

Abstract
Tipifarnib (R115777), an inhibitor of human protein farnesyltransferase (PFT), is shown to be a highly potent inhibitor of Trypanosoma cruzi growth (ED50 = 4 nM). Surprisingly, this is due to the inhibition of cytochrome P450 sterol 14-demethylase (CYP51, EC 1.14.13.70). Homology models of the T. cruzi CYP51 were used for the prediction of the binding modes of the substrate lanosterol and of Tipifarnib, providing a basis for the design of derivatives with selectivity for TcCYP51 over human PFT.
Trypanosoma cruzi (T. cruzi) is the etiologic agent of Chagas disease, a chronic illness that frequently leads to fatal cardiomyopathy. An estimated 13 million people in Latin America are chronically infected with T. cruzi (www.who.int/tdr/dw/chagas2003.htm), and, unfortunately, no safe and effective anti-protozoan drugs are available to cure them. We have been interested in exploiting the T. cruzi protein farnesyltransferase (PFT) as a drug target1, since numerous PFT inhibitors exist as a result of programs to make anti-cancer drugs2. We found that the human PFT inhibitor, Tipifarnib (also known as R115777, Fig. 1), has an IC50 of ~75 nM against the T. cruzi PFT enzyme. Unexpectedly, the concentration of Tipifarnib that caused 50% growth inhibition of T. cruzi amastigotes in culture was considerably lower (4 nM). This suggested that a mechanism of action separate from inhibition of T. cruzi PFT was at play. Since we had previously observed that a variety of imidazole containing compounds had potent anti-T. cruzi activity by blocking the sterol 14-demethylase enzyme (TcCYP51)3, we hypothesized that the imidazole containing compound, Tipifarnib, might be acting by a similar mechanism. In fact, this appeared to be the case as demonstrated by blockage of sterol biosynthesis in T. cruzi cells by this compound at the level of the sterol 14-demethylase activity (Fig. 2).
Figure 1.
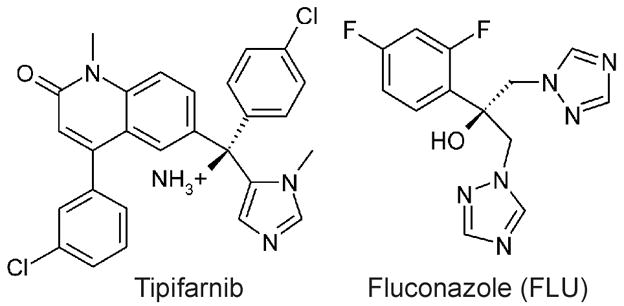
Structures of Tipifarnib and Fluconazole
Figure 2.
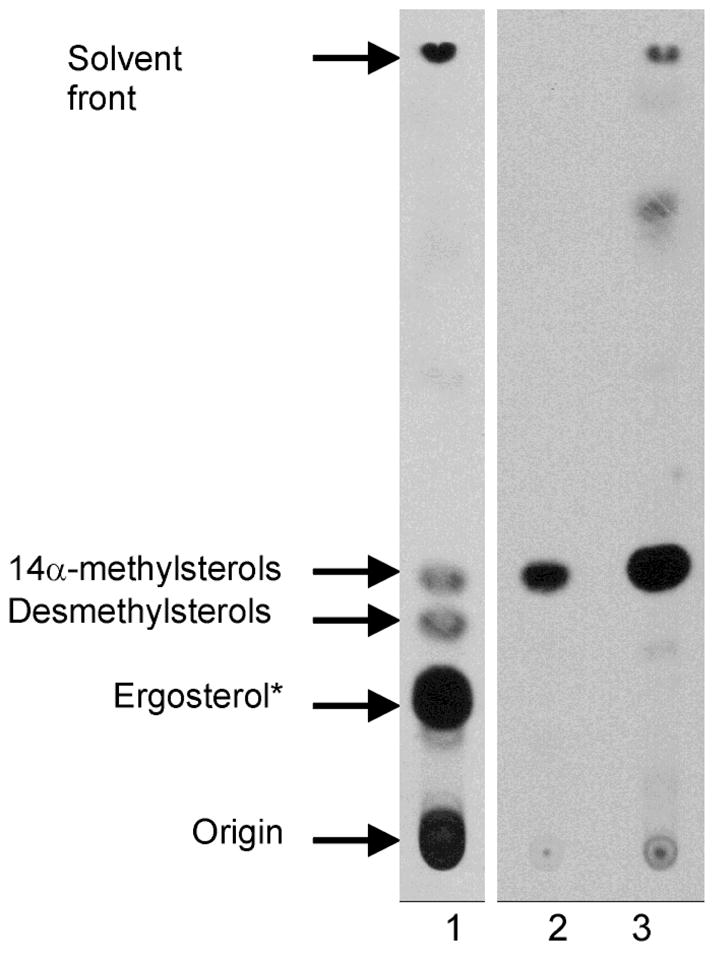
Inhibition of sterol biosynthesis in T. cruzi cells treated with Tipifarnib at the level of sterol 14-demethylase activity. T. cruzi epimastigotes were grown for 24 h with 100 ∝Ci of 3H-mevalonolactone which is incorporated into endogenous sterol production3. Neutral lipids were extracted and separated by thin layer chromatography. Lane 1, T. cruzi grown without drug. Lane 2, T. cruzi grown with Tipifarnib. Lane 3, T. cruzi grown with ketoconazole (a known inhibitor of T. cruzi CYP51). *In addition to ergosterol, T. cruzi produces other C4, C14-demethylated sterols4
In order to elucidate the binding mode of Tipifarnib with the TcCYP51, Tipifarnib was docked into the binding site of two TcCYP51 homology models. Homology modeling was done with the program MODELLER4, version 6v2, based on the structures of CYP51 from Mycobacterium tuberculosis (MtCYP51) containing two different azole inhibitors5 (PDB codes: 1E9X, 1EA1). The alignment of the TcCYP51 and MtCYP51 sequences for the structure prediction was taken from an alignment of 14 CYP51 sequences, as described elsewhere6. The overall sequence identity between Mt- and modeled TcCYP51 amounts to 29% (the predicted membrane spanning segment of TcCYP51 was not considered in the models), 50% (18 out of 36) of the binding site residues are identical. Two different sets of 5 models each were calculated. The residues 95–102 (Mt numbers) were modeled on the basis of 1E9X in one set of models and on the basis of 1EA1 in a second set to account for the structural differences of the two template structures in the region of the BC-loop and the C-helix, which are located at the opening of the binding site to the solvent. From each set the best model, according to the MODELLER molecular objective function, was used for further work.
To validate the model, the substrate lanosterol was docked into the binding sites of the two homology models. The idea was that if the model was valid, a binding mode of lanosterol should be predicted showing the 14-methyl group in a location favorable for the heme-catalyzed oxidation reaction. Lanosterol was chosen because Phe78 of MtCYP51, a key residue for substrate specificity, is replaced by an isoleucine in TcCYP51, suggesting lanosterol as the substrate of TcCYP517. Two different random starting orientations of lanosterol were used for the docking searches by manually placing the molecule into the binding site cavity. Then MCDOCK of the FLO/QXP program suite8, version 0602, was used to extensively search for the overall best binding geometry (10,000 cycles of Metropolis Monte Carlo search for each starting orientation) in the 1E9X- as well as the 1EA1- based homology model of TcCYP51. Precautions had to be taken to account for the uncertainty of the model coordinates in the BC-loop/C-helix regions resulting from the structural flexibility of this region of the protein5. For this purpose the sidechain conformations of selected residues of this loop were considered flexible during the docking calculations: These were Met123[Arg96], Arg124[Lys97], Leu127[100], Asn128[His101] for the 1E9X based model and Arg122[95], Met123[Arg96], Gln126[Met99], Leu127[100] for the 1EA1 based model (corresponding residues of MtCYP51 are given in backets throughout the text).
Without any restraints directing the search, 13 out of the 50 best predicted placements (25 per binding site model) show the 14-methyl group within a distance to the heme iron atom that is considered to be productive with respect to the oxidation of this group, i.e. within the range from 4.2 to 5.5 Å9. The binding mode of lanosterol in these 13 productive placements is basically identical – the rmsd of the two most different geometries amounts to 1.5 Å. This binding mode was the only one found in a separate docking search when a restraint was applied to keep the distance between the iron atom and the 14-methyl close to 4.85 Å (i.e. the mean of the limits of the productive distance). This binding geometry shows high similarity with that of estriol in MtCYP51 that has been published during our investigations7 (Fig. 3). In the estriol as well as the predicted lanosterol binding mode, the hydroxyl-substituent of the A-ring is located in a hydrophilic region formed by the NH of residue 357[322] and the backbone carbonyl oxygen atoms of the residues 358[323], 459[432], and 460[433]. The estriol-OH forms a H-bond with the C=O of residue 460[433], whereas the backbone carbonyl oxygen of Met358[323] is the most likely H-bond acceptor for the hydroxyl-group of lanosterol in TcCYP51. As stated by Produst et al.7, such minor differences of the binding modes of estriol and lanosterol may result from the structural differences of these compounds (Fig. 3). However, the ring system of lanosterol occupies the same space in the binding site as estriol, with the 14-methyl group of lanosterol pointing into a cleft formed by Ala291[256], His294[259] and Leu356[321], towards the heme iron atom. The acyclic “tail” of lanosterol is directed towards the BC-loop and the C-helix, most notably residues 122[95] and 123[96]. A similar binding mode of lanosterol was predicted by different docking methods for MtCYP5110 and for the CYP51 from Candida albicans9,11 as well as Aspergillus fumigatus11. In case of Candida albicans the hydroxyl group was reported to form a hydrogen bond to the sidechain of a Ser rather than backbone groups9. However, the agreement between the experimentally determined binding mode of estriol, that is believed to reflect the substrate binding mode in MtCYP517, with the predicted binding mode of lanosterol in TcCYP51 supports the suitability of our model of TcCYP51 for structure based drug design.
Figure 3.

The predicted binding mode of lanosterol (green) in TcCYP51 as compared to estriol in MtCYP51 (yellow; 1X8V; stereo figure). TcCYP51 sidechains located within 4.0 Å of lanosterol are depicted, residues mentioned in the text are labeled. The hydrogen bond between the lanosterol hydroxyl group and the backbone of Met358 is indicated (dashed line). Estriol forms a H-bond with Met433 of MtCYP51, which corresponds to Met460 of TcCYP51. (The helix between residue 284 and 301 was omitted to allow this view, as were the sidechains of the residues Ala291 and His294.)
In order to determine the binding mode of Tipifarnib in TbCYP51, the protocol that was successfully used for the docking of lanosterol was applied to dock the inhibitor into the binding sites of both homology models. We assumed that the imidazole group binds to the heme iron atom. The imidazole nitrogen was therefore placed at the location occupied by the iron-bound nitrogen of fluconazole in 1EA15 and tethered to the iron atom by the assignment of a distance restraint prior to the docking calculations. For both models of TcCYP51 the same binding geometry of Tipifarnib was predicted. The comparison with the binding mode of fluconazole (FLU) reveals several similarities (Fig. 4): The aromatic ring of the quinolinone ring system that is directly attached to the chiral carbon atom of Tipifarnib, and the difluorophenyl of FLU occupy the same region of the sterol binding pocket. Both, the p-Cl-phenyl ring of Tipifarnib and the triazole of FLU that is not bound to the heme iron atom, point to the hydrophilic pocket where the sterol hydroxyl group binds. However, in both cases this pocket remains largely unoccupied. Most notably, the hydroxyl group of FLU and the amino function of Tipifarnib are found at identical locations within the active site. In the crystal structure with FLU, a water molecule mediates a hydrogen bond between the hydroxyl group and a heme propionate. This water molecule was not included in our original docking calculations with Tipifarnib. Inclusion of this water molecule in a new docking simulation did not change the predicted binding mode of the inhibitor (Fig. 4). The conformation of the C-helix in the 1EA1-based homology model allows the formation of an additional hydrogen bond between the carbonyl group of Tipifarnib and the backbone NH of Met123[Arg96]. It should be noted that the chlorine of the p-Cl-phenyl of Tipifarnib would clash with the sidechain of Phe78 of MtCYP51 (d = 1.8 Å, after superposition of 1EA1 with the TcCYP51 homology model) and the m-Cl-phenyl ring would overlap with the sidechain of Gln72. In TcCYP51, Gln72 and Phe78 are replaced by Pro99 and Ile105, respectively, which significantly enlarges the binding site in these regions. As pointed out by Podust et al.7, the presence of the large Phe78 sidechain seems to lead to preferred processing of obtusifoliol, which carries only one methyl group at position 4 of the sterol ring system, as opposed to 4α,4β-dimethylated sterols like lanosterol (Fig. 3). The recent finding12 that the CYP51 of Trypanosoma brucei is obtusifosiol specific, is in agreement with this role of Phe78.
Figure 4.

The predicted binding mode of Tipifarnib (green) in TcCYP51 as compared to fluconazole (yellow; 1EA1; stereo). TcCYP51 sidechains located within 4.5 Å of Tipifarnib are depicted. The inhibitor forms hydrogen bonds with Met123 and, via a water molecule, with a heme propionate (water oxygen atoms are indicated as red spheres, H-bonds as dashed yellow lines). The 4 water molecules in the upper right indicate the location of the hydrophilic pocket being utilized for the design of Tipifarnib derivatives with specificity for CYP51 over PFT. Pro99 and Ile105 replace Gln72 and Phe78 of MtCYP51, respectively. Due to clashes with these two residues, MtCYP51 is predicted to have low affinity for Tipifarnib.
The binding mode was inspected in order to identify portions of the inhibitor that are suitable for structural modifications in the course of the lead optimization process. From clinical trials with Tipifarnib as anticancer drug, as e.g. described in13, it can be concluded that side effects might be a liability for this compound. The adverse effects presumably result from the persistent inhibition of the target enzyme, human protein-farnesyltransferase (PFT), during the treatment. We therefore decided to pursue a strategy for the optimization of Tipifarnib as agent against Chagas disease that minimizes its potency as PFT inhibitor. The comparison of the X-ray structure of mammalian PFT:Tipifarnib complex14 with the binding mode in TcCYP51 allows for the rational design of new derivatives for this purpose. In the template structures for the homology modeling, parts of the BC-loop and the C-helix are poorly defined7. In order to make reliable predictions, it is appropriate to focus first on those parts of the inhibitor which contact well defined and structurally conserved regions of the binding site. This is in particular the case for the p-Cl-phenyl ring pointing to the binding pocket of the sterol hydroxyl-group. The structure of PFT with Tipifarnib strongly suggests that large substituents in the meta- or para-position of this ring will be detrimental to potency vs. PFT. The binding mode in TcCYP51 on the other hand suggests that such substituents can be used to fill the pocket occupied by the A-ring of the sterol substrate which is not filled by Tipifarnib. Currently derivatives of Tipifarnib with modifications targeting this pocket are being synthesized.
Based upon good agreement of the predicted lanosterol binding mode with the binding geometry of estriol in MtCYP51 and the unambiguousness of the calculated Tipifarnib binding mode in TcCYP51 we expect our model to be a firm basis for structure based design of Tipifarnib derivatives. This, together with its excellent pharmacokinetic properties15,16 and its high potency versus T. cruzi cells, make Tipifarnib a highly promising lead compound for the development of new drugs against Chagas disease.
Acknowledgments
O. Hucke is a fellow of the German Academy of Natural Scientists Leopoldina (BMBF-LPD 9901/8-77). F. Buckner is supported by NIH grant AI 48043
References
- 1.Buckner FS, Eastman RT, Nepomuceno-Silva JL, Speelmon EC, Myler PJ, et al. Cloning, heterologous expression, and substrate specificities of protein farnesyltransferases from Trypanosoma cruzi and Leishmania major. Mol Biochem Parasitol. 2002;122:181–188. doi: 10.1016/s0166-6851(02)00099-3. [DOI] [PubMed] [Google Scholar]
- 2.Sebti SM, Adjei AA. Farnesyltransferase inhibitors. Sem Oncol. 2004;31:28–39. doi: 10.1053/j.seminoncol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Buckner F, Yokoyama K, Lockman J, Aikenhead K, Ohkanda J, et al. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc Natl Acad Sci USA. 2003;100:15149–15153. doi: 10.1073/pnas.2535442100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sali A, Blundell TL. Comparative Protein Modeling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 5.Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14 alpha-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci USA. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckner FS, Joubert BM, Boyle SM, Eastman RT, Verlinde CLMJ, et al. Cloning and analysis of Trypanosoma cruzi lanosterol 14 alpha-demethylase. Mol Biochem Parasitol. 2003;132:75–81. doi: 10.1016/j.molbiopara.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Podust LM, Yermalitskaya LV, Lepesheva GI, Podust VN, Dalmasso EA, et al. Estriol bound and ligand-free structures of sterol 14 alpha-demethylase. Structure. 2004;12:1937–1945. doi: 10.1016/j.str.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 8.McMartin C, Bohacek RS. QXP: Powerful, rapid computer algorithms for structure-based drug design. J Comput-Aided Mol Des. 1997;11:333–344. doi: 10.1023/a:1007907728892. [DOI] [PubMed] [Google Scholar]
- 9.Ji HT, Zhang WN, Zhou YJ, Zhang M, Zhu J, et al. A three-dimensional model of lanosterol 14 alpha-demethylase of Candida albicans and its interaction with azole antifungals. J Med Chem. 2000;43:2493–2505. doi: 10.1021/jm990589g. [DOI] [PubMed] [Google Scholar]
- 10.Podust LM, Stojan J, Poulos TL, Waterman MR. Substrate recognition sites in 14 alpha-sterol demethylase from comparative analysis of amino acid sequences and X-ray structure of Mycobacterium tuberculosis CYP51. J Inorg Biochem. 2001;87:227–235. doi: 10.1016/s0162-0134(01)00388-9. [DOI] [PubMed] [Google Scholar]
- 11.Sheng CQ, Zhang WN, Zhang M, Song YL, Ji HT, et al. Homology modeling of lanosterol 14 alpha-demethylase of Candida albicans and Aspergillus fumigatus and insights into the enzyme-substrate interactions. J Biomol Struct Dyn. 2004;22:91–99. doi: 10.1080/07391102.2004.10506984. [DOI] [PubMed] [Google Scholar]
- 12.Lepesheva GI, Nes WD, Zhou WX, Hill GC, Waterman MR. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry. 2004;43:10789–10799. doi: 10.1021/bi048967t. [DOI] [PubMed] [Google Scholar]
- 13.Kurzrock R, Albitar M, Cortes JE, Estey EH, Faderl SH, et al. Phase II study of R115777; a farnesyl transferase inhibitor, in myelodysplastic syndrome. J Clin Oncol. 2004;22:1287–1292. doi: 10.1200/JCO.2004.08.082. [DOI] [PubMed] [Google Scholar]
- 14.Reid TS, Beese LS. Crystal structures of the anticancer clinical candidates R1 15777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggest a mechanism of FTI selectivity. Biochemistry. 2004;43:6877–6884. doi: 10.1021/bi049723b. [DOI] [PubMed] [Google Scholar]
- 15.Crul M, de Klerk GJ, Swart M, van’t Veer LJ, de Jong D, et al. Phase I clinical and pharmacologic study of chronic oral administration of the farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 2002;20:2726–2735. doi: 10.1200/JCO.2002.09.116. [DOI] [PubMed] [Google Scholar]
- 16.Lebowitz PF, Eng-Wong J, Widemann BC, Balis FM, Jayaprakash N, et al. A phase I trial and pharmacokinetic study of tipifarnib, a farnsyltransferase inhibitor, and tamixofen in metastatic breast cancer. Clin Cancer Res. 2005;11:1247–1252. [PubMed] [Google Scholar]