, two-sided, exact inference) (Hollander and Wolfe, 1973). A nonparametric method was used because of the small sample sizes. The p values for group differences were corrected for multiple comparisons using the Bonferroni's method (Bland and Altman, 1995). The level of statistical significance was set at 0.00625 (0.05/8 brain regions).
Histological analysis
Amyloid histology and Aβ immunohistochemistry were performed on Formalin-fixed tissue sections obtained from the Drexel University College of Medicine brain bank, as described in detail previously (Ikonomovic et al., 2004). Briefly, tissue sections were stained with a mouse monoclonal antibody 10D5 (Aβ1–16, 1:1500; Elan Pharmaceuticals, San Francisco, CA) using a signal intensification method with imidazole acetate buffer and DAB/nickel ammonium sulfate. For 6-CN-PiB and X-34 fluorescent histochemistry, slides were incubated with 6-CN-PiB (45 min) or X-34 (10 min), washed in dH2O, differentiated in 80% ethanol/0.2% NaOH and PBS, respectively, washed in dH2O, and coverslipped (Ikonomovic et al., 2006).
Results
Clinical characterization
No subject showed active psychiatric disorders or neurological abnormalities other than the cognitive problems described below. The specific PS1 mutation, neuropsychological test scores, clinical diagnosis, and apolipoprotein E (ApoE) genotype of each subject are shown in Table 1 in which subjects are coded according their mutation and age [e.g., CY1(35), in which “CY” refers to the C410Y mutation, “1” is an arbitrary subject number, and “35” represents the age when scanned]. Quantitative ratings of extrapyramidal symptoms according to CERAD (Morris et al., 1989) or the Unified Parkinson's disease rating scale (UPDRS) (Fahn and Elton, 1987) are shown in Table 1. All subjects had either no symptoms or clinically insignificant symptoms, and no subject was judged to have symptoms that would suggest Parkinson's disease.
PS1 mutation carriers CY1(35), CY3(38), and CY4(42) scored entirely within the normal range on all neuropsychological tests (i.e., within 1 SD of control norms). Subjects CY2(37) and CY5(45) had mild difficulty on visuoconstructive tests, with CY5(45) also exhibiting mild attentional difficulty, whereas performance on all other tests was normal. They did not meet clinical criteria for AD or MCI (Petersen, 2004; Gauthier et al., 2006). Subject AP1(35) (in which “AP” refers to the A426P mutation and the rest is as above) showed mild memory deficits and mild difficulties with word generation and confrontation naming. Subject AP2(43) exhibited mild difficulty on tests of memory and visuoconstructive ability. The test performances for AP1(35) and AP2(43) met criteria for “Amnestic MCI–Multiple Domain” (Petersen, 2004; Gauthier et al., 2006). Subject AP4(46) exhibited moderate difficulty on tests of memory, with normal performance on other tests, a pattern that characterizes “Amnestic MCI–Single Domain.” Subjects AP3(45) and AP5(49) both met clinical criteria for AD (McKhann et al., 1984; Lopez et al., 2000). Both exhibited memory and language impairments, but their most severe deficits were evident on tests of visuoconstructive ability and executive functioning. The family reported that AP1(35) had a documented learning disorder in high school and suspected that AP3(45) and AP5(49) did as well, but they were never formally tested. All test performances of the CY-C(35) control subject were within normal limits. The AP-C(47) control subject showed only very mild difficulty on the visuoconstructive test but was well within normal limits overall.
Pattern of PiB distribution in sporadic AD and controls
Sporadic AD subjects (n = 12) and controls (n = 18) from the general population were studied for comparison with the PS1 mutation carriers. Transaxial PiB images from a typical mild AD subject (77 years of age; MMSE of 27) and a typical normal control (NC−; 72 years of age; MMSE of 30) are shown in Figure 1. As described previously (Klunk et al., 2004; Lopresti et al., 2005; Price et al., 2005), the AD patient had marked PiB retention in cortical areas known to show heavy plaque load in postmortem studies (Thal et al., 2002). PiB retention in the AD patient also was high in the striatum, an area known to contain extensive fibrillar amyloid deposits in AD (Braak and Braak, 1990; Brilliant et al., 1997). PiB retention was particularly elevated in anterior and ventral areas of the striatum in sporadic AD subjects (see Fig. 3). In contrast, no increased PiB retention was observed in cerebellum or white matter, areas known to contain little if any fibrillar amyloid deposition in sporadic AD (Joachim et al., 1989). Typically, very little cortical PiB retention was present in cognitively normal controls, and only small amounts of residual, nonspecific PiB retention was seen in white matter in amounts identical to those observed in AD (Fig. 1, NC−) (Klunk et al., 2004). In a few cognitively normal controls, we observed varying amounts of PiB retention in cortical areas. An example of one such “mildly PiB-positive normal control” (NC+) can be seen in Figure 1. This subject was scanned at baseline and 2 years later and showed progressive PiB retention, predominantly in the left frontal lobe. The coregistered MRI shows the outline of the striatum, and the PiB–PET image shows very little PiB retention in striatum in this NC+ subject.
Figure 3.

Detailed regional distribution of striatal PiB retention in a PS1C410Y mutation carrier and a subject with sporadic AD. Areas with high levels of PiB retention (SUVRpons >1.3) are superimposed over coronal, sagittal and transaxial MR images. The corresponding unobstructed MR images are shown to the right of each PiB overlay to aid in anatomical definition. PiB retention in an asymptomatic PS1 mutation carrier, CY2(37), is shown in the left six columns and that in a mild, sporadic AD subject (MMSE of 25) is shown on the right. Every other slice (2.4 mm) in the coronal, sagittal, and transaxial planes is shown through the entire distribution of PiB retention in the striatum. Full sagittal and transaxial slices are included at the bottom to show the marked striatal focality in the PS1 mutation carrier compared with the extensive cortical involvement in the sporadic AD subject.
Pattern of PiB distribution in PS1 mutation carriers
A noncarrier cousin of the PS1C410Y carriers [CY-C(35)] showed PiB retention that was indistinguishable from typical control subjects (Fig. 2). However, the five PS1C410Y carriers showed a pattern of PiB retention that was very different from any pattern observed previously in control or AD subjects but was remarkably similar among the five PS1C410Y carriers (Fig. 2). Most notable was the significantly high and focal PiB retention in the striatum coupled with a relative lack of PiB retention in the cortical areas typically affected by AD (see Fig. 1). Most of the PS1C410Y carriers did show moderately increased PiB retention in the neocortical areas and thalamus as well but not to the degree observed in the striatum (see Fig. 5). Details of the pattern of amyloid deposition in a PS1C410Y carrier compared with a sporadic AD case are shown in Figure 3. The deposition in the PS1C410Y carrier covers almost the complete extent of the caudate and putamen, but the globus pallidus appeared to be unaffected except for some diffusion of signal from the putamen. In sporadic AD, high levels of PiB retention were always observed in the anterior and ventral portions of the striatum, but the more dorsal and posterior portions of the striatum were not as involved as they were in the PS1 mutation carriers. The example shown in Figure 3 represents the most severely affected striatum in the sporadic AD group. It is very clear that the focality of the striatal PiB retention, as expressed by the striatal/neocortical ratio, in sporadic AD cases (1.09 ± 0.14; range of 0.92–1.37) was very different from that observed in the PS1 mutation carriers (1.76 ± 0.27; range of 1.38–2.24; p < 0.00001).
As in the PS1C410Y family, the noncarrier sibling from the PS1A426P kindred [AP-C(47)] had PiB retention typical of control subjects (Fig. 4). The five PS1A426P carriers showed focal striatal PiB retention similar to the PS1C410Y carriers, although the extent of retention was somewhat less (Fig. 4). Comparable with the PS1C410Y kindred, increased PiB retention also was observed in the neocortex and thalamus of the PS1A426P carriers but to a lesser degree than the increase observed in sporadic AD (Figs. 3, 4). AP3(45) and AP5(49) both showed relatively little neocortical PiB retention for subjects with clinical AD in both the images and the quantitative data (Fig. 5).
Figure 4.
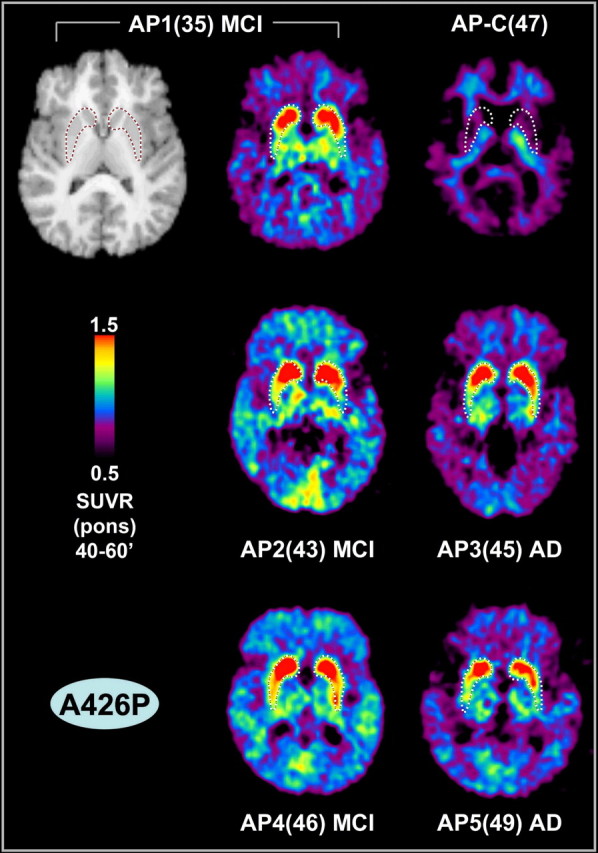
PS1A426P mutation carriers: PiB–PET and MRI images. Transaxial PiB–PET SUVRpons images (40–60′) from five siblings who carry the PS1A426P mutation and a sibling without the mutation [AP-C(47)] are shown. The clinical diagnoses of the mutation carriers are noted. The MRI of the AP1(35) MCI subject is shown as an example. A tracing of the location of the striatum from each subject's MRI scan is superimposed on the respective PiB image, but only the MRI for the AP1(35) MCI subject is shown (top left).
Quantitative analyses of PiB retention
The site of data acquisition (Pittsburgh or MGH) and scanner type (Seimens or GE) did not appear to have an impact because the quantitative data (Fig. 5) were very consistent across the two sites. When PiB retention was expressed quantitatively, several patterns emerged that fell into four groups (Fig. 5), as follows.
Large increase in AD and smaller increase in PS1 mutation carriers
In the neocortical regions in which there was a large increase in PiB retention in sporadic AD subjects compared with controls (i.e., frontal, precuneus/posterior cingulate, and parietal cortices), there was a much smaller increase in PiB retention in the PS1 mutation carriers. For example, PiB retention in the parietal cortex of the PS1 mutation carriers was significantly smaller than that in the sporadic AD subjects (p = 3.1 × 10−6). The parietal PiB retention in sporadic AD subjects was 196 ± 27% of the control value (p = 3.8 × 10−8), but the parietal PiB retention in the PS1 mutation carriers was only 127 ± 10% of the control value, although this smaller increase was still significant (p = 4.5 × 10−6). This also was true of frontal and precuneus/posterior cingulate cortex.
Large increase in AD and larger increase in PS1 mutation carriers
In contrast to the neocortical areas discussed above, PiB retention in the anterior/ventral striatum (predominantly anterior portions of caudate and putamen) (Fig. 3) in sporadic AD subjects was 212 ± 23% of the control value (p = 3.8 × 10−8) but was even higher (246 ± 32%; p = 2.4 × 10−7) in the PS1 mutation carriers. Interestingly, approximately half of the PS1 mutation carriers [CY1(35), CY2(37), CY3(38), AP2(43), and AP4(46)] had higher PiB retention in the anterior/ventral striatum than any AD subject (Fig. 5).
Moderate increase in both AD and PS1 mutation carriers
In brain areas in which PiB retention in AD subjects was moderately increased over controls (such as the occipital and mesial temporal cortex and thalamus), PiB retention in the PS1 mutation carriers also was increased over controls to a degree similar to that observed in AD subjects.
Little or no increase in both AD and PS1 mutation carriers
In the cerebellum, in which AD subjects show little if any increase in PiB retention over controls, the PS1 mutation carriers showed a small but significant increase in PiB retention over controls. In white matter areas such as pons (SUV without ratio; see Materials and Methods) and subcortical white matter, there was no significant difference between controls and either sporadic AD subjects or PS1 mutation carriers (data not shown).
Histological analysis of postmortem tissue
To determine whether there was histological evidence of heavy amyloid deposition in the striatum (relative to cortex) of PS1 mutation carriers, we examined postmortem striatal tissue sections from the affected parent (i.e., mutation carrier who developed clinical AD) of two PS1C410Y carriers included in this study. The results were comparable in the affected parent of both carriers, and photomicrographs of processed tissue sections from one of the affected parents are shown in Figure 6. Near-adjacent tissue sections were immunostained with the 10D5 anti-Aβ antibody (Fig. 6A,D) or histostained with a highly fluorescent derivative of PiB (Mathis et al., 2003) (6-CN-PiB) (Fig. 6B,E) and the highly fluorescent Congo red derivative X-34 (Styren et al., 2000; Ikonomovic et al., 2006) (Fig. 6C,F). Figure 6 shows that numerous Aβ-immunoreactive and 6-CN-PiB- or X-34-positive plaques are distributed densely in the striatum, which is free of other (i.e., neuritic) amyloid pathology. Striatal Aβ-immunoreactive plaques had a diffuse appearance, but the staining with the β-sheet stain X-34 indicates that they contained substantial amounts of fibrillar amyloid. Aβ-immunoreactive and 6-CN-PiB/X-34-positive plaques also were present in the adjacent neocortex, in which they appeared as well defined circular deposits that were of considerably lower densities compared with striatal plaques (Fig. 6D–F are representative of cortical areas with the greatest concentrations of plaques). Occasional neurofibrillary tangles were observed with X-34 histostaining in the neocortex but not in the striatum. Although Lippa et al. (1996) previously noted that the overall grade of amyloid angiopathy was slightly greater in eoFAD than in sporadic AD, there was little or no cerebrovascular amyloid in the limited tissue samples available for examination for these two particular PS1 cases, although a previous postmortem study reported mild-to-moderate amyloid angiopathy in the frontal cortex of PS1 C410Y mutation carriers (Mann et al., 2001).
Figure 6.
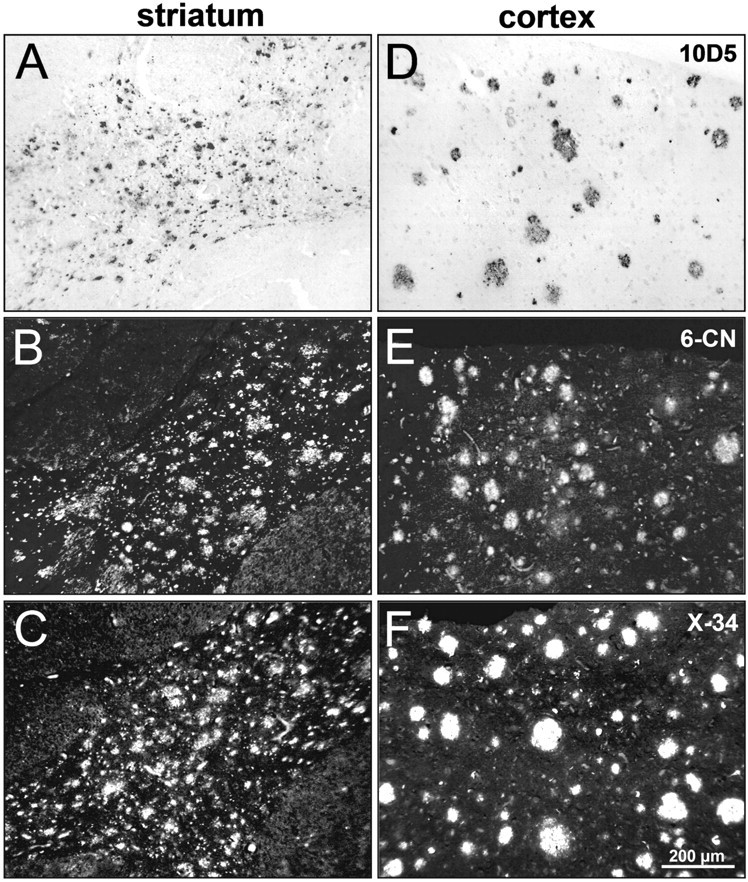
Plaque deposits in the striatum and adjacent neocortex from one parent of a PS1C410Y mutation carrier. Paired photomicrographs of striatal and cortical regions are shown from three representative tissue sections processed for immunohistochemistry with 10D5 anti-Aβ antibody (A, D) or 6-CN-PiB (B, E) and X-34 (C, F) histochemistry. Plaques are abundant and densely distributed throughout the striatum (A–C). In cortical gray matter (D–F; pial surface toward the top), plaques are also present but at much lower densities. Scale bar, 200 μm.
Discussion
This report represents the initial findings from what is designed to be a longitudinal study to delineate the natural history of amyloid deposition in eoFAD. To our knowledge, this is the first report of amyloid imaging in asymptomatic carriers of PS1 mutations, some of whom are more than 10 years younger than the age at which onset of cognitive symptoms would be expected (e.g., 48 years for the PS1C410Y mutation carriers). The findings suggest that amyloid deposition in these two families begins in the striatum well before the onset of cognitive symptoms.
Previous studies using structural MRI and [18F]fluorodeoxyglucose (FDG) PET determination of cerebral metabolic rate for glucose have shown changes in presymptomatic and symptomatic eoFAD subjects. Fox and colleagues (Schott et al., 2003; Ridha et al., 2006) reported that changes in hippocampal atrophy rate were evident 5.5 years before a diagnosis of AD. Two early reports showed decreased cerebral metabolism by FDG–PET in symptomatic eoFAD subjects (Cutler et al., 1985; Guze et al., 1992), and three groups have shown evidence of temporoparietal hypometabolism in asymptomatic individuals at risk for eoFAD (Kennedy et al., 1995; Rossor et al., 1996; Wahlund et al., 1999; Matsushita et al., 2002).
In the present study, the 10 subjects from two unrelated families, carrying two different PS1 mutations, unexpectedly showed a strikingly similar, focal deposition of amyloid deposition that appears to begin in the striatum. Initially, this striatal amyloid deposition appears to be clinically silent, at least in the PS1C410Y family. Overall, there did not appear to be a recognizable pattern of amyloid deposition that distinguished the PS1 mutation carriers with clinical AD from the less impaired or asymptomatic subjects. The data currently available from sporadic AD patients, MCI patients, and cognitively normal older subjects who show low levels of PiB retention (e.g., NC+ of this study) do not suggest that amyloid deposition typically begins in the striatum early in the course of sporadic AD (Mintun et al., 2006). In these older subjects, the earliest deposition appears to be in the frontal cortex and the precuneus/posterior cingulate region (Mintun et al., 2006). These same areas show small increases in most of the PS1 mutation carriers in this study.
The postmortem pattern of amyloid deposition in PS1 mutation carriers with clinical dementia has been reported to be very similar to the pattern observed in sporadic AD (Lippa et al., 1996). Another postmortem study suggested that PS1 mutation carriers have similar loads of Aβ40-containing plaques and greater loads of Aβ42(43)-containing plaques in their frontal cortex compared with sporadic AD cases (Mann et al., 2001). Both of these previous studies included deceased individuals from the PS1C410Y kindred included in this study. However, neither of these studies included the striatum in their analysis. Our limited analysis of striatal and nearby cortical tissue suggests that the number of plaques in the cortex never reaches the level observed in the striatum of PS1C410Y mutation carriers.
Taking the data from this study together with the previous postmortem studies, we can hypothesize a natural history of amyloid deposition in the PS1C410Y mutation carriers. In these individuals, amyloid deposition appears to begin in the striatum before the mid-30s, well before the onset of symptoms. Amyloid deposition in the striatum progresses rapidly and may reach an early plateau. Sometime after the striatal amyloid becomes well established, neocortical amyloid deposition begins in a pattern that is similar to that seen in the first stages of sporadic AD. By the time of death, the neocortical amyloid pathology reaches or exceeds that typical for sporadic AD but never reaches the density observed in the striatum. It may be that cognitive symptoms do not occur in PS1C410Y mutation carriers until neocortical plaque pathology becomes substantial (and perhaps neurofibrillary tangle pathology as well).
In the PS1A426P mutation carriers, it is more difficult to form a hypothesis about the natural history of amyloid deposition because of the absence of postmortem tissue from late-stage cases. However, the initial events in the striatum appear to be similar to the PS1C410Y mutation carriers. What is unclear is whether the PS1A426P mutation carriers ever reach a level of cortical amyloid deposition that would be typical of sporadic AD. Even the oldest, PS1A426P mutation carrier, who already carries a clinical diagnosis of AD, had not yet acquired a pattern of amyloid deposition that is typical of sporadic AD. In a previous study, a sporadic AD-like pattern was often already present in late-onset MCI subjects (Lopresti et al., 2005). Therefore, doubt remains about whether the PS1A426P mutation will ultimately lead to the typical pattern of PiB retention seen in sporadic AD or whether the pattern will remain atypical throughout the clinical course. Another possibility that must be entertained is that AD-like neocortical plaque deposition is present in the symptomatic PS1A426P mutation carriers but is not detected by PiB–PET in the same manner as neocortical plaque deposits in sporadic AD.
It is not yet known whether these two PS1 mutation kindreds are representative of eoFAD mutation carriers in general. To date, there is one other report of amyloid imaging using PiB in a symptomatic patient with eoFAD. Theuns et al. (2006) reported widespread retention of PiB, typical of that observed in sporadic AD, in a 57-year-old patient (MMSE of 18) with a novel K724N mutation in the C-terminal intracytosolic fragment of APP. The subject showed no disproportionate PiB retention in the striatum. Although the regional distribution of amyloid deposition in this APPK724N mutation carrier before the onset of symptoms is not known, the typical sporadic AD-like pattern of amyloid deposition in this subject raises the question of whether other eoFAD carriers will show the early striatal amyloid deposition observed in this study.
Although the focal nature and very early appearance of this striatal pathology was surprising in these eoFAD cases, the postmortem presence of striatal amyloid in sporadic AD has been described previously. Extensive amyloid deposition has been reported to occur in the striatum and thalamus of virtually all AD patients (Braak and Braak, 1990; Suenaga et al., 1990; Brilliant et al., 1997; Thal et al., 2002), and we observed substantial PiB retention in the striatum of sporadic AD patients in our previous in vivo studies (Fig. 1) (Klunk et al., 2004; Price et al., 2005). Although neuritic plaques have been observed in ventral striatum (Suenaga et al., 1990), most striatal plaques are not neuritic (Suenaga et al., 1990; Brilliant et al., 1997). Despite this poorly understood paucity of neuritic changes in the striatum, striatal plaques appear to be fibrillar as evidenced by the fact that they are stained well by the fibril-specific Congo red derivative X-34 (Styren et al., 2000). Our postmortem study of the two parents of PS1C410Y carriers included in this study also revealed marked subcortical amyloid deposits, although by the time of death, cortical amyloid deposits had become extensive as well (Lippa et al., 1996). However, as observed in the present in vivo PiB study, the postmortem striatal deposits were disproportionately high in the PS1C410Y mutation carriers.
In addition to the lack of neuritic dystrophy, striatal amyloid deposits in both sporadic and eoFAD (despite being fibrillar) are not typically associated with cell loss and do not cause extrapyramidal symptoms early in the clinical course. However, Parkinsonism has been reported in C410Y mutation carriers late in the course of the disease (Mann et al., 2001). This is reminiscent of the lack of tissue reaction observed around the diffuse plaques observed in the cerebellum. However, although the lack of toxic effects of cerebellar amyloid may be simply attributable to lack of extensive β-sheet conformation (Styren et al., 1998), another explanation must be sought for the lack of apparent amyloid toxicity in the striatum in which the deposits are primarily fibrillar. The presence or absence of tissue factors and the type of neurons and circuitry present may explain the differences in tissue reactivity to Aβ deposits between cortical areas and the striatum. A full understanding of these factors may provide important clues for the development of therapies that could help protect cortical neurons from the toxic effects of Aβ.
It will be important to address the cause of the very early amyloid deposition in the striatum of the PS1 mutation carriers. Although the reason is not yet known, we also have seen disproportionate retention of PiB in the striatum of an APP mutation carrier and a nondemented Down syndrome subject (our unpublished observations), although both of these cases had more cortical involvement than any of the PS1 mutation carriers included in this report. These early-onset forms of AD all share overproduction of Aβ (particularly the 42 amino acid form) as a proposed mechanism of Aβ deposition (Younkin, 1997), whereas decreased clearance might be more important in late-onset AD (Whitaker et al., 2003). It may be that the cellular milieu of the striatum is particularly prone to amyloid deposition under these conditions of overproduction. Although the relevance to human AD is not known, it should be noted that striatal amyloid deposition has been described in a PS1/APP double-transgenic mouse model (Perez et al., 2005).
The AP3(45) and AP5(49) subjects, who carry a clinical diagnosis of AD, both had lower PiB retention in frontal, temporoparietal, and precuneus cortices than most very mildly impaired sporadic AD subjects. This leads to a question regarding the driving force of the cognitive symptoms in these subjects. It is possible that a soluble form of amyloid that is not detected by PiB could be driving the cognitive deficits in these subjects (Klein, 2006). In addition, there may be insoluble but less ordered fibrillar forms of amyloid in these PS1 mutation carriers. This has been observed in Tg2576 and PS1/APP transgenic mice that have heavy amyloid loads but bind PiB very poorly (Klunk et al., 2005). Finally, although unlikely in subjects with a PS1 mutation, it is possible that amyloid deposition is not driving the cognitive deficits. Whatever the explanation, it is clear that the cortical PiB retention of these two PS1A426P carriers with a clinical diagnosis of mild or moderate AD is very different from that observed in sporadic mild-moderate AD.
The findings presented here may have important implications for clinical trials of anti-amyloid therapies. Autopsy reports on three cases from the AN-1792 trial support the concept that anti-Aβ immunotherapy can remove Aβ deposits from human brain (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). However, non-Aβ pathology remained in these cases, and the overall clinical response seen in the AN-1792 immunotherapy trial was modest (Gilman et al., 2005). This suggests that, to attain its optimal clinical effect, anti-amyloid therapy may need to be initiated at the very earliest stages of amyloid deposition, before extensive neurofibrillary changes, synapse loss, and neuronal death. To achieve this goal, it may be necessary to start anti-amyloid therapy at preclinical stages. The PS1 mutation carriers present a nearly ideal situation for the testing of amyloid-removing therapies. There is strong evidence that altered amyloid metabolism is the cause of dementia in these individuals, and early reversal of this deposition should have a significant impact on the cognitive decline that their genetic status ensures will occur in the absence of any intervention. The present amyloid imaging studies have identified an early focal target for anti-amyloid therapy in the striatum. In the C410Y kindred, this occurs at least a decade before the expected onset of symptoms. Any reversal of amyloid accumulation at this stage might represent a successful treatment effect, and amyloid imaging studies could provide a relatively rapid assessment of the efficacy.
Footnotes
This work was supported by National Institutes of Health Grants R01 AG018402, P50 AG005133, K02 AG001039, R01 AG020226, R01 MH070729, K01 MH001976, R37 AG025516, and P01 AG025204; Alzheimer's Association Grant TLL-01-3381; and United States Department of Energy Grant DE-FD02-03 ER63590. These funding agencies had no role in the design or interpretation of results or preparation of this manuscript. We thank Dr. R. Nebes and E. Halligan and the staff at the University of Pittsburgh Alzheimer's Disease Research Center (C. McConaha, E. Eror, L. Macedonia, and M. Oakley) and PET facility (S. Hulland, J. Ruszkiewicz, P. McGeown, D. Ratica, K. Malone, S. Kendro, N. Flatt, and J. Gallo) for their efforts in conducting and analyzing these studies. We are indebted to our subjects and their families for the selfless contributions that made this work possible. GE Healthcare holds a license agreement with the University of Pittsburgh based on the technology described in this manuscript. Drs. Klunk and Mathis are coinventors of PiB and, as such, have a financial interest in this license agreement. GE Healthcare provided no grant support for this study and had no role in the design or interpretation of results or preparation of this manuscript. All other authors have no conflicts of interest with this work. Drs. Klunk, Mathis, Price and DeKosky had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- Archer HA, Edison P, Brooks DJ, Barnes J, Frost C, Yeatman T, Fox NC, Rossor MN. Amyloid load and cerebral atrophy in Alzheimer's disease: an 11C-PIB positron emission tomography study. Ann Neurol. 2006;60:145–147. doi: 10.1002/ana.20889. [DOI] [PubMed] [Google Scholar]
- Becker JT, Boller F, Saxton J, McGonigle-Gibson KL. Normal rates of forgetting of verbal and non-verbal material in Alzheimer's disease. Cortex. 1987;23:59–72. doi: 10.1016/s0010-9452(87)80019-9. [DOI] [PubMed] [Google Scholar]
- Benton AL. Differential behavioral effects in frontal lobe disease. Neuropsychologia. 1968;6:53–60. [Google Scholar]
- Benton AL, Sivan AB, Hamsher KD, Varney NR, Spreen O. A clinical manual. Ed 2. New York: Oxford UP; 1994. Contributions to neuropsychological assessment. [Google Scholar]
- Beyreuther K, Dyrks T, Hilbich C, Monning U, Konig G, Multhaup G, Pollwein P, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in Alzheimer's disease and Down syndrome. Prog Clin Biol Res. 1992;379:159–182. [PubMed] [Google Scholar]
- Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkowski JG, Benton AL, Spreen O. Word fluency and brain damage. Neuropsychologia. 1967;5:135–140. [Google Scholar]
- Braak H, Braak E. Alzheimer's disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49:215–224. [PubMed] [Google Scholar]
- Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer's disease. Neuropathol Appl Neurobiol. 1997;23:322–325. [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschke H. Cued recall in amnesia. J Clin Neuropsychol. 1984;6:433–440. doi: 10.1080/01688638408401233. [DOI] [PubMed] [Google Scholar]
- Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Martin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, Penet C, Puel M, Pasquier F, Ledoze F, Bellis G, Calenda A, Heilig R, Martinez M, Mallet J, Bellis M, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer's disease. Hum Mol Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer's disease. Hum Mutat. 1998;11:183–190. doi: 10.1002/(SICI)1098-1004(1998)11:3<183::AID-HUMU1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Cutler NR, Haxby JV, Duara R, Grady CL, Moore AM, Parisi JE, White J, Heston L, Margolin RM, Rapoport SI. Brain metabolism as measured with positron emission tomography: serial assessment in a patient with familial Alzheimer's disease. Neurology. 1985;35:1556–1561. doi: 10.1212/wnl.35.11.1556. [DOI] [PubMed] [Google Scholar]
- Drzezga A, Grimmer T, Henriksen G, Wester HJ, Kurz A, Schwaiger M. Imaging of cerebral amyloid plaque load and glucose metabolism in semantic dementia and Alzheimer's disease. J Nucl Med. 2006;47(Suppl 1):73P. [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease. An [11C]PIB and [18F]FDG PET study. Neurology. 2006;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Langstrom B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2805–2807. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, Larossa GN, Spinner ML, Klunk WE, Mathis CA, Dekosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fahn S, Elton RL. Unified Parkinson's disease rating scale. In: Fahn S, Marsden CD, Calne D, Goldstein M, editors. Recent developments in Parkinson's disease. Florham Park, NJ: Macmillan Health Care Information; 1987. pp. 153–164. [Google Scholar]
- Ferrer I, Boada R, Sanchez G, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein M, Folstein S, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, Belleville S, Brodaty H, Bennett D, Chertkow H, Cummings JL, de Leon M, Feldman H, Ganguli M, Hampel H, Scheltens P, Tierney MC, Whitehouse P, Winblad B. Mild cognitive impairment. Lancet. 2006;367:1262–1270. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Guze BH, Hoffman JM, Mazziotta JC, Baxter LR, Jr, Phelps ME. Positron emission tomography and familial Alzheimer's disease: a pilot study. J Am Geriatr Soc. 1992;40:120–123. doi: 10.1111/j.1532-5415.1992.tb01930.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Perl D, Purohit D, Marin D, Khan K, Lantz M, Davis K. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer's disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Hollander M, Wolfe DA. New York: Wiley; 1973. Nonparametric statistical methods. [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Isanski BA, Mathis CA, DeKosky ST, Klunk WE. X34, a histofluorescent marker of abnormal protein aggregates with amyloid structure, in neuropathological studies of preclinical and clinical Alzheimer's disease. Methods Enzymol. 2006;412:123–144. doi: 10.1016/S0076-6879(06)12009-1. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Selkoe DJ. The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis Assoc Disord. 1992;6:7–34. doi: 10.1097/00002093-199205000-00003. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer's disease. Am J Pathol. 1989;135:309–319. [PMC free article] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Wilson IA, Nagren K, Helin S, Bruck A, Oikonen V, Kailajarvi M, Scheinin M, Viitanen M, Parkkola R, Rinne JO. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67:1575–1580. doi: 10.1212/01.wnl.0000240117.55680.0a. [DOI] [PubMed] [Google Scholar]
- Kennedy AM, Rossor MN, Frackowiak RS. Positron emission tomography in familial Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9:17–20. doi: 10.1097/00002093-199505000-00005. [DOI] [PubMed] [Google Scholar]
- Klein WL. Synaptic targeting by Aβ oligomers (ADDLS) as a basis for memory loss in early Alzheimer's disease. Alzheimers Dement. 2006;2:43–55. doi: 10.1016/j.jalz.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-β in Alzheimer's disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakides T, Silbert PL, Kakulas BA. Cerebral amyloid angiopathy and intracerebral hemorrhage with special reference to the pons. Clin Neuropathol. 1994;13:71–76. [PubMed] [Google Scholar]
- Lippa CF, Saunders AM, Smith TW, Swearer JM, Drachman DA, Ghetti B, Nee L, Pulaski-Salo D, Dickson D, Robitaille Y, Bergeron C, Crain B, Benson MD, Farlow M, Hyman BT, George-Hyslop SP, Roses AD, Pollen DA. Familial and sporadic Alzheimer's disease: neuropathology cannot exclude a final common pathway. Neurology. 1996;96:406–412. doi: 10.1212/wnl.46.2.406. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler AH, Volkow ND, Wang G-J, Ding Y-S, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Klunk W, Saxton J, Hamilton RL, Kaufer DI, Sweet RA, Cidis Meltzer C, Wisniewski S, Kamboh MI, DeKosky ST. Res evaluation and diagnosis of probable Alzheimer's disease over the last two decades. I. Neurology. 2000;55:1854–1862. doi: 10.1212/wnl.55.12.1854. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, Dekosky ST, Price JC. Simplified Quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–1972. [PubMed] [Google Scholar]
- Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1linked Alzheimer's disease. Am J Pathol. 2001;158:2165–2175. doi: 10.1016/s0002-9440(10)64688-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Arai H, Okamura N, Ohmori T, Takasugi K, Matsui T, Maruyama M, Iwatsubo T, Higuchi S. Clinical and biomarker investigation of a patient with a novel presenilin-1 mutation (A431V) in the mild cognitive impairment stage of Alzheimer's disease. Biol Psychiatry. 2002;52:907–910. doi: 10.1016/s0006-3223(02)01386-0. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of the Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995;19:541–547. doi: 10.1097/00004728-199507000-00006. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Moonis M, Swearer JM, Dayaw MP, St George-Hyslop P, Rogaeva E, Kawarai T, Pollen DA. Familial Alzheimer disease: decreases in CSF Abeta42 levels precede cognitive decline. Neurology. 2005;65:323–325. doi: 10.1212/01.wnl.0000171397.32851.bc. [DOI] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, Berg L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer's disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Pastor P, Goate AM. Molecular genetics of Alzheimer's disease. Curr Psychiatry Rep. 2004;6:125–133. doi: 10.1007/s11920-004-0052-6. [DOI] [PubMed] [Google Scholar]
- Perez SE, Lazarov O, Koprich JB, Chen EY, Rodriguez-Menendez V, Lipton JW, Sisodia SS, Mufson EJ. Nigrostriatal dysfunction in familial Alzheimer's disease-linked APPswe/PS1ΔE9 transgenic mice. J Neurosci. 2005;25:10220–10229. doi: 10.1523/JNEUROSCI.2773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Sharma V, Anderson L, Nemens E, Alonso ME, Orr H, White J, Heston L, Bird TD, Schellenberg GD. Missense mutations in the chromosome 14 familial Alzheimer's disease presenilin 1 gene. Hum Mutat. 1998;11:216–221. doi: 10.1002/(SICI)1098-1004(1998)11:3<216::AID-HUMU6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, Dekosky ST, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, O'Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- Reitan RM. Validity of the trail-making tests as an indication of organic brain damage. Percept Motor Skills. 1958;8:271–276. [Google Scholar]
- Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- Rossor MN, Kennedy AM, Frackowiak RS. Clinical and neuroimaging features of familial Alzheimer's disease. Ann NY Acad Sci. 1996;777:49–56. doi: 10.1111/j.1749-6632.1996.tb34400.x. [DOI] [PubMed] [Google Scholar]
- Saxton J, Ratcliff G, Newman A, Belle S, Fried L, Yee J, Kuller L. Cognitive test performance and presence of subclinical cardiovascular disease in the cardiovascular health study. Neuroepidemiology. 2000;19:312–319. doi: 10.1159/000026270. [DOI] [PubMed] [Google Scholar]
- Schott JM, Fox NC, Frost C, Scahill RI, Janssen JC, Chan D, Jenkins R, Rossor MN. Assessing the onset of structural change in familial Alzheimer's disease. Ann Neurol. 2003;53:181–188. doi: 10.1002/ana.10424. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop PH. Molecular genetics of Alzheimer disease. Semin Neurol. 1999;19:371–383. doi: 10.1055/s-2008-1040852. [DOI] [PubMed] [Google Scholar]
- Styren SD, Kamboh MI, DeKosky ST. Expression of differential immune factors in temporal cortex and cerebellum: the role of alpha-1-antichymotrypsin, apolipoprotein E, and reactive glia in the progression of Alzheimer's disease. J Comp Neurol. 1998;396:511–520. [PubMed] [Google Scholar]
- Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J Histochem Cytochem. 2000;48:1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- Suenaga T, Hirano A, Llena JF, Yen SH, Dickson DW. Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer's disease. Acta Neuropathol (Berl) 1990;80:280–286. doi: 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Theuns J, Marjaux E, Vandenbulcke M, Van Laere K, Kumar-Singh S, Bormans G, Brouwers N, Van den Broeck M, Vennekens K, Corsmit E, Cruts M, De Strooper B, Van Broeckhoven C, Vandenberghe R. Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum Mutat. 2006;27:888–896. doi: 10.1002/humu.20402. [DOI] [PubMed] [Google Scholar]
- Verhoeff NP, Wilson AA, Takeshita S, Trop L, Hussey D, Singh K, Kung HF, Kung MP, Houle S. In vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12:584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- Villemagne V, Ng S, Gong S, Ackermann U, Pike K, Savage G, Klunk W, Mathis C, Masters C, Rowe C. 11C-PIB PET imaging in the differential diagnosis of dementia. J Nucl Med. 2006;47(Suppl 1):74P. [Google Scholar]
- Wahlund LO, Basun H, Almkvist O, Julin P, Axelman K, Shigeta M, Jelic V, Nordberg A, Lannfelt L. A follow-up study of the family with the Swedish APP 670/671 Alzheimer's disease mutation. Dement Geriatr Cogn Disord. 1999;10:526–533. doi: 10.1159/000017200. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale. Ed 3. New York: The Psychological Corporation; 1997. [Google Scholar]
- Welsh-Bohmer KA, Butters N, Mohs RC. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). V. A normative study of the neuropsychological battery. Neurology. 1994;44:609–614. doi: 10.1212/wnl.44.4.609. [DOI] [PubMed] [Google Scholar]
- Whitaker C, Eckman C, Almeida C, Feinstein D, Atwood C, Eckman E, Crutcher K, Hersh L, Leissring M, LaVoie M, Ertekin-Taner N, Shapiro P, Takahashi R, Yamin R, Mansourian S, Estus S, Lesne S, Turner T, Farris W, Stroebel G. Live discussion: amyloid-beta degradation: the forgotten half of Alzheimer's disease. J Alzheimers Dis. 2003;5:491–497. [PubMed] [Google Scholar]
- Wilson AA, Garcia A, Chestakova A, Kung HF, Houle S. A rapid one-step radiosynthesis of the beta-amyloid imaging radiotracer N-methyl-[C-11]2-(4′-methylaminophenyl)-6hydroxybenzothiazole ([C-11]-6-OH-BTA-1) J Label Compd Radiopharm. 2004;47:679–682. [Google Scholar]
- Xia W, Ostaszewski BL, Kimberly WT, Rahmati T, Moore CL, Wolfe MS, Selkoe DJ. FAD mutations in presenilin-1 or amyloid precursor protein decrease the efficacy of a gamma-secretase inhibitor: evidence for direct involvement of PS1 in the gammasecretase cleavage complex. Neurobiol Dis. 2000;7:673–681. doi: 10.1006/nbdi.2000.0322. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The APP and PS1/2 mutations linked to early onset familial Alzheimer's disease increase the extracellular concentration and A beta 1–42 (43) Rinsho Shinkeigaku. 1997;37:1099. [PubMed] [Google Scholar]
- Zhou Y, Resnick S, Ye W, Fan H, Holt D, Klunk W, Mathis C, Dannals R, Wong D. Evaluation of reference tissue models for quantification of [11C] Pittsburgh compound B dynamic PET studies. J Nucl Med. 2006;47(Suppl 1):117P. [Google Scholar]
- Ziolko SK, Weissfeld LA, Klunk WE, Mathis CA, Hoge JA, Lopresti BJ, Dekosky ST, Price JC. Evaluation of voxel-based methods for the statistical analysis of PIB PET amyloid imaging studies in Alzheimer's disease. NeuroImage. 2006;33:94–102. doi: 10.1016/j.neuroimage.2006.05.063. [DOI] [PubMed] [Google Scholar]