

Abstract
MET, a receptor protein tyrosine kinase activated by hepatocyte growth factor (HGF), is a crucial determinant of metastatic progression. Recently, we have identified p53 as an important regulator of MET-dependent cell motility and invasion. This regulation occurs via feedforward loop suppressing MET expression by miR- 34-dependent and -independent mechanisms. Here, by using Dicer conditional knockout, we provide further evidence for microRNA-independent MET regulation by p53. Furthermore, we show that while MET levels increase immediately after p53 inactivation, mutant cells do not contain active phosphorylated MET and remain non-invasive for a long latency period at contrary to cell culture observations. Evaluation of mouse models of ovarian and prostate carcinogenesis indicates that formation of desmoplastic stroma, associated production of HGF by stromal cells and coinciding MET phosphorylation precede cancer invasion. Thus, initiation mutation of p53 is sufficient for preprogramming motile and invasive properties of epithelial cells, but the stromal reaction may represent a critical step for their manifestation during cancer progression.
Key words: feedforward loop, HGF, invasion, MET, metastasis, motility, p53 mutation
Introduction
MET receptor protein tyrosine kinase is a key regulator of cell motility and invasion.1,2 Not surprisingly, aberrant activation of MET has been associated with increased metastatic propensity of cancer cells as well as poor prognosis of cancer patients.3 Stimulation of MET is largely accomplished by its ligand, hepatocyte growth factor (HGF), also known as scatter factor.4,5 Upon HGF binding to the extracellular domain of MET, tyrosine residues Y1234 and 1235 are phosphorylated, leading to activation of the autocatalytic domain. Subsequently, the MET C-terminal binding domain (Y1349/1356) is phosphorylated, further stimulating downstream targets, such as metalloproteases, osteopontin, plasminogen activator and integrins.6 HGF has been originally identified as a fibroblast-derived epithelial morphogen,5,7 and HGF-MET pathway is regarded to be a prototypical example for stromal-epithelial interactions during developmental morphogenesis, wound healing, organ regeneration and cancer progression.1,2,8
Recently we have reported that MET-dependent cell motility and invasion are controlled by tumor suppressor p53.9 p53 executes its control by two mechanisms: (1) transactivation of miR-34 genes that target MET 3′UTR10–12 and (2) inhibition of SP1-activating binding to MET promoter.9 Since both mechanisms lead to suppression of MET expression, this type of regulation can be classified as a coherent type 3 feedforward loop or type II circuit based on previously proposed classifications of signaling pathways.13,14
Since genes encoding miR-34 family were first identified as direct targets of p53 transcriptional activation,10,15–19 a number of other microRNAs were additionally reported to be regulated by p53.20 Thus, it remains uncertain if p53-dependent expression of other microRNAs in addition to miR-34 may have additional impact on MET regulation.
We have reported that elevated levels of MET and associated increases in cell motility and invasion were observed in cell culture within first 24–72 h after CreloxP mediated p53 inactivation.9 Addition of HGF further stimulated motility and invasion of p53-deficient cells ex vivo.9 Elevated levels of MET were also observed in the ovarian surface epithelium (OSE) in vivo 72 h after p53 inactivation. However, in the mouse model of epithelial ovarian cancer (EOC) based on conditional inactivation of p53 and Rb in OSE, invasion by mutant cells is rarely observed before 100–120 d after the initiation, and EOC develops after a long latency period, with median survival time of 227 d.21,22 These results indicate that immediate implementation of the phenotypical traits associated with alterations in p53 and Rb pathways is either prevented by some compensatory mechanisms or requires some additional exogenous stimuli similar to those present in cell culture medium.
Here, we explore the feedforward loop regulation of MET in the microRNA-free cell system. Furthermore, by using autochthonous mouse models of high-grade serous EOC and prostate carcinoma, we provide evidence that stromal-epithelial interactions may play a crucial role in cancer pathogenesis by activating HGF-MET signaling and thereby facilitating motility and invasion of p53 mutant cells.
MicroRNA-Dependent Regulation of MET Mainly Depends on miR-34 Family but is Insufficient for Complete MET Control
Previously, we have shown that p53 has a miR-34-independent regulation of MET expression in miR-34-deficient background.9 To test whether there are other microRNAs targeting MET, in addition to miR-34 family, we performed a bioinformatics search using microRNA data sets obtained from OSE cells within two passages after p53 inactivation.18 Besides miR-34 family, none of the microRNAs downregulated after p53 inactivation had a seed sequence predicted to target MET. While the mechanism of microRNA action is believed to be either mRNA degradation or destabilization by 3′UTR binding, some small dsRNAs and microRNAs have been identified to activate promoter activities through complementary sequence binding.23,24 However, none of microRNAs upregulated after p53 inactivation18 had a seed sequence predicted to bind MET promoter region or 5′UTR. These results further indicate that the microRNA-dependent component of MET regulation by p53 is mainly represented by members of miR-34 family, at least in OSE cells.
To further confirm microRNA-independent MET regulation by p53, primary OSE cells were derived from Dicerfl/fl, p53fl/fl and Dicerfl/flp53fl/fl mice. Consistent with the lack of mature microRNAs, including miR-34, in Dicer-null cells,25 Dicer inactivation led to an increase of Met expression by 1.7-fold (Fig. 1A). However, concomitant inactivation of p53 and Dicer resulted in further significant elevation of Met levels, similar to levels after p53 inactivation alone. At the same time, reconstitution of p53 expression in p53- and Dicer-null OSE cells resulted in downregulation of MET expression (Fig. 1B). Thus, our findings support our earlier observations of existence of miR-34-independent regulation of MET by p53.
Figure 1.

miR-34-independent regulation of MET in microRNA-free environment. (A) Met expression in primary OSE cells isolated from Dicerfl/fl, p53fl/fl and Dicerfl/flp53fl/fl mice. Met mRNA expression (mean ± SD, n = 3) was measured by qRT-PCR 48 h after infection with Ad-Cre or Ad-LacZ. (B) MET expression in p53-/-Dicer-/- OSE cells 48 h after transfection with either empty vector (pORF) or pORF-hp53.
MET Activation Coincides with Accumulation of Desmoplastic Stroma and Expression of HGF by Connective Tissue Cells
To determine the time of MET activation, we have performed immunohistochemical detection of functionally active phosphorylated MET (P-MET) at various time after initiation of carcinogenesis in the mouse model of high-grade serous EOC associated with p53 and Rb deficiency.21 In this model cancer is induced by a single administration of the adenovirus expressing Cre into the ovarian bursa of adult mouse, and tumors develop relatively synchronously.21 Thus, this model is well-suited for stage-by-stage studies of carcinogenesis. Importantly, p53 mutations are detected in 96% cases of high-grade serous EOC26 and reported to be the early or initiating event in this EOC type.27,28 Taken together with the fact that alterations of RB pathways occur in 67% of cases of high-grade serous EOC,26,29 this mouse model is highly relevant to the pathogenesis of human EOC.
Consistent with our previous reports in references 22 and 30, early atypical cells have been consistently detected beginning 60 d after p53 and Rb inactivation. Desmoplastic stroma accumulates under dysplastic lesions within 160–220 d following by development of highly invasive and metastatic carcinomas. Expression of functionally active P-MET was below detectable level in normal OSE, while it was easily detectable in invasive adenocar-cinomas (Fig. 2). Interestingly, P-MET was barely detectable in early atypical cells at 60 d after EOC induction, even so that inactivation of both p53 and Rb in over 80% of OSE has been confirmed by microdissection-PCR genotyping as well as EGF and LacZ reporters.9,21,22 At the same time, P-MET was easily detected in dysplastic OSE above desmoplastic stroma. Appearance of P-MET coincided with elevated expression of HGF in stromal cells forming desmoplastic response. In an agreement with this observation, HGF from fibroblasts has been shown to activate MET in epithelial cancer cells.31 Interestingly, while diffuse expression of HGF was observed in the majority of desmoplastic cells, its particularly high levels were associated with cells of macrophage/histiocyte morphology. Together with our recent cell culture studies showing that addition of HGF stimulates motility and invasion of p53-deficient OSE cells,9 these results suggest that migratory and invasive features of p53 mutant cells are facilitated by desmoplastic stroma during cancer progression.
Figure 2.

Expression of phosphorylated MET (P-MET) during OSE carcinogenesis. Normal OSE (Control, arrow) and preinvasive neoplastic cells (60 d, arrow) above the regular stroma express negligible levels of P-MET. Preinvasive OSE (170 d, arrow) above desmoplastic stroma (arrowhead) and invasive neoplastic OSE cells (270 d, arrow) contain detectable amount of P-MET. Note expression of HGFα (HGF) and F4/80 in macrophages (60 d, arrowheads) and desmoplastic cells and macrophages/histiocytes (170 and 270 d, arrowheads). Hematoxylin-eosin (H&E) and ABC Elite method, hematoxylin counterstaining (P-MET, HGF and F4/80). Calibration bar, all images, 50 µm.
p53 and/or Rb Inactivation do not Upregulate HGF Expression in OSE Cells
Previously it has been reported that human, rat and bovine OSE also expresses HGF.32,33 At the same time, expression of HGF was rarely observed in the OSE cells from women with no family histories of ovarian cancer.34 Notably, even in the OSE of women with familial history of ovarian cancer or ovarian cancer cell lines, the levels of HGF were far lower than those in fibroblasts. By using our previous microarray data,9,35 we have found that mouse OSE have only negligible levels of Hgf, which were significantly lower than those in primary mouse fibroblasts (Fig. 3). Importantly, no significant elevation of Hgf expression was observed in OSE cells after p53, Rb or p53/Rb inactivation (Fig. 3), while a modest decrease of Hgf expression was observed in fibroblasts lacking both p53 and Rb.
Figure 3.
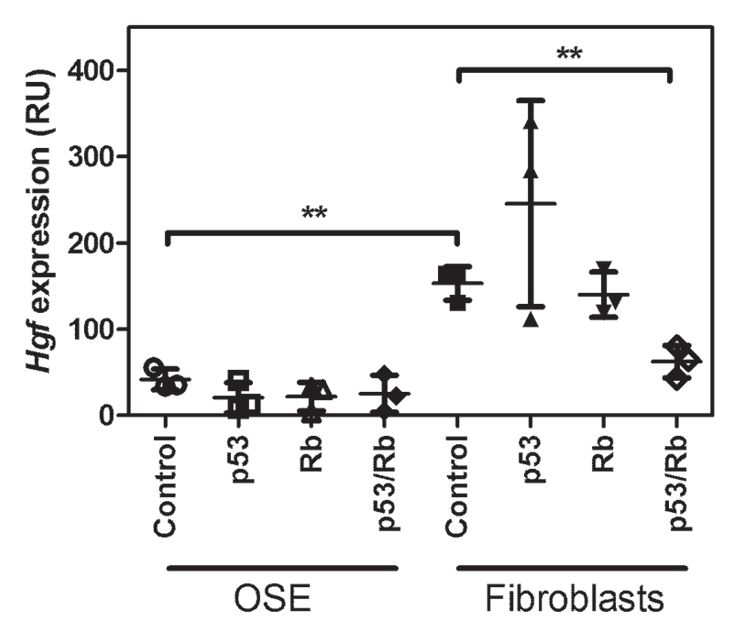
Hgf expression in OSE cells and fibroblasts after p53, Rb or p53/Rb inactivation. Signal intensity of Hgf expression (mean ± SD, n = 3) was measured by Affymetrix GCOS software and normalized by scaling each GeneChip to a target signal of 500. **p < 0.01.
P-MET is Associated with Advanced Stages of Prostate Cancer
To test if our observations can be applicable to other types of cancer, we evaluated a mouse model of prostate cancer previously established by us. In this model, conditional inactivation of p53 and Rb in the prostate epithelium results in rapidly progressing metastatic carcinomas.36 Notably, while atypical lesions were present in both proximal and distal regions of prostatic ducts, only the former progressed to overt cancers.37 Our present immunohistochemical studies indicate that only proximal neoplastic lesions exhibit elevated MET expression (Fig. 4), which may explain why progression rate of proximal vs. distal region-derived neoplasms greatly depends on microenvironment.37 Consistent with these results, particularly high levels of P-MET are observed in the neoplastic cells positioned in proximity to the desmoplastic stromal cells expressing HGF (Fig. 4).
Figure 4.

Expression (arrows) of total (MET) and phosphorylated (P-MET) MET and HFGα (HGF) in prostate neoplastic lesions. Carcinomas (Ca) and prostate intraepithelial neoplasms (PIN), are located, respectively, in the proximal and distal regions of prostatic ducts of mice with prostate epithelium-specific inactivation of p53 and Rb. Note absence of MET expression in PIN and HGFα location in stromal (arrow) but not epithelial (arrowhead) cells. ABC Elite method, hematoxylin counterstaining. Calibration bar, 50 µm.
Discussion
The biological meaning of coherent feed-forward loop regulation remains insufficiently understood.38,39 It has been suggested that such a type of regulation contributes to filtering out fluctuations in input stimuli, while allowing rapid biological responses and surveillance mechanisms necessary to regulate a leaky transcription of target genes.13 The functional redundancy of coherent feedforward loops might also facilitate signal transduction or provide selective advantages during the process of evolution.40
Recently we have shown that the degree of p53/miR-34/MET signaling network's impact on cell motility and invasion depends on the extent of alterations in its individual components.9 Mutant p53 proteins commonly contain point mutations in DNA binding domain, which renders them unable to transactivate miR-34-encoding genes. However, such mutants are still able to bind SP1 and thereby repress MET promoter activity, albeit less efficiently than wild-type p53 (Fig. 5). Complete lack of p53 abolishes both mechanisms of MET regulation, leading to its maximal expression and associated metastasis-related cancer traits, such as cell motility and invasion. Consistent with these findings, it has been reported that patients with p53-null ovarian cancer have worse prognosis than those with other p53 status in their cancers.41–43 Furthermore, the poorest prognosis was also reported for patients with p53-null cancers of the lung44 and breast.45–47 Notably, MET overexpression has been reported to be associated with poor prognosis of patients with these types of cancer as well.48–50 Therefore, our observations that p53 has a feedforward loop regulation of MET expression may provide a mechanistic link between two independently reported prognostic factors, p53-null status and MET overexpression.
Figure 5.

Proposed model of MET-dependent cancer invasion preprogrammed by early alterations of p53-regulated feedforward loop and triggered by stromal cell-derived HGF. Feedforward loop regulation of MET by wild-type p53 consists of miR-34-dependent and -independent mechanisms. p53 protein with point mutations in its p53 DNA binding domain is unable to transactivate MET targeting miR-34. However, it still binds SP1, thereby repressing MET promoter, albeit to a limited extent. In contrast, lack of p53 due to null mutations results in incapacitations of both miR-34 transactivation and MET promoter repression, thereby leading to highest levels of MET. Although p53 mutations result in increased expression of MET, cells become highly motile and invasive only after MET activation by phosphorylation (P-MET). This phosphorylation is triggered by MET ligand HGF produced by cells of newly forming desmoplastic stroma. Thus, while alterations in p53/miR-34/MET network preprogram cells for increased motility and invasion, microenvironment may play a crucial role in triggering those properties during cancer progression.
Detailed understanding of molecular mechanisms of feedforward loop regulation of MET may reshape our approach on cancer therapeutics. While inhibition of MET has been considered as an important drug target and has been under clinical trials,51 restoration of wild-type p53 in ovarian cancer patients has failed.52 Our recent results9 suggest that in the case of patients with p53 mutations retaining MET promoter repressive role, removal of mutant p53 should be avoided. In addition to miR-34 reintroduction, considered as a promising therapeutic approach, intervention of SP1 function might be beneficial for cancer treatment. Recently, tolfenamic acid, a small-molecule SP1 inhibitor that is able to induce degradation of proteins of SP1, has been identified.53,54 As expected, this inhibitor downregulates MET expression in cancer cells, including ovarian cancer cells.55,56
Carcinogenesis, including its metastatic stages, is usually described as a multistage process driven by sequential accumulation of genetic alterations responsible for expression of phenotypical traits beneficial for selection of the most autonomous and, by extension, most malignant cell clones, assuring further progression.57–61 However, there are multiple evidences that the initiating mutation is continuously required for the maintenance of malignant phenotype (reviewed in ref. 62 and 63). Reconsolidation of the concept of cancer as a multigenic disease and tumor suppression by re-introduction or elimination of a single gene is complicated, because many tumors are genomically unstable, and separation of critical alterations from “genetic noise” may be a daunting task in advanced stages of the disease. However growing evidence points to the possibility that some neoplastic cells may already have metastatic ability during the early stages of carcinogenesis.64–67 Furthermore, interactions between neoplastic cells and stroma may further affect carcinogenesis (reviewed in refs. 68 and 69). Our findings support this recent shift in paradigm of sequential cancer progression, indicating that an initiating mutation, such as p53 inactivation, may preprogram advanced cancer traits, such as cell motility and invasion. At the same time, microenvironmental cues, as opposed to genetic or epigenetic alterations within the mutant cells, are critical for triggering preprogrammed invasive traits (Fig. 5). These observations are of particular interest, because the role of stroma in cancer pathogenesis receives increasing attention.70 In the ovary, inflammatory stromal reaction is regarded as an essential part of the ovulatory process in the normal ovary71 and, thereby, may provide a missing link between inflammation and EOC pathogenesis.72 Importantly, our results in the prostate cancer model suggest a common critical role of stromal-epithelial interactions in carcinogenesis associated with alterations in p53/miR-34/MET signaling network.
Taken together, our recent and current studies have shed additional light on the role of feedforward loop regulation and stromal-epithelial interactions in cancer pathogenesis. Further elucidation of these mechanisms should facilitate development of personalized therapeutic approaches.
Materials and Methods
Experimental animals.
Mice with floxed copies of p53, Rb1 (Rb) and Dicer1 allele genes and information for their genotyping were described in references 21, 25, 73 and 74. Modeling of EOC and prostate carcinoma associated with p53 and Rb deficiency has been described by us in references 21 and 36. All mice were maintained identically following recommendations of the Cornell Institutional Laboratory Animal Use and Care Committee.
Immunohistochemical study.
Paraffin sections of 4% paraformaldehyde fixed tissue were stained according to modified the avidin-biotin-peroxidase technique.35,75 The antibodies to MET (1:50; Santa Cruz Biotechnolog), Phospho-MET Tyr1349 (1:50 Cell Signaling Technology), HGFα (1: 150, H145, Santa Cruz Biotechnology), and F4/80 (1:200; Serotec), were incubated for 1 h at room temperature.
Cell culture.
Primary mouse OSE cells were isolated and cultured as previously described in references 18, 21 and 30. In order to establish a p53/Dicer-deficient cell line, primary OSE cells were prepared from the ovary of p53fl/flDicerfl/fl mice, infected with Ad-Cre and passaged upon reaching confluence for 30 passages under the same conditions as other mouse OSE cell lines.
Quantitative real-time RT-PCR.
Total RNA was isolated using Qiazol (Qiagen) according to manufacturer's protocol. Total RNA was reverse transcribed using SuperScript III (Invitrogen) and oligo-dT primers. The PCR reactions were done in a 30-µl volume in a 96-well plate using pre-developed FAM TaqMan probes (Applied Biosystems). Mouse Gapdh was used as endogenous reference control (pre-developed TaqMan assay from Applied Biosystems). Cycling parameters were as follows: 95°C for 10 min. followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Increase in real-time fluorescence was measured by an ABI7500 qPCR system and relative fold changes were calculated using the 2−ΔΔCt method.76
Protein gel blot analysis.
Cell lysates were prepared using RIPA buffer (TRIS-HCl 50 mM, pH 7.4; NP-40 1%; Na-deoxicholate 0.25%; NaCl 150 mM; EDTA 1 mM; PMSF 1 mM; Aprotinin, leupeptin, pepstatin; 1 µg/ml each; Na3VO4 1 mM; NaF 1 mM), separated by 12% SDS-PAGE and transferred to PVDF membrane (Millipore). The membrane was incubated overnight at 4°C with antibodies to detect MET (SP 260, Santa Cruz Biotechnology, 1:1,000 dilution), p53 (FL 393, Santa Cruz Bitechnology, 1:1,000 dilution) and GAPDH (Advanced Immunohistochemical Inc.; 1:3,000 dilution) followed by incubation for 1 hr at room temperature with horseradish per-oxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and developed using chemiluminescence substrate (SuperSignal West Pico from Pierce).
Bioinformatics.
For prediction of microRNAs binding to 3′UTR of MET, Targetscan 5.2 (www.targetscan.org), Pictar (pictar.mdc-berlin.de/) and miRanda (www.microrna.org) database were searched. For prediction of microRNAs binding to 5′UTR and upstream promoter region of MET, miR-Walk (www.ma.uniheidelberg.de) database was used.
Statistical analysis.
Statistical tests used were two-sided Student's t-tests, using Prism 5.01 software (GraphPad, Inc.).
Acknowledgments
This work was supported by National Institutes of Health Grants R01 CA96823, R01 CA112354 and The Marsha Rivkin Center for Ovarian Cancer funding (to A.Y.N.) and Graduate Research Assistantship awarded by the College of Veterinary Medicine, Cornell University (to C.I.H.).
References
- 1.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 2.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 3.Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be MET? Trends Mol Med. 2005;11:284–292. doi: 10.1016/j.molmed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10:2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weidner KM, Arakaki N, Hartmann G, Vandekerckhove J, Weingart S, Rieder H, et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA. 1991;88:7001–7005. doi: 10.1073/pnas.88.16.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferracini R, Longati P, Naldini L, Vigna E, Comoglio PM. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J Biol Chem. 1991;266:19558–19564. [PubMed] [Google Scholar]
- 7.Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 1991;67:901–908. doi: 10.1016/0092-8674(91)90363-4. [DOI] [PubMed] [Google Scholar]
- 8.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 9.Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Korner S, Wang W, et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci USA. 2011;108:14240–14245. doi: 10.1073/pnas.1017536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Migliore C, Petrelli A, Ghiso E, Corso S, Capparuccia L, Eramo A, et al. MicroRNAs impair MET-mediated invasive growth. Cancer Res. 2008;68:10128–10136. doi: 10.1158/0008-5472.CAN-08-2148. [DOI] [PubMed] [Google Scholar]
- 12.Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 2010;16:1119–1128. doi: 10.1158/1078-0432.CCR-09-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 16.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 17.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67:8433–8438. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 19.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Wong MY, Yu Y, Walsh WR, Yang JL. microRNA-34 family and treatment of cancers with mutant or wild-type p53. Int J Oncol. 2011;38:1189–1195. doi: 10.3892/ijo.2011.970. [DOI] [PubMed] [Google Scholar]
- 21.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63:3459–3463. [PubMed] [Google Scholar]
- 22.Williams RM, Flesken-Nikitin A, Ellenson LH, Connolly DC, Hamilton TC, Nikitin AY, et al. Strategies for high-resolution imaging of epithelial ovarian cancer by laparoscopic nonlinear microscopy. Transl Oncol. 2010;3:181–194. doi: 10.1593/tlo.09310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105:1608–1613. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci USA. 2006;103:17337–17342. doi: 10.1073/pnas.0607015103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi R, O'Carroll D, Pasolli HA, Zhang Z, Dietrich FS, Tarakhovsky A, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet. 2006;38:356–362. doi: 10.1038/ng1744. [DOI] [PubMed] [Google Scholar]
- 26.TCGA-Network, author. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berchuck A, Kohler MF, Marks JR, Wiseman R, Boyd J, Bast RC., Jr The p53 tumor suppressor gene frequently is altered in gynecologic cancers. Am J Obstet Gynecol. 1994;170:246–252. doi: 10.1016/s0002-9378(94)70414-7. [DOI] [PubMed] [Google Scholar]
- 28.Leitao MM, Soslow RA, Baergen RN, Olvera N, Arroyo C, Boyd J. Mutation and expression of the Tp53 gene in early stage epithelial ovarian carcinoma. Gynecol Oncol. 2004;93:301–306. doi: 10.1016/j.ygyno.2004.01.043. [DOI] [PubMed] [Google Scholar]
- 29.Corney DC, Flesken-Nikitin A, Choi J, Nikitin AY. Role of p53 and Rb in ovarian cancer. Adv Exp Med Biol. 2008;622:99–117. doi: 10.1007/978-0-387-68969-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flesken-Nikitin A, Toshkov I, Naskar J, Tyner KM, Williams RM, Zipfel WR, et al. Toxicity and biomedical imaging of layered nanohybrids in the mouse. Toxicol Pathol. 2007;35:806–812. doi: 10.1080/01926230701584239. [DOI] [PubMed] [Google Scholar]
- 31.Kankuri E, Cholujova D, Comajova M, Vaheri A, Bizik J. Induction of hepatocyte growth factor/scatter factor by fibroblast clustering directly promotes tumor cell invasiveness. Cancer Res. 2005;65:9914–9922. doi: 10.1158/0008-5472.CAN-05-1559. [DOI] [PubMed] [Google Scholar]
- 32.Parrott JA, Skinner MK. Expression and action of hepatocyte growth factor in human and bovine normal ovarian surface epithelium and ovarian cancer. Biol Reprod. 2000;62:491–500. doi: 10.1095/biolreprod62.3.491. [DOI] [PubMed] [Google Scholar]
- 33.Uzumcu M, Pan Z, Chu Y, Kuhn PE, Zachow R. Immunolocalization of the hepatocyte growth factor (HGF) system in the rat ovary and the anti-apoptotic effect of HGF in rat ovarian granulosa cells in vitro. Reproduction. 2006;132:291–299. doi: 10.1530/rep.1.00989. [DOI] [PubMed] [Google Scholar]
- 34.Wong AS, Pelech SL, Woo MM, Yim G, Rosen B, Ehlen T, et al. Coexpression of hepatocyte growth factor-Met: an early step in ovarian carcinogenesis? Oncogene. 2001;20:1318–1328. doi: 10.1038/sj.onc.1204253. [DOI] [PubMed] [Google Scholar]
- 35.Choi J, Curtis SJ, Roy DM, Flesken-Nikitin A, Nikitin AY. Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am J Pathol. 2010;177:2645–2658. doi: 10.2353/ajpath.2010.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66:7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 2007;67:5683–5690. doi: 10.1158/0008-5472.CAN-07-0768. [DOI] [PubMed] [Google Scholar]
- 38.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38:20–24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- 40.Kafri R, Springer M, Pilpel Y. Genetic redundancy: new tricks for old genes. Cell. 2009;136:389–392. doi: 10.1016/j.cell.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 41.Shahin MS, Hughes JH, Sood AK, Buller RE. The prognostic significance of p53 tumor suppressor gene alterations in ovarian carcinoma. Cancer. 2000;89:2006–2017. doi: 10.1002/1097-0142(20001101)89:9<2006::AID-CNCR18>3.3.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Rose SL, Robertson AD, Goodheart MJ, Smith BJ, DeYoung BR, Buller RE. The impact of p53 protein core domain structural alteration on ovarian cancer survival. Clin Cancer Res. 2003;9:4139–4144. [PubMed] [Google Scholar]
- 43.Sood AK, Sorosky JI, Dolan M, Anderson B, Buller RE. Distant metastases in ovarian cancer: association with p53 mutations. Clin Cancer Res. 1999;5:2485–2490. [PubMed] [Google Scholar]
- 44.Hashimoto T, Tokuchi Y, Hayashi M, Kobayashi Y, Nishida K, Hayashi S, et al. p53-null mutations undetected by immunohistochemical staining predict a poor outcome with early-stage non-small cell lung carcinomas. Cancer Res. 1999;59:5572–5577. [PubMed] [Google Scholar]
- 45.Lai H, Ma F, Trapido E, Meng L, Lai S. Spectrum of p53 tumor suppressor gene mutations and breast cancer survival. Breast Cancer Res Treat. 2004;83:57–66. doi: 10.1023/B:BREA.0000010699.53742.60. [DOI] [PubMed] [Google Scholar]
- 46.Olivier M, Langerod A, Carrieri P, Bergh J, Klaar S, Eyfjord J, et al. The clinical value of somatic Tp53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006;12:1157–1167. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 47.Rossner P, Jr, Gammon MD, Zhang YJ, Terry MB, Hibshoosh H, Memeo L, et al. Mutations in p53, p53 protein overexpression and breast cancer survival. J Cell Mol Med. 2009;13:3847–3857. doi: 10.1111/j.1582-4934.2008.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–1679. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- 49.Tolgay Ocal I, Dolled-Filhart M, D'Aquila TG, Camp RL, Rimm DL. Tissue microarray-based studies of patients with lymph node negative breast carcinoma show that met expression is associated with worse outcome but is not correlated with epidermal growth factor family receptors. Cancer. 2003;97:1841–1848. doi: 10.1002/cncr.11335. [DOI] [PubMed] [Google Scholar]
- 50.Masuya D, Huang C, Liu D, Nakashima T, Kameyama K, Haba R, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer. 2004;90:1555–1562. doi: 10.1038/sj.bjc.6601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 52.Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003;4:415–422. doi: 10.1016/S1470-2045(03)01139-2. [DOI] [PubMed] [Google Scholar]
- 53.Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis and Sp protein degradation. J Natl Cancer Inst. 2006;98:855–868. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
- 54.Colon J, Basha MR, Madero-Visbal R, Konduri S, Baker CH, Herrera LJ, et al. Tolfenamic acid decreases c-Met expression through Sp proteins degradation and inhibits lung cancer cells growth and tumor formation in orthotopic mice. Invest New Drugs. 2011;29:41–51. doi: 10.1007/s10637-009-9331-8. [DOI] [PubMed] [Google Scholar]
- 55.Papineni S, Chintharlapalli S, Abdelrahim M, Lee SO, Burghardt R, Abudayyeh A, et al. Tolfenamic acid inhibits esophageal cancer through repression of specificity proteins and c-Met. Carcinogenesis. 2009;30:1193–1201. doi: 10.1093/carcin/bgp092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basha R, Ingersoll SB, Sankpal UT, Ahmad S, Baker CH, Edwards JR, et al. Tolfenamic acid inhibits ovarian cancer cell growth and decreases the expression of c-Met and survivin through suppressing specificity protein transcription factors. Gynecol Oncol. 2011;122:163–170. doi: 10.1016/j.ygyno.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 57.Fialkow PJ. Clonal origin of human tumors. Biochim Biophys Acta. 1976;458:283–321. doi: 10.1016/0304-419x(76)90003-2. [DOI] [PubMed] [Google Scholar]
- 58.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 59.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 60.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 61.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 62.Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2:251–265. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 63.Nikitin AY, Connolly DC, Hamilton TC. Pathology of Ovarian Neoplasms in Genetically Modified Mice. Comp Med. 2004;54:26–28. [PubMed] [Google Scholar]
- 64.Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14:283–284. doi: 10.1016/j.ccr.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 65.Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841–1844. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8:227–239. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immuno-surveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010;120:2030–2039. doi: 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell. 2005;7:499–500. doi: 10.1016/j.ccr.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 69.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–520. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 70.Schauer IG, Sood AK, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13:393–405. doi: 10.1593/neo.101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Espey LL. Current status of the hypothesis that mammalian ovulation is comparable to an inflammatory reaction. Biol Reprod. 1994;50:233–238. doi: 10.1095/biolreprod50.2.233. [DOI] [PubMed] [Google Scholar]
- 72.Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91:1459–1467. doi: 10.1093/jnci/91.17.1459. [DOI] [PubMed] [Google Scholar]
- 73.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 74.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 75.Nikitin AYu, Lee WH. Early loss of the retino-blastoma gene is associated with impaired growth inhibitory innervation during melanotroph carcinogenesis in Rb+/− mice. Genes Dev. 1996;10:1870–1879. doi: 10.1101/gad.10.15.1870. [DOI] [PubMed] [Google Scholar]
- 76.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]