

Abstract
Finding new uses for old drugs is a strategy embraced by the pharmaceutical industry, with increasing participation from the academic sector. Drug repurposing efforts focus on identifying novel modes of action, but not in a systematic manner. With intensive data mining and curation, we aim to apply bio- and cheminformatics tools using the DRUGS database, containing 3,837 unique small molecules annotated on 1,750 proteins. These are likely to serve as drug targets and antitargets (i.e., associated with side effects, SE). The academic community, the pharmaceutical sector and clinicians alike could benefit from an integrated, semantic-web compliant computer-aided drug repurposing (CADR) effort, one that would enable deep data mining of associations between approved drugs (D), targets (T), clinical outcomes (CO) and SE. We report preliminary results from text mining and multivariate statistics, based on 7,684 approved drug labels, ADL (Dailymed) via text mining. From the ADL corresponding to 988 unique drugs, the “adverse reactions” section was mapped onto 174 SE, then clustered via principal component analysis into a 5x5 self-organizing map that was integrated into a Cytoscape network of SE-D-T-CO. This type of data can be used to streamline drug repurposing and may result in novel insights that can lead to the identification of novel drug actions.
Keywords: drug discovery, drug side effects, drug targets, Principal Component Analysis, text mining
1. Computer Aided Drug Repurposing
The pharmaceutical industry is subject to an “innovation deficit” [1], which is often expressed as the widening gap between productivity (new molecular entities, NMEs, approved each year) and the annual R&D budget. The number of NMEs approved has declined from mid-40s in the early nineties [2], to under 15 in recent years. The price of drug innovation estimates [3] place the cost of a new drug anywhere between $500 million to over $2 billion, depending on the therapy area and the developing firm [4]. These trends indicate a sharp decline in research productivity across the entire pharmaceutical sector, with the exception of biologics. Currently, major pharmaceutical houses seek to increase short-term profitability via mergers and acquisitions, drastic reductions in research personnel and an increased outsourcing effort. It is therefore not surprising that the National Institutes of Health (NIH) is emerging as a leader not only in the arena of early drug discovery via MLI, the Molecular Libraries Initiative [5], but also in the area of translational medicine via the Clinical and Translational Science Awards (CTSA) initiative [6]. Academic investigators are now more effective in “de-risking” compounds of industrial interest [7].
1.1 Examples
Genomics, pharmacovigilance and side-effects evaluation [8,9], screening drug libraries against neglected diseases [10], data mining for drug side-effects [11] and finding novel targets using in silico tools [12] are equally valid strategies to identify novel uses for old drugs. The concept of drug repurposing [13] is not novel to the pharmaceutical industry: One of the oldest semi-synthetic drugs, acetylsalicylic acid, an anti-inflammatory drug formulated as 500-mg tablets launched in 1896 as Aspirin, was recently re-positioned as daily-dose “baby Aspirin” (75-mg tablets in Europe, 81-mg tablets in the US), for cardiovascular disease prevention [14]. Pfizer combines cetirizine, a histamine H1 receptor antagonist (approved in 1987 as Zyrtec) with pseudoephedrine (a sympathomimetic approved in 1975 as Novated, now discontinued) as a new drug, “Zyrtec-D 12 hour” (launched in 2001), which contains cetirizine 5 mg and pseudoephedrine 120 mg per tablet, for the symptomatic relief of seasonal allergies [15]. Caffeine, a naturally-occurring CNS stimulant [16] used in combination with multiple API to increase alertness and diuresis [17], was approved as “Cafcit®” (caffeine citrate, injection for intravenous administration) in 2000 by the U.S. Food and Drug Administration (FDA), and in 2007 by the European Medicines Agency (EMA) for the short-term treatment of apnea of prematurity in newborn infants between 28 and 33 weeks gestational age [18]. Other examples, including duloxetine and thalidomide, are reviewed elsewhere [13].
1.2 Critical Barriers
Described in section 505(b)(2) [19] of the Federal Food, Drug, and Cosmetic Act, the process of drug repurposing is made possible by the Drug Price Competition and Patent Term Restoration Act of 1984 (also known as the Hatch-Waxman Amendments [20]), which enables the applicant for a new drug application (NDA) to reference investigations of safety and effectiveness where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Section 505(b)(2) offers patent protection (hence market monopoly) for NMEs, new dosage forms (e.g., “baby Aspirin”), in administration routes (e.g., oral vs. intra-venous caffeine citrate), new indications, and for new NME combinations (e.g., Zyrtec-D). While the expectation is that fewer clinical studies are required for repositioning a drug, this has no impact when the drug is repurposed for a medical condition that previously lacked drug therapy. The burden is even higher when therapeutic agents already exist, i.e., the petitioner needs to prove the therapeutic advantage offered by repurposed drugs. Although the process is expected to last considerably less compared to an all-new NME effort [13], the applicant must nevertheless conduct clinical trials with respect to efficacy (e.g., for new indications), as well as safety (e.g., for higher doses). This financial burden blocks drug repurposing efforts because clinical research can quickly reach the multi-year, multi-million dollar range.
1.3 CTSA
Having recognized the gap between basic and clinical science, the NIH launched efforts to bridge the repurposing “valley of death” by fostering translational research via the CTSA (Clinical and Translational Science Awards) initiative [6]. CTSA has an online collection of research volunteers [21], an index of CTSA technologies and intellectual properties [22], as well as a portal partnering academics and pharmaceutical companies for no-longer developed molecules [23].
1.4 Viability
Against the backdrop of increased difficulties in taking NMEs into the clinic, one ought to consider the merits of “drug repurposing” (also termed drug repositioning, or drug re-profiling) as a viable option. First, our level of knowledge in the polypharmacology [24, 25] of drugs has reached a good degree of maturity [26], because of an increased in-depth profiling effort, in particular for novel drugs. More importantly, our level of knowledge, addressing data completeness gaps [27], is increasing for the older drugs as well: This is, to a large extent, due to the availability of screening data in public sources such as PubChem [28] for out-of-patent drugs, as incorporated for example in the Prestwick Chemical Library [29].
1.5 Approach
We envision a computer-aided drug repurposing (CADR) platform as being a semantic-web service that would rely on factual associations between drugs, targets and clinical outcomes. The CADR platform would provide in-depth integration for these four categories:
drugs (D), i.e., the active pharmaceutical ingredients (API) and their active metabolites, with initial focus on small molecule APIs;
targets (T), macromolecules perturbed by API that lead to a clinical outcome;
positive clinical outcomes (CO), i.e., the intended therapeutic effects of drugs, as specified on the approved drug labels (ADL) under “Indications”
negative clinical outcomes, often referred to as “adverse events” or drug side effects, SE.
To establish such factual associations, a two-pronged approach is needed: (i) deep data mining of D-T interactions, including indexing, cross-referencing, processing and curation of the molecular, pharmacological and biochemical aspects of drug-target interactions; and (ii) text mining of ADL and clinical research documents using controlled vocabularies, which would be used to extensively process the “adverse events” and “indications” sections of medical package inserts or on-line repositories such as DailyMed [30]. This approach is conceptually built on prior work, which inferred novel drug targets starting from a combination of chemical and phenotypic side-effect similarities [11].
1.6 Potential
The CADR platform, while built upon open-access resources such as DrugBank [31] and DailyMed, can provide improvements in two directions: (i) Semantic web [32] compliance, which aims to provide structured drug-related information (D-T-CO and D-T-SE relationships), with associated sets of inference rules in the form of RDF (Resource Description Framework) triples that computers will use to conduct automated reasoning; and (ii) systematic mapping of SE (at the symptom level wherever possible) with targets and antitargets [33], that would overlap symptoms related to unmet clinical needs (e.g., for rare and neglected diseases [7]) with SE and CO relationships. As it increases its coverage, the CADR platform may lead to systemic analyses of both clinical and basic science data, and may reduce the impact of the accidental discovery (i.e., serendipity). This prospective review describes preliminary steps taken towards assembling the requisite elements for a viable CADR platform: First, we address efforts in developing an exhaustive knowledge base for D-T interactions. Then we discuss preliminary results based from SE data modeling, as extracted from ADL. Finally, network-based associations between D-T pairs and clinical outcomes are evaluated from the perspective of side-effect inter-relationships.
2 Data collection and analysis
2.1 Small molecule API interaction annotations (D-T)
With the final goal being data completeness [27], we identified and curated information from multiple databases referring to API in order to create a comprehensive repository of drugs, beginning with small molecules. The focus of the DRUGS database is to capture and integrate target bioactivity information for all small molecule drugs, i.e., unique API that have obtained marketed drug status for human use, regardless of country of approval. In its current form, DRUGS has 3,837 unique chemicals (December 2010). DRUGS was primarily built using data from WOMBAT-PK [34], PDSP [35] and DrugBank, with additional information collected from publications [36, 37]. DRUGS stores accurate chemical structures which were independently verified across several sources including ADL and SciFinder [38], generic names and common synonyms. Chemical structures were subject to standardization (salt removal, charge neutralization, and aromatization) prior to identity searching. For unique targets, we used UniProt [39] identifiers and ontologies to uniquely map the proteins in DRUGS, observing compatibility with the disease chemical biology approach, ChemProt [40]. As of December 2010, DRUGS had 3,837 unique API, 19,593 D-T interactions, and 1,750 unique targets.
2.2 Numerical Values
However, not all of the D-T are ascertained to be clinically relevant, nor have the D-T values been validated for having an affinity that is linked to a clinical outcome. Because massive amounts of D-T interaction data are sometimes available from on-line resources such as IUPHAR-DB [41,42], ChEMBL [43], PDSP and PubChem, in particular for older drugs, this may result in a plurality of information that is sometimes contradictory: e.g., the same API is indexed on the same target from the same species, but numerical values differ by 2 orders of magnitude or more. Furthermore, numerical values attributed to biological activities are also subject to “temporal drift”: For example, in the 1960s propranolol had an affinity of ∼31 nM for the beta-adrenergic receptor [44] (only one was known), but is now annotated with affinities of 2 nM, 5 nM and 600 nM for the β1,β2 and (β3 adrenergic receptors, respectively [41], in addition to the serotonergic 5-HT1a receptor (30 nM) [34]. Both accuracy of detection and our understanding of targets improve with time.
Therefore, we have implemented a process to eliminate duplicated D-T-bioactivity pairs, giving higher priority to expert-curated (e.g., IUPHAR-DB or “PDSP certified”) data wherever possible. Median values for bioactivity data (excluding highest and lowest value) were used wherever 5 or more values for the same biological end-point were available. A confidence score (e.g., 1.0 for highly trusted sources, and 0.5 for lack of numerical data) was implemented. For the purpose of this report, the DRUGS database relied on data from IUPHAR-DB, PDSP and WOMBAT-PK, which were converted into a format amenable for further processing via in-house data conversion and cheminformatics tools (i.e., JChem [45] and OpenEye [46] software).
2.3 Visual mapping
Earlier efforts resulted in the integration of 739 molecules (out of ∼2300) from IUPHAR-DB into iPHACE [47, 48], a web-based tool built around in extenso pharmacological annotations from IUPHAR-DB and PDSP that has the capability to visualize the interaction on many drugs on many targets (Fig. 1). The IUPHAR website has linked individual records back into iPHACE [49].This technology, currently extended to DRUGS, will help us evaluate the complexity of data parsing and extraction as well as the degree of automation achievable for future updates.
Figure 1.
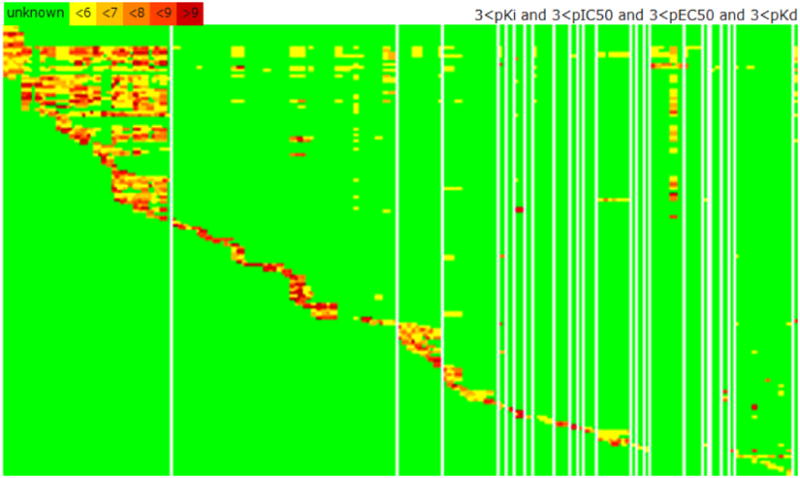
Visual summary of the IUPHAR/PDSP databases, showing 4,033 interactions for 736 drugs (rows) and 178 targets (columns). The heatmap is color coded: bioactivity is higher from yellow to dark red; green indicates absent data. Targets are clustered by family (e.g., G-protein coupled Class A amine receptors are in the far left column). The density of bioactivity in the top left corner reflects the promiscuity of this class of receptors, which includes dopaminergic, muscarinic and serotonergic receptors.
2.4 Approved drug labels availability
There are currently a number of on-line resources that contain relevant information related to package inserts and ADL: DailyMed and the (related) U.S. FDA [50], the EMA [51], the World Health Organization, WHO [52], the Australian Therapeutic Goods Authority, TGA [53] – which are open access, as well as the for-fee resources Physician Desk Reference, PDR [54], Martindale [55], and the American Hospital Formulary Service (AHFS) [56], among others. The process of deep data mining for ADL starts with data capture and mapping for API indexed in DRUGS. Priority is given to “clinical pharmacology”, “indications and usage”, “contraindications”, “adverse reactions” and “description”. However, as other sections may contain pertinent information, they are typically stored (unprocessed) for later use. Particular care needs to be given to standardize, catalog and process clinical outcomes and drug side effects via extensively annotated vocabularies. Compatibility with the side-effect resource (SIDER) [57] is likely to build on SE frequency for the available D-SE pairs. The CADR platform is likely to take advantage of this D-SE mapping to enable inferences combining drug and target information over an enormous, presently sparsely mapped space of drug-target-clinical outcome assertions that would not otherwise be possible.
2.5 ADL text mining
We processed all Dailymed (XML format) records (May 2010 version) in order to evaluate (i) the number of unique drugs present and (ii) the relationship between these drugs and SE. Dailymed entities contain a surprising number of API duplicates, e.g., over 90 drug entries contain “estradiol”. We performed de-duplication in order to simplify, structure and streamline this dataset. We flagged duplicates both at the API level, e.g., where API names are identical, and at the generic drug name level. De-duplication by active moiety can be illustrated for gentamicin: while its correct chemical name is “gentamicin C1 sulfate”, the following synonyms were identified in DailyMed: “Gentamicin Sulfate in Sodium Chloride”, “Gentamicin Sulfate in Sodium Chloride Injection”, “GENTAK”, “Isotonic Gentamicin Sulfate”, “Gentamicin Sulfate” and “GENTAMICIN SULFATE”. These trade names are listed in DailyMed as separate drugs (which they are), but cannot be easily mapped onto a unique API.
Our two-step procedure reduced the dataset from 7,684 to 1,768 unique entities, or 77% reduction. We further removed 261 animal drug products as well as allergenic therapeutics lacking specific chemical API information (e.g. “Cat pelt” and other animal extracts). This process yielded 1,329 entries. After manual curation, the dataset was found to include 1,021 small molecules, of which 20 were duplicates not detected via automation (e.g., “acetate hydrocortisone” and “hydrocortisone acetate”); 243 small molecule mixtures (e.g., simvastatin & niacin), 3 undefined mixtures (omega-3-acid ethyl esters, perflutren and sinecatechins), 28 proteins, 26 monoclonal antibodies, three non-drugs with therapeutic use (sodium acetate, sodium bicarbonate and tromethamine), one parasite extract (Trichophyton) and one insect extract (Sitotroga), respectively.
The final XML files for 988 small molecule drugs (which is what DailyMed contains) were processed with the Python xml.dom.minidom package for SE word frequency and association using the text mining TM package from the R statistical software [58]. Term-frequency vectors, term-document matrices, and distance matrices were generated and used to analyze SE similarity and groupings. In particular, we subjected the frequency matrix containing 174 SE columns for 988 rows (drugs) to PCA, principal component analysis PCA [59] using the Simca package [60]. Data were then visualized using a self-organizing map [61] via Spotfire [62]. Each SE was manually associated with a specific tissue or organ, wherever possible (see also Fig. 2).
Figure 2.
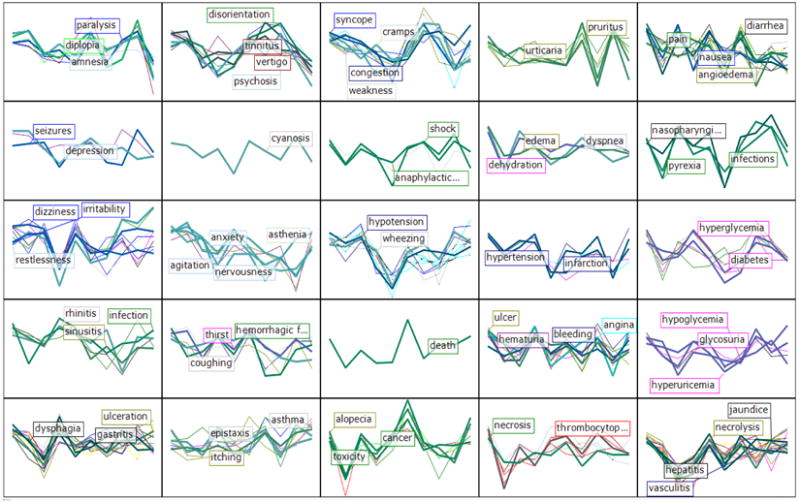
Self-organizing map (SOM) for 174 SE, based on the 10-PCA model derived from frequency of occurrence in DailyMed ADL from 988 small molecule drugs; the SOM clustering was mapped onto 25 cells. Colors encode tissue information; most frequent are skin-mucosa (brown; 27), CNS (azure blue; 17), digestive tract (black; 15), “metabolic imbalances” (magenta; 15), respiratory (pink, 13) and vascular (dark purple; 11). SE labels are representative of each cluster.
3 Associating Drugs, Targets and Clinical Outcomes
The D-SE sparse matrix (29,263 SE occurrences, or 16.81% occupancy) yielded a 10-dimensional model (missing data were attributed a 0 value). The cumulative fraction of the variation of the X variables explained by the 10-PC model, R2VX(cum) = 0.365, with a cross-validated cumulative predicted fraction of the variation of the X variables, Q2 VX(cum) = 0.171. Additional principal components produced eigen values under the 5% tolerance limit and were therefore not considered. Although clearly incomplete in terms of SE coverage, we wanted to examine the biomedical relevance of the potential D-SE associations uncovered by this model. The PCA model is graphically summarized in Figure 2, color-coded by tissue or organ: 14 such categories, plus “systemic” were added to the set, but not used in the PCA model.
The emerging clusters indicate that ADL co-occurrence is far from random. For example, rhinitis, sinusitis and infection are related (column 1, row 4, or “cluster_1_4”); ulcer, hematuria, angina and bleeding (cluster 4_4) are close to “death” (the singleton cluster_3_4), which in turn neighbors alopecia, cancer and toxicity (cluster_3_5). Yet other members of cluster_3_5, e.g., stomatitis, fever and lacrimation, relate to some members of cluster_2_5, such as itching, connectivitis, eruptions and itching, or to the more severe asthma and allergic. Toxic and immunotoxic reactions that manifest on the dermal and mucosal layers or in the digestive tract include nausea, diarrhea, vomiting, angioedema, rashes, dyspepsia and flatulence, and are co- clustered with joint-and-muscular pain symptoms such as arthralgia, myalgia, headache, pain, as well as cough (cluster_5_1). Cardiovascular and respiratory SE associations include arrhythmia, bradycardia, tachycardia, fibrillation, hypotension and phlebitis, and bronchospasm, wheezing and apnea, respectively, as well as sedation (cluster_3_3). All eight blood-located SE are in the bottom row (except thrombosis, row 4), whereas most of the CNS and mental SE are on the top left part of this SOM.
Two of the three ophthalmic SE, diplopia (double vision) and photophobia respectively, but not lacrimation, co-occur with CNS and mental SE, namely coma, paralysis, convulsions, amnesia, confusion and ataxia (cluster_1_1). While clinically associated with the eye organ, diplopia and photophobia are not really ophthalmic dysfunctions; rather, it is our perception that is altered; therefore, their clustering association to the CNS/mental neighborhood is quite appropriate. Though based on a limited data set, we conclude that, at least in part, SE occurrences can be explained by drug compartmentalization, i.e., the drug is more likely to cause side effects in the organ/tissue where it is more likely to accumulate. This result, albeit intuitive, is quite surprising, since it stems from a generic text analytics tool that lacks medical context. It matches observations from co-occurrence pharmacovigilance processing of electronic health records for seven drugs [63].
3.1 Limitations of the PCA model
For all its potential merit, this SE-based PCA model is by no means directly usable within the CADR platform: First, automated text mining yielded a rather limited (174) set of adverse reactions, which is significantly smaller than the side effects from SIDER [57]. Second, the PCA model covers only ∼36.5% of the relationship between these adverse events and the drugs included in this model, based on an already sparse matrix. Finally, the relationship between drugs and side effects depends on dosage, which requires more in-depth analysis of ADL data. Some of these aspects (SE incidence, dosage and relative risk) were detailed elsewhere [64]. Despite its limitations, the SE drug matrix allows us to conclude that text occurrences from the ADL “adverse reactions” section co-emerge in clusters by (possible) mechanism of action and topicality (i.e., organ or tissue where the effect occurs).
3.2 Exploring the SE-D-T-CO relationship
The availability of tools that enable biomedical data visualization such as Cytoscape [65,66], can be used to associate D-SE data with target information via biological network analyses. Through its plug-in architecture, we extended Cytoscape to display and analyze SE-D-T-CO networks for small molecule API. Integrating the results from the ADL-based D-SE PCA, we generated a comprehensive network, where each D-SE cluster (shown in Fig. 2) was related to the other clusters based on the inter-cluster relationship given by p-scores (from PCA), and by using available D-T-CO information from WOMBAT-PK. This Cytoscape visualization was intentionally designed to maintain the organ-based topicality observed earlier, while at the same time highlighting the possible associations between often-unrelated (at least from a clinical standpoint) drugs and their modes of action.
Knowledge mining of D-T-CO data requires controlled and structured information, where drugs and targets can be nouns, their relationship can be described as immediate interactions, and pharmacokinetics and pharmacodynamics can be described via more comprehensive associations (i.e., clinical outcomes). The complexity of this interdependence is conceptualized in Figure 3: Each node of this Cytoscape plot is a network in itself, and nodes are maintained separated for visual clarity. The network visualized here is based on 307 drugs, which were selected based on their high affinity (better than 1 μM) for each of the the targets within the network node. Two of these nodes are shown in Figures 4 and 5, and discussed further. Cytoscape session files, complete with network images for each node given in Figure 5, as well as instructions on how to upload them in Cytoscape, are available as Supporting Information.
Figure 3.
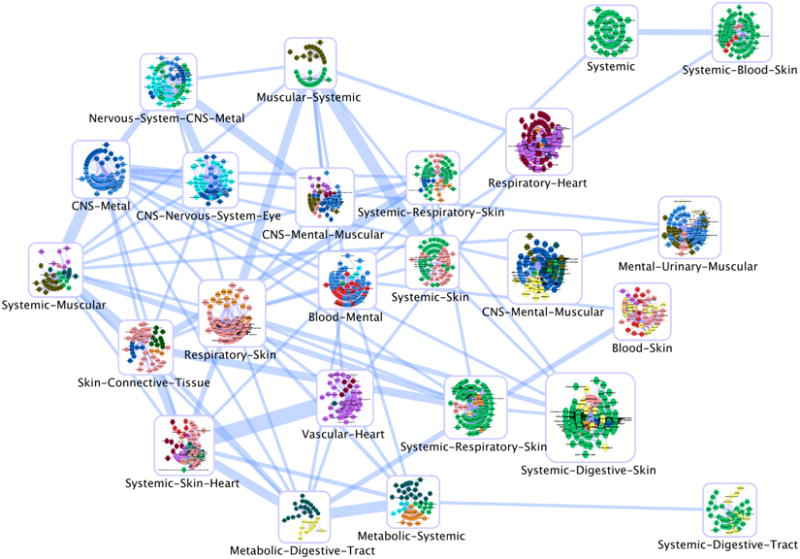
The SE-based D-T-CO network, showing the inter-dependence between drugs, targets and clinical outcomes. Color codes are as follows: Blood, red; CNS, blue; connective tissue, green; digestive tract, yellow; eye, medium purple; heart, dark red; mental, light blue; metabolic, dark cyan; muscular, olive; nervous system, cyan; respiratory, orange; skin and mucosa, pink; systemic, lime green; urinary, light yellow; vascular, magenta. Edge thickness in this network is based on the 10 dimensional PC model, where the centroids of the 25 SOM clusters was used to calculate the Euclidean distances between clusters. This was further projected on two dimensions to map the relative position among clusters
Figure 4.
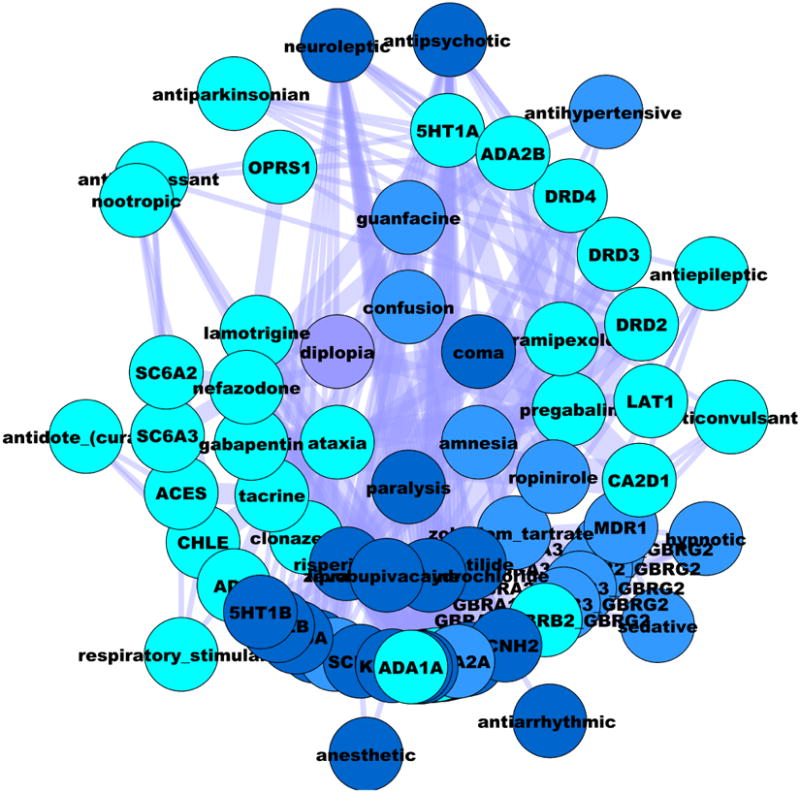
The mental-CNS-deficiency network based on SE (inner layer), the associated drugs (inner middle layer), their confirmed targets (outer middle layer), and intended clinical outcomes (outer layer). Edge thickness in this network is proportional with the strength of the DT interaction. Color codes are discussed in Figure 3.
Figure 5.
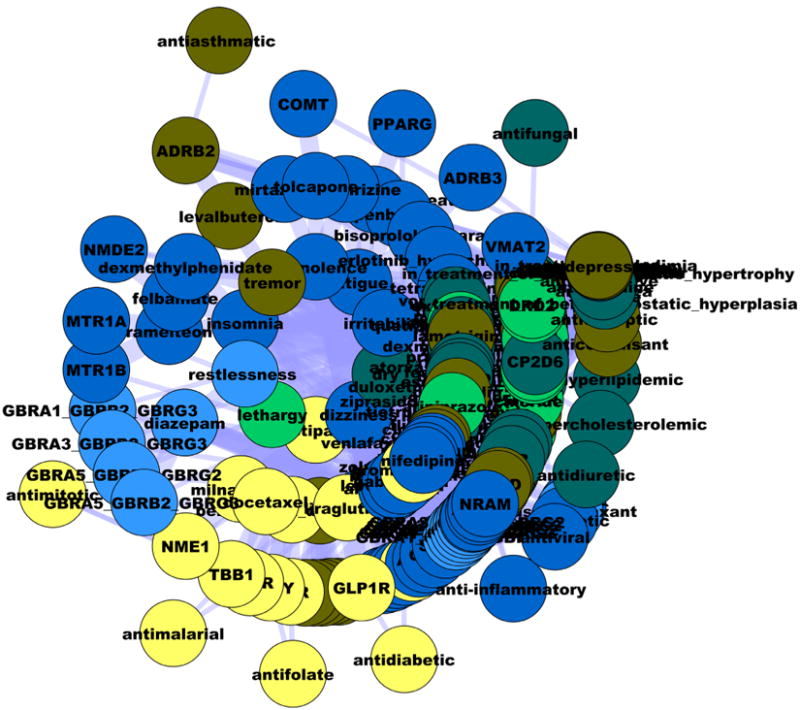
The CNS-mental-disorder network, which includes non-CNS acting drugs in addition to CNS drugs. Layer position and edges are similar to Fig 4. Color codes are discussed in Figure 3.
The complexity of these relationships is further illustrated in Figure 4, which shows the SE-D-T-CO network based on Cluster_1_1, focused on drugs associated with diplopia (e.g., lamotrigine, zonisamide, phenythoin, pregabalin and topiramate). This network primarily includes drugs and targets associated with anticonvulsant, antiparkinsonian & nootropic activities, further supporting the earlier observation that drug-induced diplopia is not an eye-related dysfunction.
Another CNS-related association, given in Figure 5, shows the SE-D-T-CO network based on Cluster_3_1, which includes CNS-related SE for non-CNS drugs (antiinflammatory, antidiuretic, immunosuppressant, anti-allergic, etc.). This network associates insomnia, dizziness and somnolence with tremor, restlessness, agitation and nervousness, respectively. Although having opposite clinical meaning, it is likely that these SE derive from interactions with the same targets, and that non-CNS drugs penetrate (at least to some extent) the blood-brain barrier.
4 Conclusions
In this report we illustrated some of the inherent difficulties in developing the required elements for a viable CADR platform. These steps are necessary, but not sufficient: First, we discussed our efforts in developing a comprehensive evidence-based system for D-T interactions; the DRUGS database attempts to collect public knowledge detailing biochemical and pharmacological interactions between drugs and (potential) targets. Then we discussed our first foray into automated text mining for side effects, one that strictly looks at word associations; these results offer some insight, supported by our 10-PC model and the SE associations.
Although in its early stages, the CADR platform illustrates how knowledge can evolve given deep data mining and tight integration. We showed that content related to clinical (e.g., DailyMed) and chemogenomic data (e.g., DRUGS) can be seamlessly processed and evaluated. Our preliminary PCA model mapped 988 unique drug ADL from DailyMed onto 174 SE. We concluded that adverse reactions can be explained by compartmentalization, i.e., the drug is more likely to cause side effects in the organ/tissue where it accumulates. Carefully associated DT-CO and DT-SE networks are likely to morph into RDF-based knowledge mining, perhaps via Cytoscape. These RDF triples can further lead to computer assertions, i.e., computer-aided drug repurposing. This may be accomplished via Chem2Bio2RDF, a semantic framework developed for linking drug-target information [67], or perhaps via deep knowledge mining processing systems [68]. Even at this preliminary stage, we have showed how D-T pairs and clinical outcomes can be associated within a recursive network-of-networks system. Such recursive system flexibility is likely to be required within the RDF framework itself: the “DT” triple (“drug A inhibits target X”) is in itself the subject of another triple, “A-inhibiting-X” causes “CO/SE”, which is probably the RDF equivalent of a phrase.
When developing DT-CO associations, evidence-based examples, where drug “A” binds to targets X1… Xn resulting in clinical outcomes CO1… COm (this is rarely a 1:1 relationship) will need to be given priority, since this allows computer assertions with relative ease. STITCH [69, 70], an online tool for the exploration of biological networks, does exactly that, i.e., it ranks D-T interactions based on scoring literature co-occurrence data starting with chemical-protein interactions. The ChemProt server [40] can also serve as D-T validation tool. However, many of these relationships are likely to require a certain degree of manual intervention. Tissue location, the presence of active metabolites and additional information related to CO and SE needs to be used for complex cases.
These are likely to result in novel insights that may lead to the identification and assertion of novel “off-target” or “off-label” drug actions. As knowledge bases asymptotically approach completeness, the CADR platform will become more amenable to deep knowledge mining and systemic analyses, integrating basic and translational science with clinical data, which may reduce the impact of the accidental discovery. It will provide to the scientific community, basic scientists and clinicians alike, a new tool to map the clinical, biological and medicinal chemistry space for small molecule drugs, effectively bridging often separate knowledge domains in a multi-disciplinary manner.
Are the factual associations assembled via the CADR platform enough to build a strong case for drug repurposing? With the expectation that it could automatically lead to an NDA, the answer is most likely negative. Such a system could rank with higher priority those applications that are more likely to result in clinically beneficial applications. However, the CADR platform is unlikely to serve as an automated drug repurposing tool in the immediate future. The plethora of DT-CO and DT-SE associations can be mined via automated reasoning, which will narrow down the search space. Yet, humans will remain center stage: Toxicity, efficacy and dosage, as well as alternate therapies (e.g., surgery) are likely to require individual decisions.
Acknowledgments
This work was supported, in part, by NIH grants 1R21GM095952-01 and 5U54MH084690-03 (TIO, OU, JJY, SLM, LAS, CGB), by the Lundbeck foundation grant R32-A3932 (SKN) and by the Villum Foundation CDSB (TIO). TIO, SKN and OU contributed equally to this work. Supporting Information, including the Cytoscape session used to create Figure 5, is available.
Footnotes
Presented at the 18th European Symposium on Quantitative Structure Activity Relationships, EuroQSAR 2010, Rhodes, Greece
Supporting Information for this article is available on the WWW under www.molinf.com
References
- 1.Drews J. Drug Discov Today. 1998;3:491–494. [Google Scholar]
- 2.Olsson T, Oprea TI. Curr Op Drug Discov Dev. 2001;4:308–313. [PubMed] [Google Scholar]
- 3.DiMasi JA, Hansen RW, Grabowski HG. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 4.Adams CP, Brantner VV. Health Affairs. 2006;25:420–428. doi: 10.1377/hlthaff.25.2.420. [DOI] [PubMed] [Google Scholar]
- 5.Austin CP, Brady LS, Insel TR, Collins FS. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 6.CTSA. http://www.ncrr.nih.gov/clinical_research_resources/clinical_and_translational_science_awards/
- 7.Collins FS. Science. 2010;327:36–37. doi: 10.1126/science.1185055. [DOI] [PubMed] [Google Scholar]
- 8.Boguski MS, Mandl KD, Sukhatme VP. Science. 2009;324:1394–1395. doi: 10.1126/science.1169920. [DOI] [PubMed] [Google Scholar]
- 9.Toney JH, Fasick JI, Singh S, Beyrer C, Sullivan DJ., Jr Science. 2009;325:1139–1140. doi: 10.1126/science.325_1339. [DOI] [PubMed] [Google Scholar]
- 10.Chong CR, Sullivan DJ., Jr Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 11.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- 12.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas Al, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KLH, Edwards DD, Shoichet BK, Roth BL. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburn TT, Thor KB. Nature Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 14.Campbell CL, Smyth S, Montalescot G, Steinhubl SR. JAMA. 2007;297:2018–2024. doi: 10.1001/jama.297.18.2018. [DOI] [PubMed] [Google Scholar]
- 15.The FDA-approved label for “Zyrtec D 12 hour” can be accessed at http://www.accessdata.fda.gov/drugsatfda_docs/label/2004/19835slr016,21150slr005,30346slr011_zyrtec_lbl.pdf
- 16.Lara DR. J Alzheimers Dis. 2010;20:S239–S248. doi: 10.3233/JAD-2010-1378. [DOI] [PubMed] [Google Scholar]
- 17.Glade MJ. Nutrition. 2010;26:932–938. doi: 10.1016/j.nut.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Henderson-Smart DJ, De Paoli AG. Cochrane Database Syst Rev. 2010;12:CD000140. doi: 10.1002/14651858.CD000432.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The FDA Guidance for the Industry with respect to 505(b)(2) is available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatorylnformation/Guidances/ucm079345.pdf
- 20.The Hatch-Waxman Amendments are at http://www.fdli.org/pubs/JournalOnline/54_2/art2.pdf
- 21.CTSA research volunteers. https://www.researchmatch.org/
- 22.CTSA IP. http://www.ctsaip.org/
- 23.CTSA Portal. http://www.ctsapharmaportal.org/
- 24.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. Nature Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 25.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Nature Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 26.Vogt I, Mestres J. Mol Inf. 2010;29:10–14. doi: 10.1002/minf.200900069. [DOI] [PubMed] [Google Scholar]
- 27.Mestres J, Gregori-Puigjané E, Valverde S, Solé RV. Nature Biotechnol. 2008;26:983–984. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 28.The PubChem portal is http://pubchem.ncbi.nlm.nih.gov/.
- 29.Prestwick Chemical Library information at http://www.prestwickchemical.fr/index.php?pa=26
- 30.DailyMed can be accessed at http://dailymed.nlm.nih.gov/dailymed/.
- 31.DrugBank is available at http://www.drugbank.ca/.
- 32.Berners-Lee T, Hendler J, Lassila O. Sci Am. 2001;284:34–3. [Google Scholar]
- 33.Vaz R, Klabunde T. Antitargets. Wiley-VCH; Weinheim; 2008. [Google Scholar]
- 34.Olah M, Rad R, Ostopovici L, Bora A, Hadaruga N, Hadaruga D, Moldovan R, Fulias A, Mracec M, TI Oprea. In: Chemical Biology: From Small Molecules to Systems Biology and Drug Design. Schreiber SL, Kapoor TM, Wess G, editors. Wiley-VCH; Weinheim: 2007. pp. 760–786. [Google Scholar]
- 35.PDSP is available at http://pdsp.med.unc.edu/.
- 36.Proudfoot JR. Bioorg Med Chem Lett. 2005;15:1087–1090. doi: 10.1016/j.bmcl.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 37.Vieth M, Sutherland JJ. J Med Chem. 2006;49:3451–3453. doi: 10.1021/jm0603825. [DOI] [PubMed] [Google Scholar]
- 38.SciFinder is available at https://scifinder.cas.org/scifinder/
- 39.The UniProt Consortium. Nucl Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taboureau O, Nielsen SK, Audouze K, Weinhold N, Edsgärd D, Roque FS, Kouskoumvekaki I, Bora A, Curpan R, Jensen TS, Brunak S, Oprea TI. Nucl Acids Res. 2011;39:D367–D372. doi: 10.1093/nar/gkq906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, Greenhill SD, Hale VA, Sharman JL, Bonner Tl, Catterall WA, Davenport AP, Delagrange P, Dollery CT, Foord SM, Gutman GA, Laudet V, Neubig RR, Ohlstein EH, Olsen RW, Peters J, Pin JP, Ruffolo RR, Searls DB, Wright MW, Spedding M. Nucl Acids Res. 2009;37:D680–D685. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The IUPHAR database is http://www.iuphar-db.org/index.jsp.
- 43.The ChemblDB databases are at http://www.ebi.ac.uk/chembldb/index.php.
- 44.Black JW, Duncan WAM, Shanks RG. Br J Pharmacol. 1965;25:577–591. doi: 10.1111/j.1476-5381.1965.tb01782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.JChem and other ChemAxon software are available from ChemAxon at http://www.chemaxon.com/product/jc_base.html.
- 46.OEChem and other OpenEye software are available from OpenEye Scientific Software at http://www.eyesopen.com/.
- 47.Garcia-Serna R, Ursu O, Oprea TI, Mestres J. Bioinformatics. 2010;26:985–986. doi: 10.1093/bioinformatics/btq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The iPHACE interface from IMIM, http://cgl.imim.es/iphace/main.php, is mirrored at UNM, http://agave.health.unm.edu/iphace/main.php.
- 49.The IUPHAR database linked to Wikipedia, iPHACE and PharmKGB in May 2010
- 50.The U.S. Food and Drug Administration on-line search interface, “Drugs@FDA” service, can be accessed at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
- 51.EMA products authorized for human use can be accessed at http://www.ema.europa.eu/htms/human/epar/a.htm.
- 52.WHO Essential Medicines are listed at http://www.who.int/topics/essential_medicines/en/.
- 53.TGA medicines are found at http://www.tga.gov.au/
- 54.PDR is available at http://www.pdr.net/
- 55.Martindale is available at http://medicinescomplete.com/mc/martindale/current/
- 56.AHFS is available at http://medicinescomplete.com/mc/ahfs/current/
- 57.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. Mol Systems Biol. 2010;6:343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feinerer I, Hornik K, Meyer D. J Stat Software. 2008;25:1–54. [Google Scholar]
- 59.Jackson JE. A users guide to principal components. Wiley; New York: 1991. [Google Scholar]
- 60.Simca software is available from Umetrics at http://www.umetrics.com/
- 61.Kohonen T. Proc IEEE. 1990;78:1464–1480. [Google Scholar]
- 62.The Spotfire data analysis module is available from TIBCO at http://spotfire.tibco.com/
- 63.Wang X, Hripcsak G, Markatou M, Friedman C. J Am Med Inform Assoc. 2009;16:328–337. doi: 10.1197/jamia.M3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garcia-Serna R, Mestres J. Expert Opin Drug Metab Toxicol. 2010;6:1253–1263. doi: 10.1517/17425255.2010.509343. [DOI] [PubMed] [Google Scholar]
- 65.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cytoscape can be downloaded from http://www.cytoscape.org.
- 67.Chen B, Dong X, Jiao D, Wang H, Zhu Q, Ding Y, Wild DJ. BMC Bioinformatics. 2010;11:255. doi: 10.1186/1471-2105-11-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mewes HW, Wachinger B, Stümpflen V. Pharmacopsychiatry. 2010;43:S2–S8. doi: 10.1055/s-0030-1249666. [DOI] [PubMed] [Google Scholar]
- 69.Kuhn M, Szklarczyk D, Franceschini A, Campillos M, von Mering C, Jensen LJ, Beyer A, Bork P. Nucl Acids Res. 2009 doi: 10.1093/nar/gkp937. 10.1093/nar/gkp937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.STITCH. http://stitch.embl.de/